

Abstract
Pediatric metabolic syndrome (MS) and its cardiometabolic components (MSCs) have become increasingly prevalent, yet little is known about the genetics underlying MS risk in children. We examined the prevalence and genetics of MS-related traits among 670 non-diabetic Mexican American (MA) children and adolescents, aged 6–17 years (49 % female), who were participants in the San Antonio Family Assessment of Metabolic Risk Indicators in Youth (SAFARI) study. These children are offspring or biological relatives of adult participants from three well-established Mexican American family studies in San Antonio, Texas, at increased risk of type 2 diabetes. MS was defined as ≥ 3 abnormalities among 6 MSC measures: waist circumference, systolic and/or diastolic blood pressure, fasting insulin, triglycerides, HDL-cholesterol, and fasting and/or 2-h OGTT glucose. Genetic analyses of MS, number of MSCs (MSC-N), MS factors, and bivariate MS traits were performed. Overweight/obesity (53 %), pre-diabetes (13 %), acanthosis nigricans (33 %), and MS (19 %) were strikingly prevalent, as were MS components, including abdominal adiposity (32 %) and low HDL-cholesterol (32 %). Factor analysis of MS traits yielded three constructs: adipo-insulin-lipid, blood pressure, and glucose factors, and their factor scores were highly heritable. MS itself exhibited 68 % heritability. MSC-N showed strong positive genetic correlations with obesity, insulin resistance, inflammation, and acanthosis nigricans, and negative genetic correlation with physical fitness. MS trait pairs exhibited strong genetic and/or environmental correlations. These findings highlight the complex genetic architecture of MS/MSCs in MA children, and underscore the need for early screening and intervention to prevent chronic sequelae in this vulnerable pediatric population.
Introduction
The twin epidemics of obesity and type 2 diabetes mellitus (T2DM) have become global public health crises (Must et al. 1999; Zimmet 2003; Smyth and Heron 2006). Linked inexorably to them, metabolic syndrome (MS) – a clustering of cardiometabolic abnormalities including obesity, insulin resistance, impaired glucose tolerance, dyslipidemia, and hypertension – strongly predicts both T2DM and cardiovascular disease (CVD), and has itself become a third pandemic, with profound impact worldwide (Reaven 1988; DeFronzo 1995; Grundy 2007; DeBoer 2011). Obesity, insulin resistance and associated inflammatory processes are major underlying mechanisms of MS (DeFronzo 1995; Cruz et al. 2005; Yang and Ming 2011), which affects an estimated 77 to 86 million adults in the US, among whom minority groups, including Mexican Americans (MAs), bear a disproportionate burden (Ford et al. 2010).
A global surge in the prevalence of obesity among children and adolescents has pushed the prevalence of MS and its correlates among youth to unprecedented levels (Goran et al. 2004; Cruz et al. 2005; Ogden et al. 2010; DeBoer 2011). An estimated 8.6 % of the National Health and Nutrition Examination Survey (NHANES) 2001–2006 sample of adolescents aged 12–19 years were found to have MS; by extrapolation, approximately 2.5 million adolescents exhibited this condition nationally (Johnson et al. 2009). Minority youths were affected disproportionately with both obesity and MS (Johnson et al. 2009; Ogden et al. 2010). The overall prevalence of MS in NHANES was highest in Hispanics [11.2 %], followed by non-Hispanic European Americans [8.9 %] and non-Hispanic African Americans [4.0 %] (Johnson et al. 2009). Once MS and its risk factors develop in childhood, they may track into adulthood, with long-term health and economic consequences, including T2DM, CVD, and their sequelae (Weiss et al. 2004; Steinberger et al. 2009; Birnbaum et al. 2011). For this reason, the identification of children and adolescents at elevated risk of developing MS and related complications is critical for initiating early interventions to minimize future morbidity and mortality (Steinberger et al. 2009).
Defining MS for children and adolescents is challenging, however (Ford and Li 2008; DeBoer 2010; Kassi et al. 2011), and the utility of MS as a clinical construct has engendered ongoing debate (Simmons et al. 2010; Tenenbaum and Fisman 2011; Golden et al. 2012). Some investigators have suggested focusing instead on individual cardiometabolic risk factors, such as obesity, insulin resistance, dyslipidemia, glucose intolerance, and hypertension (Schutte et al. 2009; Steinberger et al. 2009; Hoffman 2009). Yet detailed knowledge of the genetic basis of cardiometabolic risk factors and phenotypic correlations between these factors in children has been extremely limited (Butte et al. 2005). Several studies, including our own, have shown that multiple correlated traits involved in MS may be influenced by common genetic (Duggirala et al. 2001; Arya et al. 2002; Farook et al. 2012) and environmental factors and their interactions.
Over the past 20 years, we have examined the genetics of obesity, T2DM, CVD, MS, and related traits in Mexican American adults in San Antonio, TX, using data from several studies of well-characterized families at increased risk of T2DM. There is evidence for a strong relationship between family history of T2DM and development of MS-related traits in children (Cruz et al. 2002; Goran et al. 2004; Valdez et al. 2007). We therefore conducted a genetic epidemiologic investigation of MS-related cardiometabolic risk factors in 673 MA children aged 6–17 years, who are the offspring or biological relatives of adult participants from three well-established MA family studies.
The major objectives of this study are: (1) to examine the prevalence of MS and related cardiometabolic risk factors in children and adolescents without T2DM, from these same T2DM-enriched MA families; (2) to examine the genetic basis of MS and related traits; (3) to investigate the genetic basis for correlations between the number of MS components (MSC-N) and measures of obesity, insulin resistance, inflammation, acanthosis nigricans and physical fitness; and (4) to assess the extent to which the clustering of MS-related risk factors is influenced by common genetic factors.
Materials and methods
Study population
The San Antonio Family Assessment of Metabolic Risk Indicators in Youth–the SAFARI study–was designed to evaluate cardiometabolic risk factors in children and teenagers, aged 6–17 years old, from predominantly lower-income extended MA families whose adult members had previously participated in one of three community-based genetic epidemiologic studies in San Antonio, TX: the San Antonio Family Diabetes/Gallbladder Study (SAFDGS), the San Antonio Family Heart Study (SAFHS), and the Veterans Administration Genetic Epidemiology Study (VAGES) (Mitchell et al. 1996; Puppala et al. 2006; Coletta et al. 2009). These children represent the youngest of multiple generations from their families to have taken part in our studies. All research procedures were approved by the Institutional Review Board of the University of Texas Health Science Center at San Antonio. Written informed consent was obtained from one or both parents of each child, and signed assent was obtained from children ≥7 years old, prior to the initiation of study assessments.
Family history, demographic, phenotypic, behavioral, and environmental data were obtained for SAFARI participants through both home and clinic questionnaires. Physical, clinical, and laboratory assessments were performed at the Children’s Center of the Texas Diabetes Institute (TDI), San Antonio, TX, USA, where blood samples were collected after a 10-h fast to measure glucose, insulin, lipids, and inflammatory markers. When possible, an oral glucose tolerance test (OGTT) was performed; plasma glucose was measured at −15 and 0 min before and at 30, 60, 90, and 120 min after an oral glucose load (1.75 g/kg body weight, to a maximum of 75 g). Plasma glucose was measured by glucose-oxidase method (Beckman Glucose Analyzer 2, Beckman, Fullerton, CA, USA). Serum samples were analyzed at Texas Biomedical Research Institute, San Antonio, using the following kits for: fasting (FI) and post-load specific serum insulin (30, 60, 90, and 120 min): RIA, Millipore, Billerica, MA, USA; total cholesterol (TC): H/P Reagent, Boehringer Mannheim, Indianapolis, IN, USA; HDL-cholesterol (HDL-C): H/P Reagent, Boehringer Mannheim, Indianapolis, IN, USA; triglycerides (TG): Stanbio Liquicolor Triglycerides, Boerne, TX, USA. LDL-cholesterol (LDL-C) was estimated using the Friedewald formula. Inflammatory markers, including serum high-sensitivity C-reactive protein (hs-CRP) (ELISA, Alpco immunoassay, Salem, NH, USA), and plasma C-peptide (Luminex, Millipore, Billerica, MA, USA) were also measured.
Anthropometric data, including height, weight, waist circumference, and systolic and diastolic blood pressure, were obtained using standard protocols. Body composition (fat mass, lean-body mass, percent body fat) was measured using dual-energy-X-ray absorptiometry (DXA Hologic). Pubertal status was assessed using Tanner staging. Presence/severity of acanthosis nigricans (AN) on the neck was evaluated using a scale previously developed and validated by our group (Burke et al. 1999). AN severity score (AN-SS) ranged from 0 to 5; the dichotomous trait AN was defined as AN-SS ≥2. A modified Harvard Step Test (Treviño et al. 2004) physical fitness score (PFS) was calculated as total duration of exercise in seconds × 100, divided by the sum of three post-exercise heart rates.
T2DM diagnosis required either fasting plasma glucose (FPG) ≥126 mg/dl or 2-h post-load plasma glucose (2-h PG) ≥200 mg/dl (WHO 1999; ADA 2002). MS was defined as presence of ≥3 of the following 6 dichotomized MSCs: increased waist circumference/abdominal obesity (≥90th percentile for age, sex, MA ethnicity) (Fernández et al. 2004); hyperinsulinemia (>75th percentile for total SAFARI cohort: ≥16.25 μIU/ml) (Goodman et al. 2004); glucose intolerance [impaired fasting glucose (IFG: FPG ≥100 and <126 mg/dl) or impaired glucose tolerance (IGT: 2-h PG ≥140 and <200 mg/dl), or both] (ADA 2003; Nathan et al. 2007); hypertriglyceridemia (≥110 mg/dl) (Cook et al. 2003); low HDL-cholesterol (≤40 mg/dl) (Cook et al. 2003); and elevated systolic and/or diastolic blood pressure (≥90th percentile for height, age and sex) (NHBPEP 2004). In addition, the number of MS components (range: 0–5 in our data) was used as a semi-quantitative trait (MSC-N). Measures of insulin resistance (HOMA-IR) and insulin sensitivity were derived using the University of Oxford Diabetes Trials Unit HOMA2-IR calculator (Matthews et al. 1985; http://www.dtu.ox.ac.uk/homacalculator) and the Matsuda composite whole-body insulin sensitivity index (ISI) (Matsuda and DeFronzo 1999), respectively. To assess early-phase insulin secretion, the insulinogenic index was calculated as the ratio of the increment in serum insulin to the increment in plasma glucose from 0 to 30 min post-load (Abdul-Ghani et al. 2009). BMI percentiles by sex and age were obtained from NHANES III. Participants below the ≥85th percentile were classified as normal, those >85th percentile and <95th percentile as overweight, those ≥95th percentile and <99th percentile as obese, and those >99th percentile as severely obese.
Statistical analyses
The genetics of MS-related quantitative traits were evaluated with a variance-components approach. In a simple model, variances or covariances between relatives as a function of the genetic relationships can be specified, and heritability (h2), defined as the proportion of phenotypic variance that is attributed to additive genetic effects, can be estimated from the components of variance (Falconer 1989; Almasy and Blangero 1998). A likelihood-ratio test was used to determine whether the heritability of a given phenotype was significant (P < 0.05). Covariates such as age, sex, age2, age × sex, age2 × sex, and pubertal status (defined using two dummy variables, with pre-pubertal status as the reference) were included in all analyses, if found to be significant. This variance-components method was extended to discrete traits such as MS, using a threshold or liability model (Duggirala et al. 1997).
Phenotypic (ρP), genetic (ρG), and environmental (ρE) correlations between MS-related continuous traits were determined using bivariate genetic analysis (Lange and Boehnke 1983). This approach can be extended to conduct bivariate analysis of quantitative and dichotomous traits (Williams et al. 1999). Phenotypic correlation (ρP) between a pair of traits is given by:
where ρP is the phenotypic correlation; ρG, the additive genetic correlation; ρE, the random environmental correlation; h21 is the heritability of trait 1; h22 is the heritability of trait 2; e21 is equal to 1−h21; and e22 is equal to 1−h22. Using likelihood-ratio tests, the significance (P < 0.05) of the phenotypic, additive genetic, and random environmental correlation was determined, respectively. Trait-specific significant covariates were included in bivariate analyses; pubertal status was included irrespective of its significance, for comparability across the trait-pairs examined. Univariate and bivariate procedures are incorporated in the program SOLAR (Almasy and Blangero 1998). We employed a principal component factor analysis (PCFA) (Arya et al. 2002) using SPSS19 to extract the underlying factors of 8 MS-related continuous traits that were used to define MS and its 6 MSCs. Factors with eigenvalues ≥1 were extracted and rotated using varimax rotation with Kaiser normalization. Factor correlations with the original trait, with factor loadings >0.40, were examined to determine which variables were important constituents of each factor. We estimated the heritability of each factor using the factor-specific scores as a quantitative trait, after adjusting for significant covariate influences.
Results
A total of 673 children and adolescents (SAFHS 373, SAFDGS 126, VAGES 174) participated in SAFARI. They came from 401 nuclear families/sibships, each of which contained an average of approximately 2 (range: 1–5) children. These sibships were embedded within the original SAFHS, SAFDGS, and VAGES extended families. SAFARI children generated a total of 3,664 relative pairs: 383 sibling pairs, 550 first-cousin pairs, 661 second-cousin pairs, and 662 third-cousin pairs (Table 1). Of the 673 examined participants, only 3 (0.45 %) met T2DM criteria. These 3 children were excluded from all analyses related to MS. Table 2 shows characteristics of the 670 children without T2DM. The mean age of the children was 11.5 years; 49.3 % were girls. In our sample, 52.7 % were overweight, obese, or severely obese (BMI ≥85th percentile); 33.6 % were either obese or severely obese (BMI ≥95th percentile); and 10.9 %, were severely obese (BMI ≥99th percentile). Prevalences of IFG, IGT, and prediabetes (IFG, IGT, or both) were 5.9 %, 12.0 %, and 13.2 %, respectively; 33.1 % exhibited AN. As reported in Table 2, data availability differed from trait to trait; data were transformed, when needed, using log-transformation or inverse normal transformation. All traits were significantly heritable (P < 0.05), except for 2-h insulin and 2-h PG, which were marginally significant. After adjusting for any significant covariate effects, significant heritability estimates ranged from 0.34 (hs-CRP) to 1.00 (AN).
Table 1.
Types and numbers of relative pairs among 673 SAFARI children and adolescents, aged 6 to 17 years old
| Type of Relative Pair | No. of Pairs |
|---|---|
| Siblings | 383 |
| Avuncular | 9 |
| Half-sibs | 86 |
| Half-avuncular | 5 |
| 1st cousins | 550 |
| 1st cousins, 1 rem | 234 |
| Half first cousins | 74 |
| 1st cousins, 2 rem | 2 |
| Half first cousins, 1 rem | 36 |
| 2nd cousins | 661 |
| 2nd cousins, 1 rem | 512 |
| Half second cousins | 178 |
| 3rd cousins | 662 |
| Half second cousins, 1 rem | 10 |
| 2nd cousins, 2 rem | 6 |
| 3rd cousins, 1 rem | 137 |
| Half third cousins | 6 |
| 4th cousins | 30 |
| Other relatives (third and fourth cousins, double third cousins and 2nd cousins, half-avuncular, and others) | 83 |
| Total | 3,664 |
Table 2.
Characteristics of 670 SAFARI children and adolescents, aged 6 to 17 years old, without diabetes, and heritability estimates for MS-related traits
| Variable* | N† | Mean ± SD or % | h2 ± SE‡ | P value | Significant covariates‡ |
|---|---|---|---|---|---|
| Girls | 670 | 49.3 | - | - | |
| Age (years) | 670 | 11.5 ± 3.5 | - | - | |
| Pre-pubertal (Tanner 1) | 635 | 49.8 | - | - | |
| Pubertal (Tanner 2–4) | 635 | 24.3 | - | - | |
| Post-pubertal (Tanner 5) | 635 | 26.0 | - | - | |
| BMI (kg/m2)§ | 670 | 22.7 ± 6.5 | 0.75 ± 0.11 | 1.1 × 10−11 | age, puberty |
| BMI z-score | 670 | 1.0 ± 1.1 | 0.73 ± 0.11 | 4.1 × 10−11 | age, sex, puberty |
| Overweight | 670 | 52.7 | 0.91 ± 0.16 | 2.1 × 10−8 | sex, age2, puberty |
| Obese | 670 | 33.6 | 0.78 ± 0.19 | 6.9 × 10−6 | sex, puberty |
| Waist circumference (mm) | 664 | 764.5 ± 179.7 | 0.63 ± 0.12 | 3.0 × 10−8 | age, age2, puberty |
| Fat mass (kg)§,|| | 634 | 16.0 ± 11.1 | 0.69 ± 0.12 | 1.8 × 10−9 | age, age2, puberty |
| Lean mass (kg)§,|| | 634 | 33.1 ± 13.4 | 0.61 ± 0.11 | 3.3 × 10−8 | age, sex, age × sex, age2 × sex, puberty |
| % Body fat|| | 634 | 30.2 ± 9.9 | 0.78 ± 0.11 | 1.6 × 10−12 | sex, age × sex, age2, age2 × sex |
| Fasting insulin (μIU/ml)§ | 626 | 13.6 ± 9.4 | 0.55 ± 0.11 | 2.0 ×10−7 | age2, puberty |
| 2-h insulin (μIU/ml)§ | 415 | 69.4 ± 65.4 | 0.21 ± 0.15 | 0.0744 | age2 |
| Fasting glucose (mg/dl)§ | 630 | 89.5 ± 7.5 | 0.39 ± 0.11 | 6.3 × 10−5 | age2 |
| 2-h glucose (mg/dl)§ | 418 | 115.4 ± 21.2 | 0.21 ± 0.14 | 0.0633 | - |
| Pre-diabetes# | 630 | 13.2 | 0.47 ± 0.25 | 0.0273 | puberty |
| HOMA-IR¶ | 622 | 2.0 ± 1.3 | 0.60 ± 0.11 | 1.8 × 10−8 | age2, puberty |
| Matsuda ISI¶ | 390 | 3.7 ± 1.7 | 0.56 ± 0.16 | 0.0001 | age2 |
| Insulinogenic index¶ | 396 | 2.1 ± 1.7 | 0.70 ± 0.15 | 9.0 × 10−6 | - |
| C-peptide (ng/ml)¶ | 514 | 1.0 ± 0.9 | 0.72 ± 0.13 | 1.0 × 10−7 | puberty |
| hs-CRP (ng/ml)¶ | 553 | 1620.1 ± 3220.6 | 0.34 ± 0.12 | 0.0009 | age, sex, age2, age2 × sex, puberty |
| HDL cholesterol (mg/dl) | 623 | 45.8 ± 10.9 | 0.64 ± 0.12 | 2.9 ×10−8 | age × sex, puberty |
| LDL cholesterol (mg/dl) | 623 | 87.1 ± 23.6 | 0.61 ± 0.12 | 1.0 × 10−7 | age, puberty |
| Total cholesterol (mg/dl) | 623 | 147.9 ± 27.1 | 0.61 ± 0.12 | 3.1 × 10−8 | puberty |
| Triglycerides (mg/dl)§ | 623 | 74.9 ± 39.8 | 0.77 ± 0.11 | 8.8 × 10−13 | age × sex |
| SBP (mm Hg) | 670 | 104.1 ± 9.7 | 0.66 ± 0.11 | 1.0 × 10−10 | age, sex, age × sex |
| DBP (mm Hg) | 670 | 63.2 ± 7.0 | 0.64 ± 0.11 | 9.7 × 10−10 | age, puberty |
| Harvard PFS | 501 | 47.7 ± 24.8 | 0.64 ± 0.14 | 2.7 × 10−6 | age, sex, age × sex, age2 |
| Acanthosis nigricans (AN) | 661 | 33.1 | 1.00 | 1.0 × 10−9 | - |
| AN severity score (AN-SS) | 660 | 1.3 ± 1.7 | 0.75 ± 0.11 | 3.6 × 10−12 | puberty |
| MS** | 625 | 18.7 | 0.68 ± 0.28 | 0.0078 | puberty |
| MSC-N** | 613 | 1.3 ± 1.4 | 0.54 ± 0.13 | 1.1 × 10−5 | puberty |
See text for definitions. BMI: body mass index; HOMA-IR: Homeostasis Model of Assessment - Insulin Resistance; Matsuda ISI: Matsuda Insulin Sensitivity Index; hs-CRP: high sensitivity C-reactive proetin; PFS: Physical fitness score;
values ± 4 SD from the mean were excluded from genetic analyses;
all traits were adjusted for significant covariate effects;
traits were log-transformed for genetic analyses;
as assessed by DXA;
traits were inverse-normalized for genetic analyses;
IFG: impaired fasting glucose (prevalence: 5.9 % [37/630]); IGT: impaired glucose tolerance (12.0 % [50/418]); pre-diabetes: IFG, IGT or both: (13.2 %);
The MSC-N analysis included 613/625 children since 12 children were lacking data for some of the examined MSCs. However, these 12 children were included in the MS analysis because they were found with 3 or more risk factors of the available data.
Prevalence of the 6 individual MS components (MSCs), based on available data, ranged from 11.9 % (elevated blood pressure) to 31.9 % (low HDL-C) (Table 3; Fig 1). Abdominal adiposity, low HDL-C, hypertriglyceridemia, and hyperinsulinemia were more prevalent in girls than in boys, who exhibited higher prevalence of glucose intolerance and elevated blood pressure. MS prevalence overall was 18.7 %, with girls (21.0 %) affected more than boys (16.6 %). MS prevalence increased substantially with rising BMI percentile category (Table 4), from 1.4 % among normal-weight to 8.9 % among overweight, 39.7 % among obese, and 67.7 % among severely obese children. Similarly, the prevalence of MSCs increased dramatically with rising BMI. In particular, the transition from overweight to obesity was associated with dramatic increases in cardiometabolic components, including a tripling of the prevalence of abdominal adiposity, 140 % greater prevalence of hyperinsulinemia, and 70 % higher prevalence of glucose intolerance. The prevalence of MS itself was quadrupled among obese, compared with overweight, children. Even among children categorized as normal-weight, however, a substantial proportion already were found with elevated blood pressure (7.9 %), hyperinsulinemia (10.7 %), low HDL-C (18.1 %), and glucose intolerance (10.8 %).
Table 3.
Prevalence of MS and its six criterion cardiometabolic components, among 670 SAFARI children and adolescents, aged 6 to 17 years old, without diabetes
| Variable | N | Prevalence (%) | ||
|---|---|---|---|---|
| Total | Girls | Boys | ||
| Abdominal obesity | 664 | 31.8 | 36.7 | 27.1 |
| Low HDL cholesterol | 623 | 31.9 | 35.2 | 28.8 |
| Hypertriglyceridemia | 623 | 13.8 | 14.5 | 13.2 |
| Hyperinsulinemia | 626 | 24.9 | 29.0 | 21.0 |
| Glucose intolerance | 630 | 13.2 | 12.1 | 14.2 |
| Elevated blood pressure | 670 | 11.9 | 10.9 | 12.9 |
| MS | 625 | 18.7 | 21.0 | 16.6 |
Figure 1.
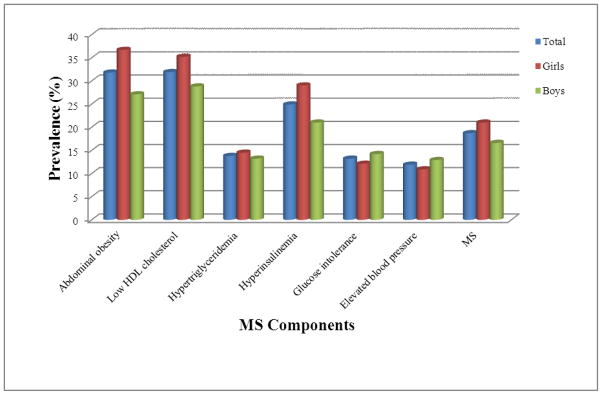
Prevalence of MS and its six cardiometabolic components, overall and by sex, among 670 SAFARI children and adolescents, aged 6 to 17 years old, without diabetes
Table 4.
Prevalence of MS and its six criterion cardiometabolic components among SAFARI children and adolescents, aged 6 to 17 years old, without diabetes, by BMI percentile category
| MS Components | MS prevalence (%) by BMI percentile: | |||||||
|---|---|---|---|---|---|---|---|---|
| <85th | ≥85th –<95th | ≥95th –<99th | >99th | |||||
| Normal | Overweight | Obese | Severely Obese | |||||
| N | % | N | % | N | % | N | % | |
| Abdominal obesity | 313 | 1.0 | 127 | 18.9 | 152 | 74.3 | 72 | 98.6 |
| Low HDL cholesterol | 288 | 18.1 | 123 | 27.6 | 146 | 47.3 | 66 | 66.7 |
| Hypertriglyceridemia | 288 | 3.5 | 123 | 16.3 | 146 | 22.6 | 66 | 34.8 |
| Hyperinsulinemia | 291 | 10.7 | 124 | 18.5 | 146 | 44.5 | 65 | 56.9 |
| Glucose intolerance | 296 | 10.8 | 124 | 10.5 | 145 | 17.9 | 65 | 18.5 |
| Elevated blood pressure | 317 | 7.9 | 128 | 9.4 | 152 | 14.5 | 73 | 28.8 |
| MS (≥ 3 MS components) | 290 | 1.4 | 124 | 8.9 | 146 | 39.7 | 65 | 67.7 |
As shown in Table 2, both MS and the number of its components exhibited by a child (MSC-N) were under strong additive genetic influences: h2 for MS = 0.68, for MSC-N = 0.54. We also examined the extent to which MSC-N correlated with measures of obesity (overweight and obese), body composition (fat mass, lean mass, and % body fat), insulin action and secretion (HOMA-IR, Matsuda ISI, insulinogenic index, and C-peptide), inflammation (hs-CRP), acanthosis nigricans (AN-SS), and physical fitness (PFS). We partitioned phenotypic correlations (ρPs) between MSC-N and the above traits into their genetic (ρG) and environmental (ρE) correlations. The ρPs between MSC-N and these traits ranged from −0.26 (Harvard PFS) to 0.73 (obese), and were highly statistically significant (Table 5). As expected, MSC-N is inversely correlated with both Matsuda ISI and PFS, while positively correlated with the other traits. All ρPs were largely and significantly influenced by common genetic factors; ρGs ranged from −0.45 (PFS) to 0.84 (obesity). Only the ρEs between MSC-N and fat mass, % body fat, HOMA-IR, and Matsuda ISI were significant; these influenced the ρPs substantially.
Table 5.
Phenotypic (ρP), genetic (ρG), and environmental (ρE) correlations between number of MS components (MSC-N) and measures of obesity and body composition, insulin resistance and insulin secretion, inflammation, acanthosis nigricans and physical fitness, among SAFARI children and adolescents, aged 6 to 17 years old, without diabetes
| Trait Paira | ρP | P value | ρG ± SE | P value | ρE ± SE | P value |
|---|---|---|---|---|---|---|
| MSC-N: Overweight | 0.66 | 5.2 × 10−37 | 0.83 ± 0.13 | 6.5 × 10−6 | 0.49 ± 0.30 | 0.2261 |
| MSC-N: Obese | 0.73 | 4.6 ×10−49 | 0.84 ± 0.11 | 1.5 × 10−5 | 0.59 ± 0.22 | 0.0711 |
| MSC-N: Fat massb | 0.64 | 6.7 × 10−52 | 0.73 ± 0.11 | 3.3 × 10−5 | 0.54 ± 0.15 | 0.0183 |
| MSC-N: Lean massc | 0.46 | 5.6 × 10−27 | 0.58 ± 0.13 | 0.0009 | 0.31 ± 0.17 | 0.1263 |
| MSC-N: %Body fat | 0.60 | 9.0 × 10−44 | 0.58 ± 0.11 | 0.0003 | 0.67 ± 0.15 | 0.0091 |
| MSC-N: HOMA-IRc | 0.56 | 5.7 × 10−42 | 0.60 ± 0.11 | 0.0007 | 0.51 ± 0.14 | 0.0180 |
| MSC-N: Matsuda ISIc | −0.56 | 1.5 × 10−28 | −0.58 ± .015 | 0.0060 | −0.53 ± 0.16 | 0.0176 |
| MSC-N: Insulinogenic indexc | 0.32 | 7.5 × 10−10 | 0.41 ± 0.19 | 0.0499 | 0.20 ± 0.23 | 0.4057 |
| MSC-N: C-peptidec | 0.32 | 1.0 × 10−11 | 0.49 ± 0.15 | 0.0055 | 0.04 ± 0.26 | 0.8937 |
| MSC-N – hs-CRPc | 0.37 | 1.6 × 10−17 | 0.59 ± 0.17 | 0.0082 | 0.22 ± 0.14 | 0.1761 |
| MSC-N: AN-SS | 0.46 | 3.4 × 10−26 | 0.54 ± 0.14 | 0.0007 | 0.37 ± 0.21 | 0.1267 |
| MSC-N: Harvard PFS | −0.26 | 2.9 × 10−8 | −0.45 ± 0.16 | 0.0063 | 0.06 ± 0.24 | 0.8078 |
All traits were adjusted for the covariate effects as described in the text;
data were log-transformed;
data were transformed using inverse normal transformation.
Using PCFA, we examined factor structures that underlie correlations among the 8 quantitative traits (WC, FI, FPG, 2-h PG, HDL-C, TG, SBP, and DBP) used to define MSCs. By comparison, we analyzed the same 8 traits from non-T2DM adults from the three parental cohorts of SAFARI. Data for both children and adults were log-transformed, for comparability. Since data for all traits are required for PCFA and 2-h PG data were not available for all children, these analyses were based on a reduced SAFARI sample (N = 412). For adults, data for all 8 traits were available for 1,803 individuals. PCFA yielded three very similar factors for children and adults (Table 6). Among factor loadings >0.40, the first extracted factor (Factor 1) exhibited high positive correlations with WC, FI, and TG, and negative correlation with HDL-C, as expected; we have interpreted this factor as an adipo-insulin-lipid factor. Factor 2 showed high positive correlations with only DBP and SBP and thus represents a blood pressure factor. Factor 3, which exhibited high positive correlations with FPG and 2-h PG, represents a glucose factor. These three factors together explained approximately 65.0 % of variation in both data sets. After adjusting for significant covariate effects, significant heritabilities (P < 0.05) were found for these factors in both children and adults, respectively: adipo-insulin-lipid factor: 0.76 and 0.58; blood pressure factor: 0.68 and 0.41; and glucose factor: 0.36 and 0.46 (Table 6).
Table 6.
Principal-component factor analyses of MS-related quantitative traits in SAFARI children and adolescents, and comparison of SAFARI factor structures with those obtained from non-diabetic adult relatives who had previously participated in SAFHS, SAFDGS, or VAGES
| Factor loadings* | ||||||
|---|---|---|---|---|---|---|
| Phenotype* | SAFARI Children† | Adults (SAFHS/SAFDGS/VAGES) ‡ | ||||
| Factor 1 | Factor 2 | Factor 3 | Factor 1 | Factor 2 | Factor 3 | |
| WC | 0.706 | 0.358 | 0.042 | 0.543 | 0.390 | 0.287 |
| HDL-C | −0.786 | −0.011 | 0.070 | −0.779 | 0.062 | 0.042 |
| TG | 0.744 | 0.056 | 0.156 | 0.706 | 0.211 | 0.096 |
| FI | 0.619 | 0.134 | 0.314 | 0.631 | 0.066 | 0.350 |
| FPG | 0.096 | 0.108 | 0.744 | 0.175 | 0.337 | 0.797 |
| 2-h PG | 0.080 | −0.041 | 0.764 | 0.076 | 0.122 | 0.836 |
| SBP | 0.194 | 0.905 | 0.094 | 0.088 | 0.845 | 0.221 |
| DBP | 0.084 | 0.906 | −0.003 | 0.104 | 0.874 | 0.030 |
| Eigenvalue | 2.1 | 1.8 | 1.3 | 2.9 | 1.2 | 1.0 |
| Variance explained (%) | 26.4 | 22.5 | 16.0 | 36.3 | 15.4 | 13.0 |
| Cumulative variance (%) | 26.4 | 48.9 | 64.9 | 36.3 | 51.6 | 64.6 |
| Construct | adipo-insulin-lipid factor | blood pressure factor | glucose factor | adipo-insulin-lipid factor | blood pressure factor | glucose factor |
| h2 ± S.E (P value) | 0.76 ± 0.15 (9.0 × 10−7) | 0.68 ± 0.14 (5.0 × 10−7) | 0.36 ± 0.15 (4.8 × 10−3) | 0.58 ± 0.05 (2.6 × 10−49) | 0.41± 0.05 (2.4 × 10−22) | 0.46 ± 0.05 (3.5 × 10−34) |
| Significant covariates | age × sex puberty | age | age2 | age sex age2 | age sex age × sex age2 | age sex |
Factor loadings in bold type are >0.40, factors in adults are shown for comparison; all traits in children and adults were log-transformed;
Sample sizes for trait-specific analyses varied from 418 for 2-h PG to 670 for SBP and DBP. However, data for all 8 traits were available for only 412 children, because of number of participants for whom OGTT could be performed (N = 418);
Only non-diabetic individuals included and sample sizes of the traits varied from 1,941 for 2-hr glucose to 2,061 for SBP and DBP. However, data for all 8 traits were available for 1,803 individuals.
In addition, we conducted bivariate genetic analyses to determine ρPs, ρGs, and ρEs among the same 8 MS-related traits in SAFARI children; Table 7 displays results for 28 trait-pairs. Except for DBP:FPG, all ρPs were statistically significant (P < 0.05); as expected, HDL-C was negatively correlated with other traits. Nine trait-pairs exhibited significant ρGs: WC with HDL-C (−0.45), TG (0.52), FI (0.53), and 2-h PG (−0.69); SBP with DBP (0.88) and TG (0.32); DBP with HDL-C (−0.28) and TG (0.34); and HDL-C with TG (−0.43); ρG is marginally significant for WC:SBP (0.25). These findings reflect potential pleiotropic influences on these MS trait-pairs. The following 5 trait-pairs were found to be under significant common non-shared environmental influences (ρEs): WC:2-h PG (0.82), TG:FI (0.58), TG:2-h PG (0.49), FPG:FI (0.32), and FI:2-h PG (0.36). The ρEs involving SBP:DBP (0.47) and FPG:2-h PG (0.26) were marginally significant. Of the trait-pairs that exhibited significant ρGs or ρEs, only ρP of the trait pair WC:2-h PG is influenced by both ρG and ρE, while the other trait pair ρPs were largely influenced by ρGs or ρEs as discussed above.
Table 7.
Phenotypic (ρP), genetic (ρG), and environmental (ρE) correlations among 8 MS-related quantitative traits used for MS factor analysis in SAFARI children and adolescents without diabetes
| Trait Pair* | ρP | P value | ρG ± SE | P value | ρE ± SE | P value |
|---|---|---|---|---|---|---|
| WC: SBP | 0.26 | 6.3 × 10−10 | 0.25 ± 0.14 | 0.0772 | 0.28 ± 0.20 | 0.1975 |
| WC: DBP | 0.19 | 5.8 × 10−6 | 0.20 ± 0.14 | 0.1566 | 0.17 ± 0.20 | 0.4083 |
| WC: HDL-C | −0.36 | 2.2 × 10−17 | −0.45 ± 0.13 | 0.0034 | −0.21 ± 0.21 | 0.3735 |
| WC: TG† | 0.41 | 6.7 × 10−22 | 0.52 ± 0.11 | 0.0002 | 0.15 ± 0.29 | 0.6285 |
| WC: FPG† | 0.10 | 0.0150 | 0.04 ± 0.18 | 0.8334 | 0.18 ± 0.17 | 0.2774 |
| WC: FI† | 0.42 | 3.0 × 10−24 | 0.53 ± 0.12 | 0.0007 | 0.27 ± 0.17 | 0.1808 |
| WC: 2-h PG† | 0.18 | 0.0004 | −0.69 ± 0.40 | 0.0221 | 0.82 ± 0.20 | 1.8 × 10−5 |
| SBP: DBP | 0.74 | 6.6 × 10−89 | 0.88 ± 0.05 | 8.8 × 10−10 | 0.47 ± 0.15 | 0.0642 |
| SBP: HDL-C | −0.12 | 0.0045 | −0.14 ± 0.14 | 0.3439 | −0.10 ± 0.22 | 0.6502 |
| SBP: TG† | 0.19 | 9.2 × 10−6 | 0.32 ± 0.12 | 0.0140 | −0.16 ± 0.32 | 0.6009 |
| SBP: FPG† | 0.14 | 0.0009 | 0.03 ± 0.17 | 0.8619 | 0.28 ± 0.16 | 0.0943 |
| SBP: FI† | 0.20 | 3.0 × 10−6 | 0.19 ± 0.14 | 0.1904 | 0.22 ± 0.18 | 0.2626 |
| SBP: 2-h PG† | 0.16 | 0.0024 | 0.10 ± 0.32 | 0.7591 | 0.24 ± 0.18 | 0.1958 |
| DBP: HDL-C | −0.14 | 0.0015 | −0.28 ± 0.14 | 0.0485 | 0.13 ± 0.22 | 0.5538 |
| DBP: TG† | 0.18 | 3.6 × 10−5 | 0.34 ± 0.13 | 0.0115 | −0.22 ± 0.31 | 0.4339 |
| DBP: FPG† | 0.07 | 0.1216 | 0.06 ± 0.17 | 0.7453 | 0.08 ± 0.16 | 0.6106 |
| DBP: FI† | 0.13 | 0.0036 | 0.14 ± 0.15 | 0.3341 | 0.10 ± 0.18 | 0.5964 |
| DBP: 2-h PG† | 0.10 | 0.0455 | 0.01 ± 0.34 | 0.9775 | 0.18 ± 0.17 | 0.2912 |
| HDL: TG† | −0.42 | 1.1 × 10−22 | −0.43 ± 0.11 | 0.0019 | −0.43 ± 0.24 | 0.1682 |
| HDL: FPG† | −0.09 | 0.0293 | −0.004 ± 0.18 | 0.9844 | −0.20 ± 0.17 | 0.2445 |
| HDL: FI† | −0.22 | 2.9 × 10−7 | −0.17 ± 0.15 | 0.2897 | −0.30 ± 0.18 | 0.1302 |
| HDL: 2-h PG† | −0.13 | 0.0142 | −0.10 ± 0.34 | 0.7608 | −0.18 ± 0.18 | 0.3111 |
| TG†: FPG† | 0.12 | 0.0042 | 0.004 ± 0.17 | 0.9818 | 0.36 ± 0.23 | 0.1136 |
| TG†: FI† | 0.30 | 1.2 × 10−11 | 0.18 ± 0.14 | 0.2032 | 0.58 ± 0.22 | 0.0280 |
| TG†: 2-h PG† | 0.18 | 0.0004 | −0.04 ± 0.31 | 0.8956 | 0.49 ± 0.24 | 0.0324 |
| FPG†: FI† | 0.19 | 7.1 × 10−6 | 0.05 ± 0.18 | 0.7976 | 0.32 ± 0.14 | 0.0280 |
| FPG†: 2-h PG† | 0.21 | 6.5 × 10−5 | 0.08 ± 0.40 | 0.8421 | 0.26 ± 0.13 | 0.0580 |
| FI†: 2-h PG† | 0.22 | 6.0 × 10−6 | −0.004 ± 0.36 | 0.9911 | 0.36 ± 0.14 | 0.0181 |
All data were adjusted for covariate effects as discussed in the text;
data were log-transformed.
Discussion
SAFARI children bear substantial cardiometabolic and related burdens: 53 % were overweight/obese; 34 %, obese; 19 % had MS; 13 %, pre-diabetes; and 33 %, acanthosis nigricans. Although several adult genetic studies of MS using family data have been conducted, comparable data are rare for pediatric populations (Butte et al. 2005). Yet such data are of special interest, because complex traits among children and adolescents are less subject to long-term aging-related influences. Our results indicate substantial additive genetic influences on MS-related traits. The moderate-to-high heritability estimates we observed (Table 2) are comparable to those available from two other pediatric studies (Butte et al. 2005; Beardsall et al. 2009).
As discrete traits, MS and its components have been examined within a number of pediatric studies, including selected samples of high-risk children and/or nationally representative data sets such as NHANES. Although its prevalence has varied with the definitions used (Cook et al. 2008; Kassi et al. 2011), MS prevalence has been shown to increase with rising BMI, and is highest among overweight and obese children and adolescents (Weiss et al. 2004; Cruz et al. 2004; Butte et al. 2005). In SAFARI data, MS increased dramatically from 1.4 % in normal-weight children to 67.7 % in severely obese children. Striking elevations in the prevalence of both MS and individual components occurred in obese, compared with overweight, children. The prevalence of glucose intolerance exhibited remarkable increases in this transition, despite corresponding but dramatically higher prevalence of hyperinsulinemia among obese, compared with overweight, children. Even the very young were affected: one third of SAFARI children with MS were less than 10 years old, and even three 6-year-olds were affected.
Comparison of MS prevalence among different study populations is complicated by differences in participants’ ages, weight, and ethnicity, and in the definition of MS. Weiss et al. (2004) found that pediatric MS prevalence varied from 38 to 49 %, depending on participant obesity level. Cook et al. (2008), analyzing data for obese adolescents in NHANES (1999–2002), reported MS prevalences ranging from 12.4 % to 44.2 %, depending upon MS definition. Among Hispanic youth in NHANES (2001–2006), the prevalence of MS was 11.2 % (Johnson et al. 2009). Among overweight Hispanic children in Viva La Familia (age 4 to 19 years), the prevalence of MS was 28 % in boys, and 27 % in girls (Butte et al. 2005). In a study of overweight Hispanic children (8–13 years), the prevalence was 30 % (Cruz et al. 2004). By comparison, the prevalence of MS in SAFARI was 18.7 %., but was higher in girls (21.0 %) than in boys (16.6 %).
In contrast to population-based studies of MS and its components, the uniqueness of the SAFARI study is its ability to disentangle the genetic architecture of cardiometabolic risk represented by MS and its components. We found evidence for strong additive genetic factors influencing the MS phenotype (h2 = 68 %), as well as overall cardiometabolic burden, as measured by number of MS components (MSC-N: h2 = 54 %) in children and adolescents. In bivariate genetic analyses of MSC-N and its correlates, we found evidence of pleiotropy: substantial contribution of common additive genetic factors to the positive phenotypic correlations between MSC-N and measures of obesity (overweight, obese, fat mass, lean mass, % body fat), insulin resistance (HOMA-IR) and secretion (insulinogenic index, C-peptide), inflammation (hs-CRP), and acanthosis nigricans, and to the negative phenotypic correlations between MSC-N and both whole-body insulin sensitivity (Matsuda ISI) and physical fitness (Harvard PFS). These correlation estimates should be interpreted with caution, however, because of redundancies between insulin resistance measures and the MS component based on fasting insulin, and between abdominal obesity and measures of overweight and obesity based on BMI percentile categories. Several other studies also have observed positive phenotypic associations with MSC-N or MS for both inflammation and acanthosis nigricans, and inverse phenotypic associations between MS prevalence and both insulin sensitivity and low physical activity (Weiss et al. 2004; Cruz et al. 2004; Shaibi et al. 2008; Ice et al. 2009; Brambilla et al. 2011; DeBoer 2011). Our bivariate genetic analyses, however, signify the strong common genetic basis for the complex associations between increasing number of MS components and the concomitant risks associated with the examined correlates of MS. In general, the main contribution to observed phenotypic correlations is genetic, and the magnitude of these genetic correlations is most pronounced for obesity-related variables, with somewhat lower genetic correlations for insulin resistance and sensitivity measures.
To detect the underlying structure among the correlated MS-related cardiometabolic traits, several studies used either factor analysis or a confirmatory factor analysis (Ford and Li 2008; Gurka et al. 2012). Reviewed by Ford and Li (2008), such studies used definitions which included a wide range of MS components (5–19) and 1–5 derived factors. Using data from 8 MS traits, our analyses yielded three factors - adipo-insulin-lipid, blood pressure, and glucose – which were essentially replicated using data from non-diabetic adult family members. Such structural patterns reveal complex interrelationships among these cardiometabolic risk factors, which are already evident very early in life in our SAFARI children. In both data sets, the factor-specific scores were influenced by substantial additive genetic factors.
We employed bivariate genetic analyses of the eight phenotypically correlated traits in SAFARI children to provide additional insights into their complex interrelationships. In general, these traits exhibited significant phenotypic correlations, some of which were predominantly influenced by genetic correlations, and others by environmental correlations. The four traits (WC, FI, TG, and HDL-C) loaded on factor 1 (adipo-insulin-lipid factor) exhibited significant genetic correlations, including negative correlations between HDL-C and certain traits. While the blood pressure measures (SBP and DBP) loaded on factor 2 were strongly genetically correlated, it is evident that they also shared common genetic influences with lipid traits, including the inverse genetic association between DBP and HDL-C. Glucose traits (factor 3: FPG and/or 2-h PG) were environmentally correlated with obesity, insulin, and lipids in these children, but the positive phenotypic correlation between WC and 2-h PG was largely influenced by negative genetic correlation and positive environmental correlation, in turn suggesting that genetic and environmental sources of variation influence these traits through different physiological mechanisms (Falconer 1989). It is plausible that the observed inverse genetic correlation between WC and 2-h PG is related to the cascade of events related to hyperinsulinemia, reactive hypoglycemia, hyperphagia, and obesity (Ludwig 2002; Chaput et al. 2008).
In recent years, numerous genome-wide association studies of individual traits related to MS have successfully localized susceptibility variants/genes for such phenotypes as obesity, T2DM, dyslipidemia, and hypertension, as well as childhood obesity; a few studies have also localized variants/genes with potential pleiotropic influences on MS (McCarthy 2010; Zeller et al. 2012; Fall and Ingelsson 2012; Norris and Rich 2012; Manco and Dallapiccola 2012). Our immediate plans are to conduct genome-wide screenings for the identification of genetic variants influencing individual MS traits - most importantly, those pleiotropically influencing multivariate MS traits in our data. Such efforts should yield significant insights into the genetic mechanisms underlying the complex metabolic pathways related to MS trait clustering patterns.
Obesity and insulin resistance are the major intertwined underlying processes of MS. Insulin resistance represents a core component of metabolic risk which precedes, and lays the groundwork for, the later development of such life-threatening diseases as T2DM and CVD. By the time glucose levels have risen to the diagnostic threshold of glucose intolerance, β-cell function has already become severely compromised (Abdul-Ghani and DeFronzo 2009). One in every 8 SAFARI participants has already reached this stage. The high rate of prediabetes in this and other studies (Abdul-Ghani and DeFronzo 2009; Li et al. 2009) strongly suggests that –by screening high-risk children for insulin resistance – lifestyle and/or pharmacologic interventions could be initiated during the closing window of opportunity in late adolescence and early adulthood, to prevent later development of T2DM and vascular dysfunction.
Limitations of our study include the following: our estimate of the prevalence of IGT/pre-diabetes may be biased, since OGTT data were available for only 62 % of SAFARI children. It is possible that heritability estimates may have been inflated since shared environmental influences were not accounted for in our analyses. Such influences, however, appear to be minimal in this study, since the children are distributed across large pedigrees, represented by a wide variety of relative pairs, and sib-pairs account for only 10 % of the relative pairs examined. In addition, SAFARI children are from families residing predominantly in lower-income neighborhoods, and higher heritability can be expected when environmental conditions are relatively uniform. It should be reiterated that no definition of MS in children and adolescents has been fully validated; in fact, there have been continued concerns about the diagnostic criteria for pediatric MS, including their usefulness for treatment purposes, and the degree to which estimates of the prevalence of MS vary with the definitions used (Ford and Li 2008; Brambilla and Pietrobelli 2009; DeBoer 2010; Kassi et al. 2011).
In conclusion, we have demonstrated dramatic increases in the prevalence of MS and its individual risk components with increasing age and adiposity. Even very young SAFARI participants, however, and those categorized as normal-weight, exhibited cardiometabolic risk indicators, including dyslipidemia, hyperinsulinemia, and glucose intolerance. Our findings highlight the complex genetic architecture of MS and its risk components in MA children, and underscore the need to identify early-warning cardiometabolic risk indicators, and to screen normal-weight, as well as overweight and/or obese, children for them (Steinberger et al. 2009; Goodman et al. 2009; May et al. 2012). Early detection of cardiometabolic risk could provide a life-changing window of opportunity in which to intervene to interrupt the cascade of metabolic dysfunction which these conditions all too often bring in their wake. Given the observed burden of MS and its risk factors in our SAFARI children, our goals for the near future include implementation of effective intervention strategies in this vulnerable population. In addition, identification of specific genetic factors underlying the phenotypic expressions of MS risk factors may increase the potential to identify genetic predictors of an individual’s response to dietary, physical activity, and other lifestyle interventions, and thus facilitate the tailoring of individualized programs for both prevention and treatment interventions. Such targeted early-life interventions could have significant clinical impact on cardiometabolic risk, not only in childhood but also in adulthood.
Acknowledgments
This study was supported by grants from the National Institutes of Health (R01 HD049051, HD041111, DK053889, DK042273, K01DK064867, P01 HL045522, DK047482, MH059490, M01-RR-01346, and HD049051-5S1 (ARRA). This work was also supported by a Veterans Administration Epidemiologic grant to R.A.D. In addition, we thank for their support the Fraternal Order of Eagles San Antonio Aerie and Auxiliary. We thank the University Health System and the Texas Diabetes Institute, San Antonio, Texas for extending their excellent facilities to our study. The AT&T Genomics Computing Center supercomputing facilities used for this work were supported in part by a gift from the AT&T Foundation and with support from the National Center for Research Resources Grant Number S10 RR029392. This investigation was conducted in facilities constructed with support from Research Facilities Improvement Program grants C06 RR013556 and C06 RR017515 from the National Center for Research Resources of the National Institutes of Health. We thank Dr. William Rogers, Dr. Rolando Lozano, Dr. Nancy Butte, Anne Adolph, Richard Granato, Margaret Fragoso, David Rupert, Rhonda Lyons, Tanya Prado, Elizabeth Sosa, Bonnie Sanchez, and Nicolas Ballí for their excellent help and assistance. Lastly, and most importantly, we are deeply indebted to the children, teenagers, parents, and extended family members of the SAFARI study, whose great enthusiasm and commitment have made this research possible.
Footnotes
Conflict of interest
The authors have no conflicts of interest to disclose.
References
- Abdul-Ghani MA, DeFronzo RA. Plasma glucose concentration and prediction of future risk of type 2 diabetes. Diabetes Care. 2009;32:S194–198. doi: 10.2337/dc09-S309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Ghani MA, Lyssenko V, Tuomi T, DeFronzo RA, Groop L. Fasting versus postload plasma glucose concentration and the risk for future type 2 diabetes: results from the Botnia Study. Diabetes Care. 2009;32:281–286. doi: 10.2337/dc08-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association. clinical practice recommendations. Diabetes Care. 2002;25(Suppl 1):S1–S147. doi: 10.2337/diacare.25.2007.s1. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. screening for type 2 diabetes. Diabetes Care. 2003;26(Suppl 1):S21–S24. doi: 10.2337/diacare.26.2007.s21. [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya R, Blangero J, Williams K, Almasy L, Dyer TD, Leach RJ, O’Connell P, Stern MP, Duggirala R. Factors of insulin resistance syndrome--related phenotypes are linked to genetic locations on chromosomes 6 and 7 in nondiabetic mexican-americans. Diabetes. 2002;51:841–847. doi: 10.2337/diabetes.51.3.841. [DOI] [PubMed] [Google Scholar]
- Beardsall K, Ong KK, Murphy N, Ahmed ML, Zhao JH, Peeters MW, Dunger DB. Heritability of childhood weight gain from birth and risk markers for adult metabolic disease in prepubertal twins. J Clin Endocrinol Metab. 2009;94:3708–3713. doi: 10.1210/jc.2009-0757. [DOI] [PubMed] [Google Scholar]
- Birnbaum HG, Mattson ME, Kashima S, Williamson TE. Prevalence rates and costs of metabolic syndrome and associated risk factors using employees’ integrated laboratory data and health care claims. J Occup Environ Med. 2011;53:27–33. doi: 10.1097/JOM.0b013e3181ff0594. [DOI] [PubMed] [Google Scholar]
- Brambilla P, Pietrobelli A. Behind and beyond the pediatric metabolic syndrome. Italian J Pediatr. 2009;35:41. doi: 10.1186/1824-7288-35-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla P, Pozzobon G, Pietrobelli A. Physical activity as the main therapeutic tool for metabolic syndrome in childhood. Int J Obes (Lond) 2011;35:16–28. doi: 10.1038/ijo.2010.255. [DOI] [PubMed] [Google Scholar]
- Burke JP, Hale DE, Hazuda HP, Stern MP. A quantitative scale of acanthosis nigricans. Diabetes Care. 1999;22:1655–1659. doi: 10.2337/diacare.22.10.1655. [DOI] [PubMed] [Google Scholar]
- Butte NF, Comuzzie AG, Cole SA, Mehta NR, Cai G, Tejero M, Bastarrachea R, Smith EO. Quantitative genetic analysis of the metabolic syndrome in Hispanic children. Pediatr Res. 2005;58:1243–1248. doi: 10.1203/01.pdr.0000185272.46705.18. [DOI] [PubMed] [Google Scholar]
- Chaput JP, Tremblay A, Rimm EB, Bouchard C, Ludwig DS. A novel interaction between dietary composition and insulin secretion: effects on weight gain in the Quebec Family Study. Am J Clin Nutr. 2008;87:303–309. doi: 10.1093/ajcn/87.2.303. [DOI] [PubMed] [Google Scholar]
- Coletta DK, Schneider J, Hu SL, Dyer TD, Puppala S, Farook VS, Arya R, Lehman DM, Blangero J, DeFronzo RA, Duggirala R, Jenkinson CP. Genome-wide linkage scan for genes influencing plasma triglyceride levels in the Veterans Administration Genetic Epidemiology Study. Diabetes. 2009;58:279–284. doi: 10.2337/db08-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S, Auinger P, Li C, Ford ES. Metabolic syndrome rates in United States adolescents, from the National Health and Nutrition Examination Survey, 1999–2002. J Pediatr. 2008;152:165–170. doi: 10.1016/j.jpeds.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Cook S, Weitzman M, Auinger P, Nguyen M, Dietz WH. Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch Pediatr Adolesc Med. 2003;157:821–827. doi: 10.1001/archpedi.157.8.821. [DOI] [PubMed] [Google Scholar]
- Cruz ML, Shaibi GQ, Weigensberg MJ, Spruijt-Metz D, Ball GD, Goran MI. Pediatric obesity and insulin resistance: chronic disease risk and implications for treatment and prevention beyond body weight modification. Annu Rev Nutr. 2005;25:435–468. doi: 10.1146/annurev.nutr.25.050304.092625. [DOI] [PubMed] [Google Scholar]
- Cruz ML, Weigensberg MJ, Huang TT, Ball G, Shaibi GQ, Goran MI. The metabolic syndrome in overweight Hispanic youth and the role of insulin sensitivity. J Clin Endocrinol Metab. 2004;89:108–113. doi: 10.1210/jc.2003-031188. [DOI] [PubMed] [Google Scholar]
- Cruz ML, Huang TT, Johnson MS, Gower BA, Goran MI. Insulin sensitivity and blood pressure in black and white children. Hypertension. 2002;40:18–22. doi: 10.1161/01.hyp.0000019972.37690.ef. [DOI] [PubMed] [Google Scholar]
- Deboer MD. Underdiagnosis of Metabolic Syndrome in Non-Hispanic Black Adolescents: A Call for Ethnic-Specific Criteria. Curr Cardiovasc Risk Rep. 2010;4:302–310. doi: 10.1007/s12170-010-0104-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoer MD. Ethnicity, obesity and the metabolic syndrome: implications on assessing risk and targeting intervention. Expert Rev Endocrinol Metab. 2011;6:279–289. doi: 10.1586/eem.11.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA. Insulin resistance and hyperinsulinemia: the link between NIDDM, CAD, hypertension and dyslipidemia. In: Schwartz CL, Born GVR, editors. New horizons in diabetes mellitus and cardiovascular disease. Current Science; London: 1995. pp. 11–27. [Google Scholar]
- Duggirala R, Blangero J, Almasy L, Arya R, Dyer TD, Williams KL, Leach RJ, O’Connell P, Stern MP. A major locus for fasting insulin concentrations and insulin resistance on chromosome 6q with strong pleiotropic effects on obesity-related phenotypes in nondiabetic Mexican Americans. Am J Hum Genet. 2001;68:1149–1164. doi: 10.1086/320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggirala R, Williams JT, Williams-Blangero S, Blangero J. A variance component approach to dichotomous trait linkage analysis using a threshold model. Genet Epidemiol. 1997;14:987–992. doi: 10.1002/(SICI)1098-2272(1997)14:6<987::AID-GEPI71>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Falconer DS. Introduction to quantitative genetics. 3. New York, NY, USA: Longman Scientific and Technical; 1989. [Google Scholar]
- Fall T, Ingelsson E. Genome-wide association studies of obesity and metabolic syndrome. Mol Cell Endocrinol. 2012 doi: 10.1016/j.mce.2012.08.018. S0303–7207:00413–3. [DOI] [PubMed] [Google Scholar]
- Farook VS, Puppala S, Schneider J, Fowler S, Chittoor G, Dyer TD, Allayee H, Cole SA, Arya R, Black MH, Curran JE, Almasy L, Buchanan TA, Jenkinson CP, Lehman DM, Watanabe RM, Blangero JJ, Duggirala R. Metabolic Syndrome is linked to Chromosome 7q21 and associated with genetic variants in CD36 and GNAT3 in Mexican Americans. Obesity. 2012;20:2083–2092. doi: 10.1038/oby.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández JR, Redden DT, Pietrobelli A, Allison DB. Waist circumference percentiles in nationally representative samples of African-American, European-American, and Mexican-American children and adolescents. J Pediatr. 2004;145:439–444. doi: 10.1016/j.jpeds.2004.06.044. [DOI] [PubMed] [Google Scholar]
- Ford ES, Li C. Defining the metabolic syndrome in children and adolescents: will the real definition please stand up? J Pediatr. 2008;152:160–164. doi: 10.1016/j.jpeds.2007.07.056. [DOI] [PubMed] [Google Scholar]
- Ford ES, Li C, Zhao G. Prevalence and correlates of metabolic syndrome based on a harmonious definition among adults in the US. J Diabetes. 2010;2:180–193. doi: 10.1111/j.1753-0407.2010.00078.x. [DOI] [PubMed] [Google Scholar]
- Golden SH, Brown A, Cauley JA, Chin MH, Gary-Webb TL, Kim C, Sosa JA, Sumner AE, Anton B. Health disparities in endocrine disorders: biological, clinical, and nonclinical factors--an Endocrine Society scientific statement. J Clin Endocrinol Metab. 97:E1579–639. doi: 10.1210/jc.2012-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman E, Daniels SR, Morrison JA, Huang B, Dolan LM. Contrasting prevalence of and demographic disparities in the World Health Organization and National Cholesterol Education Program Adult Treatment Panel III definitions of metabolic syndrome among adolescents. J Pediatr. 2004;145:445–451. doi: 10.1016/j.jpeds.2004.04.059. [DOI] [PubMed] [Google Scholar]
- Goodman E, Li C, Tu YK, Ford E, Sun SS, Huang TT. Stability of the factor structure of the metabolic syndrome across pubertal development: confirmatory factor analyses of three alternative models. J Pediatr. 2009;155:S5, e1–8. doi: 10.1016/j.jpeds.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goran MI, Bergman RN, Avila Q, Watkins M, Ball GD, Shaibi GQ, Weigensberg MJ, Cruz ML. Impaired glucose tolerance and reduced β-cellfunction in overweight Latino children with a positive family history for type 2 diabetes. J Clin Endocrinol Metab. 2004;89:207–212. doi: 10.1210/jc.2003-031402. [DOI] [PubMed] [Google Scholar]
- Grundy SM. Metabolic syndrome: a multiplex cardiovascular risk factor. J Clin Endocrinol Metab. 2007;92:399–404. doi: 10.1210/jc.2006-0513. [DOI] [PubMed] [Google Scholar]
- Gurka MJ, Ice CL, Sun SS, Deboer MD. A confirmatory factor analysis of the metabolic syndrome in adolescents: an examination of sex and racial/ethnic differences ardiovasc. Diabetol. 2012;11:128. doi: 10.1186/1475-2840-11-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman RP. Metabolic syndrome racial differences in adolescents. Curr Diabetes Rev. 2009;5:259–265. doi: 10.2174/157339909789804332. [DOI] [PubMed] [Google Scholar]
- Ice CL, Murphy E, Minor VE, Neal WA. Metabolic syndrome in fifth grade children with acanthosis nigricans: results from the CARDIAC project. World J Pediatr. 2009;5:23–30. doi: 10.1007/s12519-009-0004-7. [DOI] [PubMed] [Google Scholar]
- Johnson WD, Kroon JJ, Greenway FL, Bouchard C, Ryan D, Katzmarzyk PT. Prevalence of risk factors for metabolic syndrome in adolescents: National Health and Nutrition Examination Survey (NHANES), 2001–2006. Arch Pediatr Adolesc Med. 2009;163:371–377. doi: 10.1001/archpediatrics.2009.3. [DOI] [PubMed] [Google Scholar]
- Kassi E, Pervanidou P, Kaltsas G, Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med. 2011;9:48. doi: 10.1186/1741-7015-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange K, Boehnke M. Extensions to pedigree analysis. IV. Covariance components models for multivariate traits. Am J Hum Genet. 1983;14:513–524. doi: 10.1002/ajmg.1320140315. [DOI] [PubMed] [Google Scholar]
- Li C, Ford ES, Zhao G, Mokdad AH. Prevalence of pre-diabetes and its association with clustering of cardiometabolic risk factors and hyperinsulinemia among U.S. adolescents: National Health and Nutrition Examination Survey 2005–2006. Diabetes Care. 2009;32:342–347. doi: 10.2337/dc08-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig DS. The glycemic index: physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. JAMA. 2002;287:2414–2423. doi: 10.1001/jama.287.18.2414. [DOI] [PubMed] [Google Scholar]
- Manco M, Dallapiccola B. Genetics of pediatric obesity. Pediatrics. 2012;130:123–133. doi: 10.1542/peds.2011-2717. [DOI] [PubMed] [Google Scholar]
- Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- May LA, Kuklina EV, Yoon PW. Prevalence of Cardiovascular disease risk factors among US adolescents, 1999–2008. Pediatrics. 2012;129:1035–1041. doi: 10.1542/peds.2011-1082. [DOI] [PubMed] [Google Scholar]
- McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363:2339–2350. doi: 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
- Mitchell BD, Kammerer CM, Blangero J, Mahaney MC, Rainwater DL, Dyke B, Hixson JE, Henkel RD, Sharp RM, Comuzzie AG, VandeBerg JL, Stern MP, MacCluer JW. Genetic and environmental contributions to cardiovascular risk factors in Mexican Americans. Circulation. 1996;94:2159–2170. doi: 10.1161/01.cir.94.9.2159. [DOI] [PubMed] [Google Scholar]
- Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282:1523–1529. doi: 10.1001/jama.282.16.1523. [DOI] [PubMed] [Google Scholar]
- Nathan DM, Davidson MB, DeFronzo RA, Heine RJ, Henry RR, Pratley R, Zinman B American Diabetes Association. Impaired fasting glucose and impaired glucose tolerance: implications for care. Diabetes Care. 2007;30:753–759. doi: 10.2337/dc07-9920. [DOI] [PubMed] [Google Scholar]
- National High Blood Pressure Education Program (NHBPEP) Working Group on High Blood Pressure in Children and Adolescents . The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114 (Suppl 2 4th report):555–576. [PubMed] [Google Scholar]
- Norris JM, Rich SS. Genetics of glucose homeostasis: implications for insulin resistance and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2012;32:2091–2096. doi: 10.1161/ATVBAHA.112.255463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden CL, Carroll MD, Curtin LR, Lamb MM, Flegal KM. Prevalence of high body mass index in US children and adolescents, 2007–2008. JAMA. 2010;303:242–249. doi: 10.1001/jama.2009.2012. [DOI] [PubMed] [Google Scholar]
- Puppala S, Dodd GD, Fowler S, Arya R, Schneider J, Farook VS, Granato R, Dyer TD, Almasy L, Jenkinson CP, Diehl AK, Stern MP, Blangero J, Duggirala R. A genomewide search finds major susceptibility loci for gallbladder disease on chromosome 1 in Mexican Americans. Am J Hum Genet. 2006;78:377–392. doi: 10.1086/500274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven GM. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- Schutte AE, Schutte R, Huisman HW, Rooyen JM, Malan L, Olckers A, Malan NT. Classifying Africans with the metabolic syndrome. Horm Metab Res. 2009;41:79–85. doi: 10.1055/s-0028-1104603. [DOI] [PubMed] [Google Scholar]
- Shaibi GQ, Cruz ML, Goran MI. Cardiorespiratory fitness is strongly related to the metabolic syndrome in adolescents: response to Janssen and Cramp. Diabetes Care. 2008;31:e8. doi: 10.2337/dc07-2133. [DOI] [PubMed] [Google Scholar]
- Simmons RK, Alberti KG, Gale EA, Colagiuri S, Tuomilehto J, Qiao Q, Ramachandran A, Tajima N, Brajkovich Mirchov I, Ben-Nakhi A, Reaven G, Hama Sambo B, Mendis S, Roglic G. The metabolic syndrome: useful concept or clinical tool? Report of a WHO Expert Consultation. Diabetologia. 53:600–605. doi: 10.1007/s00125-009-1620-4. [DOI] [PubMed] [Google Scholar]
- Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. 2006;12:75–80. doi: 10.1038/nm0106-75. [DOI] [PubMed] [Google Scholar]
- Steinberger J, Daniels SR, Eckel RH, Hayman L, Lustig RH, McCrindle B, Mietus-Snyder ML. American Heart Association Atherosclerosis, Hypertension, and Obesity in the Young Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular Nursing; and Council on Nutrition, Physical Activity, and Metabolism. Progress and challenges in metabolic syndrome in children and adolescents: a scientific statement from the American Heart Association Atherosclerosis, Hypertension, and Obesity in the Young Committee of the Council on Cardiovascular Disease in the Young; Council on Cardiovascular Nursing; and Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2009;119:628–647. doi: 10.1161/CIRCULATIONAHA.108.191394. [DOI] [PubMed] [Google Scholar]
- Tenenbaum A, Fisman EZ. “The metabolic syndrome... is dead”: These reports are an exaggeration. Cardiovasc Diabetol. 2011;10:11. doi: 10.1186/1475-2840-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treviño RP, Yin Z, Hernandez A, Hale DE, Garcia OA, Mobley C. Impact of the Bienestar school-based diabetes mellitus prevention program on fasting capillary glucose levels: a randomized controlled trial. Arch Pediatr Adolesc Med. 2004;158:911–917. doi: 10.1001/archpedi.158.9.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez R, Greenlund KJ, Khoury MJ, Yoon PW. Is family history a useful tool for detecting children at risk for diabetes and cardiovascular diseases? A public health perspective. Pediatrics. 2007;120:S78–86. doi: 10.1542/peds.2007-1010G. [DOI] [PubMed] [Google Scholar]
- World Health Organization (WHO) Obesity: preventing and managing the global epidemic report of a WHO consultation. World Health Organ Tech Rep Ser. 1999;894:1–253. [PubMed] [Google Scholar]
- Weiss R, Dziura J, Burgert TS, Tamborlane WV, Taksali SE, Yeckel CW, Allen K, Lopes M, Savoye M, Morrison J, Sherwin RS, Caprio S. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;350:2362–2374. doi: 10.1056/NEJMoa031049. [DOI] [PubMed] [Google Scholar]
- Williams JT, Van Eerdewegh P, Almasy L, Blangero J. Joint multipoint linkage analysis of multivariate qualitative and quantitative traits. I. Likelihood formulation and simulation results. Am J Hum Genet. 1999;65:1134–1147. doi: 10.1086/302570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Ming XF. CD36: the common soil for inflammation in obesity and atherosclerosis? Cardiovasc Res. 2011;89:485–486. doi: 10.1093/cvr/cvq406. [DOI] [PubMed] [Google Scholar]
- Zeller T, Blankenberg S, Diemert P. Genomewide association studies in cardiovascular disease--an update 2011. Clin Chem. 2012;58:92–103. doi: 10.1373/clinchem.2011.170431. [DOI] [PubMed] [Google Scholar]
- Zimmet P. The burden of type 2 diabetes: are we doing enough? Diabetes Metab. 2003;29:6, S9–18. doi: 10.1016/S1262-3636(03)72783-9. [DOI] [PMC free article] [PubMed] [Google Scholar]