

Abstract
The processes of bone resorption and bone formation are tightly coupled in young adults, which is crucial to maintenance of bone integrity. We have documented that osteoclasts secrete chemotactic agents to recruit osteoblast lineage cells, contributing to coupling. Bone formation subsequent to bone resorption becomes uncoupled with aging, resulting in significant bone loss. During bone resorption, osteoclasts release and activate transforming growth factor beta 1 (TGF-β1) from the bone matrix; thus, elevated bone resorption increases the level of active TGF-β in the local environment during aging. In this study, we examined the influences of TGF-β1 on the ability of osteoclasts to recruit osteoblasts. TGF-β1 increased osteoclast expression of the chemokine CXCL16 to promote osteoblast migration. TGF-β1 also directly stimulated osteoblast migration, however, this direct response was blocked by conditioned medium from TGF-β1-treated osteoclasts due to the presence of leukemia inhibitory factor (LIF) in the medium. CXCL16 and LIF expression was dependent on TGF-β1 activation of Smad2 and Smad3. These results establish that TGF-β1 induces CXCL16 and LIF production in osteoclasts, which modulate recruitment of osteoblasts to restore the bone lost during the resorptive phase of bone turnover.
Keywords: osteoclast, coupling, migration, TGF-β, CXCL16, LIF
1. Introduction
It has been recognized for many years that bone resorbed by osteoclasts is replaced through bone deposition by osteoblasts, a process termed coupling. Once adult height is reached, bone resorption is tightly linked to precisely replace the removed bone in both location and amount. However, the process breaks down with aging, resulting in a net loss of bone. Because of a significant portion of aging adults loose sufficient bone to be at risk for debilitating fracture, it is crucial to resolve the factors that drive coupling of bone resorption to subsequent bone formation and how local factors can influence this process. Recruitment of bone forming progenitors cells to active resorption sites is a necessary early step in coupling. We showed that osteoclasts release chemotactic agents to enhance mesenchymal cell migration [1]. During periods of high bone turnover, elevated bone resorption releases and activates bone matrix-bound TGF-β [2–4]. In humans, mutations in TGF-β that influence its activity have profound impacts on bone development. Loeys-Doetz syndrome, which is caused by activating mutations in the TGF-β receptors, leads to cleft palate, scoliosis, and craniosynostosis [5]. Defects in TGF-β sequestration cause Marfan’s Syndrome, which is manifested in an unusually tall stature with long limbs and long, slender fingers [6]. Mutations that constitutively activate TGF-β1 cause inherited Camurai-Engelmann Disease [7]. This is primarily manifested by a thickening of the bone diaphysis due to increased bone formation. Moreover, QTL mapping revealed that a TGF-β latency binding protein is a strong candidate gene located on chromosome 7 associated with BMD differences between mouse strains [8]. Taken together, these data confirm that TGF-β has important roles in bone metabolism.
There are three mammalian TGF-β isoforms, which are similar in amino acid sequences [9]. TGF-β1 is the most abundant form in mineralized tissues, although all three isoforms are found bound to the bone matrix in inactivating complexes [9]. The three TGF-β isoforms activate a single receptor complex, which is expressed in most tissues. Thus, tight control of TGF-β activation is required to control biological functions. We showed osteoclasts secrete cathepsins to activate latent TGF-β [2]. Thus active TGF-β is present when osteoclasts are actively resorbing bone. TGF-β was reported to enhance migration in both transformed and normal cells, including osteoblastic cells [10–18]. However, there are reports that TGF-β can also inhibit migration of normal and transformed cells, indicating complex and potentially tissue-specific influences of TGF-β on migration [19–21]. Tang et al [18] documented that TGF-β released during bone resorption is a chemotactic agent that recruits mesenchymal cells to the sites of resorption to begin the replacement phase of bone turnover. Because of the elevated levels of TGF-β present at sites of bone resorption and our evidence that osteoclasts secrete chemotactic factors, we examined the influences of TGF-β on osteoclast support of mesenchymal cell migration.
2. Materials and Methods
All chemicals were from Sigma Chemical Co., St Louis, MO unless indicated elsewise.
2.1. Osteoclast culture
All protocols were approved by the Mayo Clinic IACUC prior to the start of the studies. Six to 12 week-old C57Bl/6 mice (Jackson Laboratories) were sacrificed and bone marrow harvested and processed as we previously reported [22]. Bone marrow macrophages were cultured in αMEM supplemented with 10% (v/v) FBS, 100 ng/mL RANKL, and 25 ng/mL M-CSF (differentiation medium) for 4 days. When the cells are re-fed on day 3, either vehicle (PBS with 0.1% BSA) or 2 ng/ml TGF-β1 (R&D) was added.
2.2. Conditioned media preparation
For the migration and protein assays, mature osteoclast conditioned media were harvested and stored at −80°C until assayed.
2.3. Osteoblast culture, migration, and quantitation
Calvarial osteoblastic cells from neonatal mice were obtained and cultured as previously described [23]. Cultures were maintained and replated for experiments in αMEM supplemented with 10% FBS (base medium). For migration experiments, 95% confluent cells were harvested and assayed using the QCM 24-well Colorimetric Cell Migration Assay (Millipore). Calvarial cells in base medium with or without pre-treatment (see below) were added to the inserts. Base or conditioned media with or without additives (as detailed below and in the figure legends) were added to the bottom chamber. The assembled assay was incubated at 37° C for 6 hours. Cell migration through the insert membrane was quantitated by staining cells with the provided reagent, cells on the upper side of the membrane were removed, and the cell stain was extracted and quantitated using absorbance at 560 nm. Inhibitor treatments are detailed below.
2.4. Treatments
2.4.1. CXCL16 treatment
Recombinant mouse CXCL16 (R&D) was reconstituted at 50 μg/ml in sterile 0.1% PBS (vehicle) and stored at −20°C until used. Vehicle or 250 ng/ml CXCL16 was added to base medium in the bottom chamber and the migration assay carried out as outlined above.
2.4.2. Inhibitor treatments
Antibody neutralization: Osteoclast conditioned media were pre-treated with either isotype control or a neutralizing antibody to TGF-β (R&D; at 12.5 μl/ml), CXCL16 (R&D; at 0.5 uμg/ml), or LIF (Sigma Aldrich; 10 uμg/ml) prior to placement in the bottom of the migration chamber. Adenoviral infections: On day 3, marrow cultures were infected at a multiplicity of 100 with empty vector, dominant negative Smad2, dominant negative Smad3 (both gifts from Dr. Rosa Serra, University of Alabama), or both. Six hours later, the media were removed and the cells re-fed with media containing either vehicle or 2 ng/ml TGF-β1 for 24 hours. Cells were harvested for RNA as described. Pharmacological kinase inhibition: Vehicle (DMSO), InSolution AKT inhibitor IV (Millipore) (which targets all AKF isoforms but not PI3K), or the MEK1/2 inhibitor U0126 (Millipore) was added to marrow cultures with the day 3 feeding and the cells were incubated at 37°C in a CO2 incubator. Fifteen minutes later, either vehicle or 2 ng/ml TGF-β1 were added for 24 hours. Cells were harvested for RNA as described.
2.5. Quantitative Real Time Polymerase Chain Reaction
Cells were rinsed with PBS and RNA harvested from mature osteoclasts using Qiagen’s RNeasy total RNA purification kit according to the product literature. Following quantitation, cDNA was synthesized and real time PCR analysis carried out as we reported [24]. Primers were:
| GENE | FORWARD | REVERSE |
|---|---|---|
| CXCL16 | 5′-CAAGACCCTAGCGCCTACAG -3′ | 5′-CCATTCACTGATGGAGACGA -3′ |
| LIF | 5′-GATCCCAGTCCCCTTAGCTC -3′ | 5′-CCTCATGATCCGACTTCGTT -3′ |
| Tubulin A1A | 5′-GAGTGCATCTCCATCCACGTT-3′ | 5′-TAGAGCTCCCAGCAGGCATT-3′ |
Fluorescence was quantified as the Ct value. The differences between the mean Ct values of the genes were denoted (Δ-Ct) and the difference between the Δ-Ct value of the Tubulin A1A was calculated as ΔΔ-Ct. The log2(ΔΔ-Ct) resulted in the relative quantification value of gene expression. Expression is calculated for each gene in response to TGF-β1 relative to expression of that gene in vehicle treated cells.
2.6. Chemokine secretion analysis
The R&D Proteome Profiler Antibody Array for Mouse Chemokines was used according to the manufacturer’s instructions. Briefly, two biological replicates treated with either vehicle or TGFβ1 were assayed using the provided membranes with capture and control antibodies spotted in duplicate on the nitrocellulose membrane. All reagents were supplied by the manufacturer with the exception of the conditioned media. Processing and analysis followed the provided protocol.
2.6.1. LIF receptor expression analysis
Eighty percent confluent calvarial cells were treated with either vehicle or 2 ng/ml TGF-β1 for 6 hours. Cell extracts were harvested and protein concentrations determined with the BioRad DC Protein Assay kit as instructed. Proteins (40 μg) were separated using 10% SDS-PAGE followed by electroblotting to Immobilon-P membranes (Millipore, Bedford, MA). Membranes were probed as described with antibodies to mouse/human CXCR6 (Acris Antibodies) [22]. Lane loading was monitored by re-probing blots for β-actin (Sigma Chemical Compahy). Signals were visualized using the ECL Plus detection system (Amersham Biosciences, Buckinghamshire, England) according to the manufacturer’s instructions.
2.6.2. LIF secretion analysis
The R&D Mouse LIF Quantikine ELISA kit was used with a standard curve to quantitate LIF levels in vehicle or TGF-β 1 treated osteoclast conditioned media according to the manufacturer’s protocol. Three biological replicates treated with either vehicle or TGF-β 1 were assayed.
2.7. Statistics
Each experiment had at least 3 replicates and was repeated at least 3 times. These data are representative of the results. Data were analyzed using a one way analysis of variance (ANOVA) as compared to controls as indicated in each figure legend and are presented as mean +/− SEM. Significance was determined at p<0.05 using KaleidaGraph software (Synergy Software, Reading PA).
3. Results
3.1. TGF-β stimulates osteoclasts to increase osteoblast migration
To examine how TGF-β impacts osteoclast-directed promotion of osteoblast migration, osteoclasts were treated with either vehicle or TGF-β 1 for 24 hours prior to collection of the conditioned media. To eliminate direct TGF-β effects on migration, an aliquot of each conditioned medium was pre-treated with a well-characterized TGF-β neutralizing antibody. Calvarial cells were placed in migration chambers for 6 hours and migration through a porous membrane was assessed (Figure 1). Conditioned medium from TGF-β1 treated osteoclasts significantly stimulated migration and neutralizing TGF-β did not alter this response. Because our parallel studies described below confirmed that the neutralizing activity of this antibody, we interrogated other mechanisms of action in the conditioned medium.
Figure 1.

Conditioned medium from TGF-β1 treated osteoclasts stimulates osteoblast migration. Conditioned media from osteoclasts treated with vehicle or 2 ng/ml TGF-β1 for 24 hours were collected. Conditioned media were treated with isotype control (Cont Ab) or TGF-β neutralizing antibody (αTGF-β) and placed in the bottom chamber. Calvarial cells in base medium were placed in the migration inserts as detailed in the methods. The chambers were incubated and analyzed by staining cells that migrated through the membrane within 6 hours as detailed in the methods section. *p<0.05 compared to vehicle treated.
3.2. TGF-β increases CXCL16 expression to enhance osteoblast migration
Because chemokines stimulate cell migration, we employed an antibody array to determine whether TGF-β enhanced osteoclast chemokine secretion. Conditioned media from vehicle and TGF-β1 treated osteoclasts were assessed. In replicate samples, CXCL16 was elevated in the TGF-β 1-treated osteoclast conditioned medium (Figure 2A, solid arrows). Osteoclasts express high levels of the chemokine CCL9/MIP1γ [25] but we did not detect an influence of TGF-β on its secretion (Figure 2A, open arrows). Unexpectedly, levels of many of the other chemokines in this antibody array were suppressed by TGF-β1 treatment. The suppressed chemokines included CCL2, 8, 12, 21, 22, 27, and 28; CXCL1, 2, 5, 9, 11, 12, and 13; CLL3 and CX3CL1 (see identities of the coordinates in Supplemental Figure 1). We reported that osteoclasts secrete the chemokine sphingosine 1 phosphate (S1P) [1]; however, we were unable to detect any effects of TGF-β1 on S1P production (data not shown).
Figure 2.
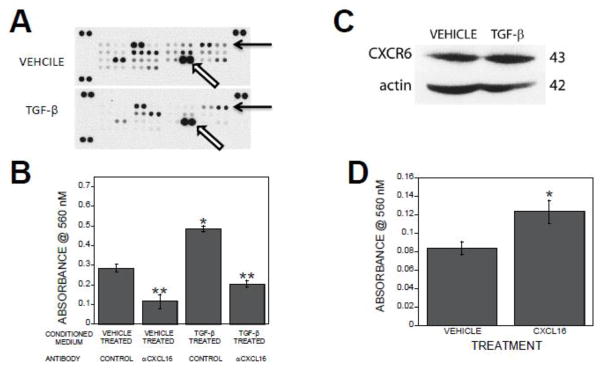
CXCL16 mediates TGF-β1 stimulation of osteoblast migration. Conditioned media from osteoclasts treated with vehicle or 2 ng/ml TGF-β1 for 24 hours were collected. (A). Media were assessed using the Proteome Profiler Antibody Array for mouse chemokines according to the provided protocol as detailed in the Methods section. Solid arrows point to the duplicate spots that probe for CXCL16 and open arrows point to CCL9/MIP1γ. (B). Conditioned media were treated with isotype control or a CXCL16 neutralizing antibody prior to placement in the bottom migration chamber. Calvarial cells in base medium were placed in the migration inserts as detailed in the methods. The chambers were incubated and analyzed by staining cells that migrated through the membrane within 6 hours as detailed in the methods section. *p<0.05 compared to vehicle treated. **p<0.05 compared to isotype control antibody treated. (C). Calvarial cells were treated with either vehicle or 2 ng/ml TGF-β1 for 6 hours. Forty μg of cell lysates were analyzed for CXCR6 and actin expression by western blotting. Apparent molecular weight in kDa is indicated on the right. (D). Base medium containing either vehicle or 250 ng/ml CXCL16 was placed in the bottom migration chamber. Calvarial cells in base medium were placed in the migration inserts as detailed in the methods. The chambers were incubated and analyzed by staining cells that migrated through the membrane within 6 hours as detailed in the methods section. *p<0.05 compared to vehicle treated.
To determine if CXCL16 effects osteoblast migration, migration assays were performed in the presence or absence of TGF-β1 and a CXCL16 neutralizing antibody. CXCL16 neutralization suppressed basal migration of osteoblasts toward vehicle treated osteoclast conditioned medium (Figure 2B). CXCL16 neutralization also abrogated the observed migration enhancement toward TGF-β1 treated osteoclast conditioned medium (Figure 2B). To verify that osteoblasts expression the cognate receptor for CXCL16, CXCR6 and determine whether TGF-β alters receptor expression during the time frame of our migration study, we examined CXCR6 protein levels. Osteoblasts treated for 6 hours with either vehicle or TGF-β expressed comparable levels of CXCR6 protein (Figure 2C). To verify a role for CXCL16 in promoting osteoblast migration, recombinant mouse CXCL16 migration influences were examined. Two hundred and fifty ng/ml of CXCL16 in base medium stimulated osteoblast migration within 6 hours of exposure (Figure 2D).
To determine whether enhanced expression of CXCL16 in TGF-β1-treated osteoclasts was due to increased CXCL16 gene expression, osteoclasts were treated with vehicle or 2 ng/ml TGF-β1 for 1 or 24 hours and gene expression was quantitated by real time PCR. Expression of CXCL16 mRNA was rapidly stimulated by TGF-β1 treatment and remained elevated for 24 hours (Figure 3A). A dose-dependent increase in CXCL16 expression was observed between 0.2 and 2 ng/ml (data not shown). Neutralizing TGF-β during osteoclast treatment suppressed the influences of TGF-β on CXCL16 gene expression, verifying the effectiveness of antibody neutralization (Figure 3B). To resolve the mechanisms of TGF-β1 modulation of osteoclast CXCL16 expression, we inhibited AKTs, MEK1/2, Smad 2, Smad 3, or both Smads. Suppression of Smad signaling suppressed TGF-β1 stimulation of CXCL16 mRNA expression (Figure 3C). Inhibition of AKT or MEK kinase pathways had no impact on TGF-β1 stimulation of CXCL16 gene expression.
Figure 3.
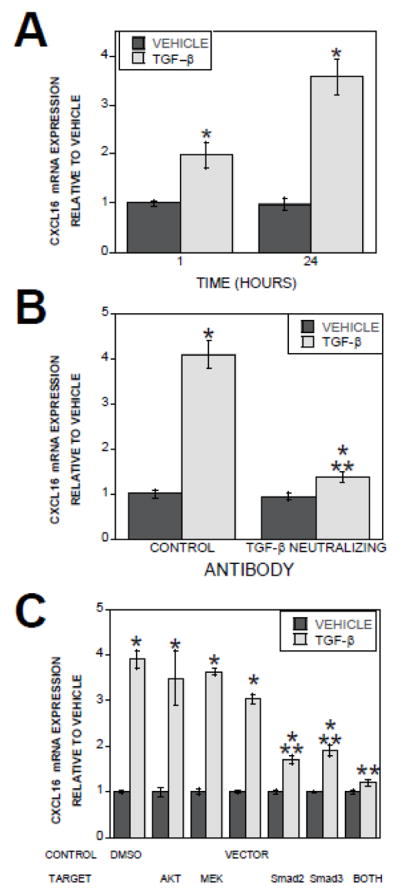
Effect of TGF-β1 on CXCL16 gene expression. (A). Marrow-derived osteoclast RNA was assessed for CXCL16 expression in vehicle and TGF-β1 treated osteoclasts by Real Time PCR. *p<0.05 compared to mRNA levels between vehicle and TGF-β1 treated osteoclasts. (B). Osteclasts were treated with vehicle or 2 ng/ml TGF-β1 and either isotype control (CONTROL) or TGF-β neutralizing antibody for 24 hours and assessed for CXCL16 expression by Real Time PCR. *p<0.05 compared to mRNA levels between vehicle and TGF-β1 treated osteoclasts. **p<0.05 comparing isotype control to TGF-β neutralizing antibody. C). Day 3 osteoclast precursors were fed and either treated with DMSO or the indicated kinase inhibitor or infected with the empty adenovirus expressing vector (VECTOR), dominant negative Smad2 (dnS2), dominant negative Smad3 (dnS3), or both dominant negative adenoviruses (both) for 24 hours. On day 4, osteoclasts were treated with vehicle or 2 ng/ml β1 for 24 hours. RNA was assessed for CXCL16 expression by Real Time PCR. *p<0.05 compared to mRNA levels between vehicle and TGF-β. **p<0.05 comparing vector to dominant negative adenovirus treated cultures.
3.3. TGF-β-induced LIF expression in osteoclasts blocks direct TGF-β osteoblast migration stimulation
Given the published data that TGF-β directly stimulates calvarial cell migration [9] and the presumed presence of residual TGF-β1 in the TGF-β treated osteoclast conditioned medium, the lack of any impact of TGF-β neutralization on migration toward TGF-β-treated osteoclast conditioned medium was unexpected. We therefore examined direct influences of adding TGF-β1 to the osteoclast conditioned media on calvarial cell migration. TGF-β1 addition to vehicle treated osteoclast conditioned medium stimulated migration (Figure 4). Surprisingly, addition of TGF-β1 to the TGF-β-treated osteoclast conditioned medium did not further enhance migration above the effect of the conditioned medium itself (Figure 4). To determine whether the TGF-β treated osteoclast conditioned medium response was the maximum stimulatory response possible or whether there was a general migration antagonist present, migration in response to BMP6 supplementation was evaluated. Addition of BMP6 to the TGF-β-treated osteoclast conditioned medium enhanced migration above the migration response to the medium (Figure 4).
Figure 4.
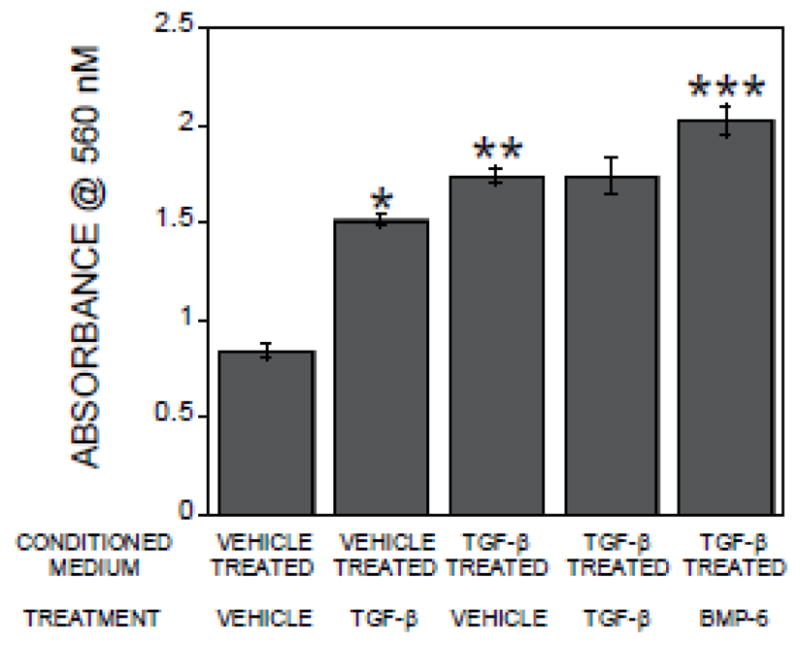
Conditioned medium from TGF-β-treated osteoclasts suppresses direct TGF-β stimulation of calvarial cell migration. Conditioned media from osteoclasts treated with vehicle or 2 ng/ml TGF-β1 for 24 hours were collected. Conditioned media were treated with vehicle, TGF-β, or BMP6 as indicated and placed in the bottom chambers as detailed in the methods. Calvarial cells in base medium were placed in the migration inserts as detailed in the methods. The chambers were incubated and analyzed by staining cells that migrated through the membrane within 6 hours as detailed in the methods section. *p<0.05 comparing vehicle treated conditioned medium with and without TGF-β addition for assay; **p<0.05 comparing vehicle treated osteoclast conditioned medium to TGF-β treated osteoclast conditioned medium; ***p<0.05 comparing TGF-β treated osteoclast conditioned medium with and without BMP6 treatment.
In many cell types, LIF inhibits migration, although studies indicated a complex array of LIF influences on many cell types including osteoblasts [26–28]. While LIF suppresses PDGF-induced random movement, or chemokinesis, of osteoblasts, it promotes PDGF-induced chemotactic responses and osteoblast differentiation [29]. Since LIF is induced by TGF-β1 in Schwann cells [30], we evaluated TGF-β1 effects on LIF gene and protein expression and if LIF expression in osteoclast conditioned medium influenced TGF-β-induced osteoblast migration. Within one hour of TGF-β1 treatment LIF message levels were elevated and stimulation was sustained over 24 hours of treatment (Figure 5A). Examination of osteoclast conditioned media for LIF protein expression levels revealed LIF accumulation in response to TGF-β after 8 and 24 hours of treatment (Figure 5B). TGF-β neutralization blocked TGF-β induction of LIF mRNA (Figure 5C). LIF neutralization enhanced migration toward either vehicle or TGF-β1 treated osteoclast conditioned media (Figure 6A). Investigation of the mechanisms by which TGF-β1 stimulated LIF gene expression revealed MEK and SMAD2/3 activation by TGF-β1 mediate stimulation of LIF gene expression (Figure 6B).
Figure 5.

TGF-β stimulates LIF expression by osteoclasts to suppress direct TGF-β effects on osteoblast migration. (A). Marrow-derived osteoclast RNA was assessed for expression of LIF expression in vehicle and TGF-β1 treated osteoclasts by Real Time PCR. *p<0.05 compared to mRNA levels between vehicle and TGF-β treated osteoclasts. (B). Conditioned media from osteoclasts treated with vehicle or 2 ng/ml TGF-β1 for 8 or 24 hours were collected and assayed for LIF protein levels. *p<0.05 compared to vehicle treated. (C). Osteclasts were treated with vehicle or 2 ng/ml TGF-β1 and either isotype control (CONTROL) or TGF-β neutralizing antibody for 24 hours and assessed for LIF expression by Real Time PCR. *p<0.05 compared to mRNA levels between vehicle and TGF-β1 treated osteoclasts. **p<0.05 comparing isotype control to TGF-β neutralizing antibody.
Figure 6.
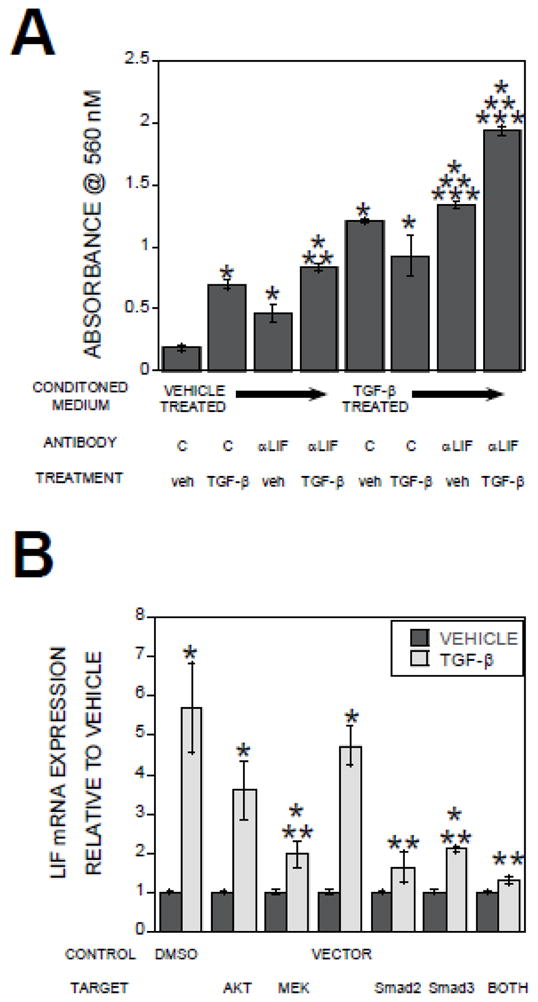
(A). Conditioned media were treated with isotype control (C) or a LIF neutralizing antibody prior to placement in the bottom migration chamber. Calvarial cells in base medium with either vehicle (veh) or 2 ng/ml TGF-β1 were placed in the migration inserts as detailed in the methods. The chambers were incubated and analyzed by staining cells that migrated through the membrane within 6 hours as detailed in the methods section. (B) Day 3 osteoclast precursors were fed and either treated with DMSO or the indicated kinase inhibitor or infected with the empty adenovirus expressing vector (VECTOR), dominant negative Smad2 (dnS2), dominant negative Smad3 (dnS3), or both dominant negative adenoviruses (both) for 24 hours. On day 4, osteoclasts were treated with vehicle or 2 ng/ml β1 for 24 hours. RNA was assessed for LIF expression by Real Time PCR. *p<0.05 compared to vehicle treated osteoclasts; **p<0.05 combined treatment compared to isotype control antibody or vehicle treated; ***p<0.05 compared to TGF-β treated osteoclasts with control antibody and vehicle.
4. Discussion
Modulation of TGF-β influences on bone cells is crucial to normal bone metabolism. During periods of high bone turnover, osteoclasts release bone-bound TGF-β and activate it, thus the impacts of TGF-β on bone turnover are important to understand in order to design therapies to slow high bone turnover rates [2–4]. Extensive studies have documented that TGF-β regulates both osteoclast differentiation and osteoblast differentiation [9, 31, 32]. Early osteoclast progenitor commitment required the presence of TGF-β [32]. Our studies revealed bi-phasic TGF-β impacts on osteoclast differentiation when present throughout differentiation in that lower concentrations promote differentiation whereas higher doses inhibit differentiation [24]. Likewise, TGF-β either promotes or inhibits osteoclast apoptosis, depending on the model system [33–35]. In bone marrow mesenchymal stem cells, TGF-β drives cell differentiation and we have shown that TGF-β promotes osteoblast commitment over adipocyte commitment in pluripotent precursors [36]. TGF-β stimulated early osteoblast progenitor proliferation although it inhibited final stages of differentiation [4, 37]. TGF-β promotes migration of osteosarcoma, mesenchymal, and calvarial cells, although not of all mesenchymal model systems [38–42]. Thus it is important to understand the complex nature of TGF-β impacts on osteoblast precursor recruitment, which is the focus of this study.
As summarized in Figure 7, TGF-β stimulates expression of the chemokine CXCL16 in osteoclasts to enhance recruitment of osteoblast lineage cells. TGF-β also increased LIF expression in osteoclasts, which suppressed direct TGF-β effects on calvarial cell migration. These complex interactions may allow for more nuanced modulation within the bone microenvironment. CXCL16 is a transmembrane protein that is shed from the cells as the result of ADAM 10-mediated release [43]. The membrane form of CXCL16 is a scavenger receptor for oxLDL [43] while the soluble form is involved in T cell recruitment (15880344). Soluble CXCL16 is a driving force in collagen-induced rheumatoid arthritis in mice and is associated with rheumatoid arthritis development in humans as well [44, 45]. CXCL16 and its cognate receptor CXCR6 play a role in recruitment of T cells to lungs as well [46]. Our discovery that LIF opposes direct TGF-β stimulation of osteoblast lineage cell migration further expands our understanding of the complex influences of TGF-β on bone metabolism. Modulation of LIF production in response to TGF-β enables a fine tuning of migration activity of osteoblast lineage cells by providing another layer of control. Moreover, LIF likely also acts in other autocrine and paracrine ways to alter bone metabolism such as by maintaining a stem cell pool in the vicinity of the resorption site [47], promoting bone formation [48], or controlling osteoclast differentiation [49]. In neurons, TGF-β and LIF act in concert to promote survival, indicating that LIF is not suppressive of all TGF-β influences [50]. This diversity in migration responses and mechanisms induced by TGF-βs is consistent with the pleotropic influences of TGF-β.
Figure 7.

Schematic of TGF-β effects on osteoclast influences on osteoblast migration. TGF-β activates SMAD signaling to stimulate CXCL16 expression and SMAD and MEK signaling to stimulate LIF expression. Secreted CXCL16 stimulates osteoblastic cell migration while secreted LIF prevents direct TGF-β stimulation of osteoblast migration.
We examined the mechanisms by which TGF-β induced CXCL16 and LIF expression and observed that Smads 2 and 3 both contribute to the increased gene expression. Unqufroren et al [51] evaluated contributions of these Smads to pancreatic ductal adenocarcinoma cell TGF-β responses and documented that Smad3, but not Smad2, mediated TGF-β stimulation of migration. In contrast, Li et al [52] demonstrated that Smads 2 and 3 oppose each other in TGF-β2 influences on human lens cell migration. In this study, Smad2 mediates the migratory response, which was enhanced when Smad3 was silenced with small interfering RNA approaches. These studies indicate that the respective roles for SMAD2 and SMAD3 in mediating TGF-β influences on bone may be cell- and tissue-dependent.
Tang et al [18] examined whether this elevation in active TGF-β influenced coupling of bone resorption to subsequent bone formation and revealed a selective role for the TGF-β1 isoform in stimulating mesenchymal cell migration. In this report, the investigators examined a mouse in which osteoblast lineage cells secrete active TGF-β and observed local elevation in the vicinity of osteoblasts, which resulted in a loss of targeted migration of osteoblasts to the bone surface. Pharmacological TGF-β inhibition restored targeted migration by blocking the osteoblast-produced TGF-β effects, which suggests that other factors such as CXCL16 and S1P, in addition to released TGF-β, influence mesenchymal cell migration to the sites of bone resorption. Because osteoclasts both secrete and activate TGF-β, systemic TGF-β inhibition may have been less effective at inhibiting osteoclast autocrine TGF-β influences in this study [2]. Thus, TGF-β stimulation of CXCL16 resulting from local TGF-β release may have contributed to the restoration of mesenchymal cell migration that was observed in the Tang study. This hypothesis will require further studies to evaluate.
Although expression of many chemokines was reduced with TGF-β treatment, our results support that the lack of a chemotactic response to direct TGF-β treatment of calvarial cells is due to TGF-β-mediated increased LIF expression rather than reduced expression of multiple chemokines. Muscle cell damage leads to increased production of TGF-β and IGF-1, which initiate the repair process [53, 54]. While TGF-β has no direct impact on muscle cell migration, it inhibits a migratory response to IGF-1 [55]. With aging, increased bone resorption causes increased local TGF-β release and activation. Whether TGF-β-mediated LIF expression induction contributes to age-related uncoupling of bone formation from bone resorption or if this inhibitory response involves TGF-β-mediated LIF induction remains to be investigated.
5. Conclusions
This study reveals another component of the complex interactions between TGF-β and cells resident in bone. Potential roles for local TGF-β release/activation during bone resorption include indirect recruitment through stimulating osteoclast CXCL16 production and indirect tempering of the direct recruitment responses to TGF-β through LIF stimulation. These diverse influences likely contribute to the pleotropic TGF-β influences on bone metabolism.
Supplementary Material
{kind=link}
Highlights.
TGF-β stimulates osteoclasts to increase production of the chemokine CXCL16, which promotes osteoblast migration.
TGF-β stimulation of CXCL16 is Smad-dependent.
TGF-β stimulates osteoclasts to increase LIF production, which blocks direct TGF-β stimulation of osteoblast migration.
TGF-β stimulation of LIF is MEK and Smad-dependent.
Acknowledgments
This work was supported by National Institutes of Health grant P01 AG004875, National Institutes of Health training grants AR056950 and DK07352, and The Mayo Foundation. We wish to acknowledge Christine Hachfeld for her assistance. The authors have no conflicts of interest.
Abbreviations
- TGF-β1
transforming growth factor beta 1
- LIF
leukemia inhibitory factor
- CED
Camurati-Engleman Disease
- QTL
Quantitative Trait Locus
- RANKL
receptor activator of NFκB ligand
- M-CSF
macrophage colony stimulating factor
- αMEM
alpha Minimal Essential Medium
- FBS
fetal bovine serum
- BSA
bovine serum albumin
- PBS
Phosphate Buffered Saline
- PDGF
platelet derived growth factor
- S1P
sphingosin 1 phosphate
- IGF-1
insulin like growth factor 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Quint P, Ruan M, Pederson L, Kassem M, Westendorf JJ, Khosla S, Oursler MJ. Sphingosine 1-Phosphate (S1P) Receptors 1 and 2 Coordinately Induce Mesenchymal Cell Migration through S1P Activation of Complementary Kinase Pathways. The Journal of biological chemistry. 2013;288:5398–406. doi: 10.1074/jbc.M112.413583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oursler MJ. Osteoclast synthesis and secretion and activation of latent transforming growth factor beta. J Bone Miner Res. 1994;9:443–52. doi: 10.1002/jbmr.5650090402. [DOI] [PubMed] [Google Scholar]
- 3.Pfeilschifter J, Seyedin SM, Mundy GR. Transforming Growth Factor Beta Inhibits Bone Resorption in Fetal Rat Long Bone Cultures. J Clinc Invest. 1988;82:680–685. doi: 10.1172/JCI113647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Filvaroff E, Erlebacher A, Ye J, Gitelman SE, Lotz J, Heillman M, Derynck R. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development. 1999;126:4267–79. doi: 10.1242/dev.126.19.4267. [DOI] [PubMed] [Google Scholar]
- 5.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature genetics. 2005;37:275–81. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 6.Matt P, Schoenhoff F, Habashi J, Holm T, Van Erp C, Loch D, Carlson OD, Griswold BF, Fu Q, De Backer J, Loeys B, Huso DL, McDonnell NB, Van Eyk JE, Dietz HC. Circulating transforming growth factor-beta in Marfan syndrome. Circulation. 2009;120:526–32. doi: 10.1161/CIRCULATIONAHA.108.841981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janssens K, Gershoni-Baruch R, Guanabens N, Migone N, Ralston S, Bonduelle M, Lissens W, Van Maldergem L, Vanhoenacker F, Verbruggen L, Van Hul W. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati-Engelmann disease. Nat Genet. 2000;26:273–5. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee A, Larson EA, Carlos AS, Belknap JK, Rotwein P, Klein RF. Congenic mice provide in vivo evidence for a genetic locus that modulates intrinsic transforming growth factor beta 1-mediated signaling and bone acquisition. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2012;27:1345–56. doi: 10.1002/jbmr.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-betal to the bone. Endocr Rev. 2005;26:743–74. doi: 10.1210/er.2004-0001. [DOI] [PubMed] [Google Scholar]
- 10.Yao J, Yang W, Liu Y, Sun YX, Jiang Q. Dexamethasone inhibits TGF-beta2-induced migration of human lens epithelial cells: implications for posterior capsule opacification prevention. Molecular medicine reports. 2012;5:1509–13. doi: 10.3892/mmr.2012.827. [DOI] [PubMed] [Google Scholar]
- 11.Yuan Y, Chen H, Ma G, Cao X, Liu Z. Reelin is involved in transforming growth factor-betal-induced cell migration in esophageal carcinoma cells. PLoS ONE. 2012;7:e31802. doi: 10.1371/journal.pone.0031802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schafer H, Struck B, Feldmann EM, Bergmann F, Grage-Griebenow E, Geismann C, Ehlers S, Altevogt P, Sebens S. TGF-betal -dependent L1 CAM expression has an essential role in macrophage-induced apoptosis resistance and cell migration of human intestinal epithelial cells. Oncogene. 2013;32:180–9. doi: 10.1038/onc.2012.44. [DOI] [PubMed] [Google Scholar]
- 13.Shang D, Liu Y, Yang P, Chen Y, Tian Y. TGFBI-promoted adhesion, migration and invasion of human renal cell carcinoma depends on inactivation of von Hippel-Lindau tumor suppressor. Urology. 2012;79:966, e1–7. doi: 10.1016/j.urology.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Yuan X, Tang J, Zhang Y. Lipopolysaccharide disrupts the directional persistence of alveolar myofibroblast migration through EGF receptor. American journal of physiology Lung cellular and molecular physiology. 2012;302:L569–79. doi: 10.1152/ajplung.00217.2011. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y, Yang Y, Gao R, Yang X, Yan X, Wang C, Jiang S, Yu L. RLIM interacts with Smurf2 and promotes TGF-beta induced U20S cell migration. Biochemical and biophysical research communications. 2011;414:181–5. doi: 10.1016/j.bbrc.2011.09.053. [DOI] [PubMed] [Google Scholar]
- 16.Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H, Yang H, Chen X, Hu Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. Journal of cell science. 2011;124:3235–46. doi: 10.1242/jcs.082875. [DOI] [PubMed] [Google Scholar]
- 17.Gatza CE, Holtzhausen A, Kirkbride KC, Morton A, Gatza ML, Datto MB, Blobe GC. Type III TGF-beta receptor enhances colon cancer cell migration and anchorage-independent growth. Neoplasia. 2011;13:758–70. doi: 10.1593/neo.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, Hu J, Feng X, Van Hul W, Wan M, Cao X. TGF-betal-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nature medicine. 2009;15:757–65. doi: 10.1038/nm.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai K, Minamiya Y, Koyota S, Ito M, Saito H, Sato Y, Motoyama S, Sugiyama T, Ogawa J. Inhibition of dendritic cell migration by transforming growth factor-beta1 increases tumor-draining lymph node metastasis. Journal of experimental & clinical cancer research: CR. 2012;31:3. doi: 10.1186/1756-9966-31-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert KE, Huang H, Mythreye K, Blobe GC. The type III transforming growth factor-beta receptor inhibits proliferation, migration, and adhesion in human myeloma cells. Molecular biology of the cell. 2011;22:1463–72. doi: 10.1091/mbc.E10-11-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romo P, Madigan MC, Provis JM, Cullen KM. Differential effects of TGF-beta and FGF-2 on in vitro proliferation and migration of primate retinal endothelial and Muller cells. Acta ophthalmologica. 2011;89:e263–8. doi: 10.1111/j.1755-3768.2010.01968.x. [DOI] [PubMed] [Google Scholar]
- 22.Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20764–9. doi: 10.1073/pnas.0805133106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Subramaniam M, Gorny G, Johnsen SA, Monroe DG, Evans GL, Fraser DG, Rickard DJ, Rasmussen K, van Deursen JM, Turner RT, Oursler MJ, Spelsberg TC. TIEG1 null mouse-derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Mol Cell Biol. 2005;25:1191–9. doi: 10.1128/MCB.25.3.1191-1199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karst M, Gorny G, Galvin RJ, Oursler MJ. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-beta regulation of osteoclast differentiation. J Cell Physiol. 2004;200:99–106. doi: 10.1002/jcp.20036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lean JM, Murphy C, Fuller K, Chambers TJ. CCL9/MIP-1 gamma and its receptor CCR1 are the major chemokine ligand/receptor species expressed by osteoclasts. Journal of cellular biochemistry. 2002;87:386–93. doi: 10.1002/jcb.10319. [DOI] [PubMed] [Google Scholar]
- 26.Kurzrock R, Estrov Z, Wetzler M, Gutterman JU, Talpaz M. LIF: not just a leukemia inhibitory factor. Endocrine reviews. 1991;12:208–17. doi: 10.1210/edrv-12-3-208. [DOI] [PubMed] [Google Scholar]
- 27.Hahn T, Levin S, Handzel ZT. Leucocyte migration inhibition factor (LIF) production by lymphocytes of normal children, newborns, and children with immune deficiency. Clinical and experimental immunology. 1976;24:448–54. [PMC free article] [PubMed] [Google Scholar]
- 28.Rocklin RE. Products of activated lymphocytes: leukocyte inhibitory factor (LIF) distinct from migration inhibitory factor (MIF) Journal of immunology. 1974;112:1461–6. [PubMed] [Google Scholar]
- 29.Chandrasekhar S, Harvey AK. Modulation of PDGF mediated osteoblast chemotaxis by leukemia inhibitory factor (LIF) Journal of cellular physiology. 1996;169:481–90. doi: 10.1002/(SICI)1097-4652(199612)169:3<481::AID-JCP8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 30.Matsuoka I, Nakane A, Kurihara K. Induction of LIF-mRNA by TGF-beta 1 in Schwann cells. Brain Res Mol Brain Res. 1997;776:170–80. doi: 10.1016/s0006-8993(97)01015-9. [DOI] [PubMed] [Google Scholar]
- 31.Zuo C, Huang Y, Bajis R, Sahih M, Li YP, Dai K, Zhang X. Osteoblastogenesis regulation signals in bone remodeling. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2012;23:1653–63. doi: 10.1007/s00198-012-1909-x. [DOI] [PubMed] [Google Scholar]
- 32.Fuller K, Lean JM, Bayley KE, Wani MR, Chambers TJ. A role for TGFbeta(l) in osteoclast differentiation and survival. Journal of cell science. 2000;113 (Pt 13):2445–53. doi: 10.1242/jcs.113.13.2445. [DOI] [PubMed] [Google Scholar]
- 33.Gingery A, Bradley E, Shaw A, Oursler MJ. Phosphatidylinositol 3-kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89:165–79. doi: 10.1002/jcb.10503. [DOI] [PubMed] [Google Scholar]
- 34.Gingery A, Bradley EW, Pederson L, Ruan M, Horwood NJ, Oursler MJ. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp Cell Res. 2008;314:2725–38. doi: 10.1016/j.yexcr.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes DE, Dai A, Tiffee JC, Li HH, Mundy GR, Boyce BF. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2:1132–6. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- 36.Kumar A, Ruan M, Clifton K, Syed F, Khosla S, Oursler MJ. TGF-beta mediates suppression of adipogenesis by estradiol through connective tissue growth factor induction. Endocrinology. 2012;153:254–63. doi: 10.1210/en.2011-1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erlebacher A, Filvaroff EH, Ye JQ, Derynck R. Osteoblastic responses to TGF-beta during bone remodeling. Mol Biol Cell. 1998;9:1903–18. doi: 10.1091/mbc.9.7.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Celotti F, Colciago A, Negri-Cesi P, Pravettoni A, Zaninetti R, Sacchi MC. Effect of platelet-rich plasma on migration and proliferation of SaOS-2 osteoblasts: role of platelet-derived growth factor and transforming growth factor-beta. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2006;14:195–202. doi: 10.1111/j.1743-6109.2006.00110.x. [DOI] [PubMed] [Google Scholar]
- 39.Makhijani NS, Bischoff DS, Yamaguchi DT. Regulation of proliferation and migration in retinoic acid treated C3H10T1/2 cells by TGF-beta isoforms. Journal of cellular physiology. 2005;202:304–13. doi: 10.1002/jcp.20128. [DOI] [PubMed] [Google Scholar]
- 40.Blanquaert F, Carpentier G, Morvan F, Caruelle JP, Barritault D, Tardieu M. RGTA modulates the healing pattern of a defect in a monolayer of osteoblastic cells by acting on both proliferation and migration. Journal of biomedical materials research Part A. 2003;64:525–32. doi: 10.1002/jbm.a.10400. [DOI] [PubMed] [Google Scholar]
- 41.Andrades JA, Han B, Becerra J, Sorgente N, Hall FL, Nimni ME. A recombinant human TGF-betal fusion protein with collagen-binding domain promotes migration, growth, and differentiation of bone marrow mesenchymal cells. Experimental cell research. 1999;250:485–98. doi: 10.1006/excr.1999.4528. [DOI] [PubMed] [Google Scholar]
- 42.Yee JA, Yan L, Dominguez JC, Allan EH, Martin TJ. Plasminogen-dependent activation of latent transforming growth factor beta (TGF beta) by growing cultures of osteoblast-like cells. Journal of cellular physiology. 1993;157:528–34. doi: 10.1002/jcp.1041570312. [DOI] [PubMed] [Google Scholar]
- 43.Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, Hartmann D, Sedlacek R, Dietrich S, Muetze B, Schuster B, Kallen KJ, Saftig P, Rose-John S, Ludwig A. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAMI10. Journal of immunology. 2004;172:6362–72. doi: 10.4049/jimmunol.172.10.6362. [DOI] [PubMed] [Google Scholar]
- 44.van Lieshout AW, Popa C, Meyer-Wentrup F, Lemmers HL, Stalenhoef AF, Adema GJ, van Riel PL, van Tits LJ, Radstake TR. Circulating CXCL16 is not related to circulating oxLDL in patients with rheumatoid arthritis. Biochemical and biophysical research communications. 2007;355:392–7. doi: 10.1016/j.bbrc.2007.01.161. [DOI] [PubMed] [Google Scholar]
- 45.van der Voort R, van Lieshout AW, Toonen LW, Sloetjes AW, van den Berg WB, Figdor CG, Radstake TR, Adema GJ. Elevated CXCL16 expression by synovial macrophages recruits memory T cells into rheumatoid joints. Arthritis and rheumatism. 2005;52:1381–91. doi: 10.1002/art.21004. [DOI] [PubMed] [Google Scholar]
- 46.Morgan AJ, Guillen C, Symon FA, Huynh TT, Berry MA, Entwisle JJ, Briskin M, Pavord ID, Wardlaw AJ. Expression of CXCR6 and its ligand CXCL16 in the lung in health and disease. Clinical and experimental allergy: journal of the British Society for Allergy and Clinical Immunology. 2005;35:1572–80. doi: 10.1111/j.1365-2222.2005.02383.x. [DOI] [PubMed] [Google Scholar]
- 47.Schraml E, Fehrer C, Brunauer R, Lepperdinger G, Chesnokova V, Schauenstein K. lin-Sca-1+ cells and age-dependent changes of their proliferation potential are reliant on mesenchymal stromal cells and are leukemia inhibitory factor dependent. Gerontology. 2008;54:312–23. doi: 10.1159/000161736. [DOI] [PubMed] [Google Scholar]
- 48.Martin TJ, Allan EH, Evely RS, Reid IR. Leukaemia inhibitory factor and bone cell function. Ciba Foundation symposium. 1992;167:141–50. doi: 10.1002/9780470514269.ch9. discussion 150–5. [DOI] [PubMed] [Google Scholar]
- 49.Bozec A, Bakiri L, Hoebertz A, Eferl R, Schilling AF, Komnenovic V, Scheuch H, Priemel M, Stewart CL, Amling M, Wagner EF. Osteoclast size is controlled by Fra-2 through LIF/LIF-receptor signalling and hypoxia. Nature. 2008;454:221–5. doi: 10.1038/nature07019. [DOI] [PubMed] [Google Scholar]
- 50.Marzella PL, Gillespie LN, Clark GM, Bartlett PF, Kilpatrick TJ. The neurotrophins act synergistically with LIF and members of the TGF-beta superfamily to promote the survival of spiral ganglia neurons in vitro. Hearing research. 1999;138:73–80. doi: 10.1016/s0378-5955(99)00152-5. [DOI] [PubMed] [Google Scholar]
- 51.Ungefroren H, Groth S, Sebens S, Lehnert H, Gieseler F, Fandrich F. Differential roles of Smad2 and Smad3 in the regulation of TGF-beta1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: control by Racl. Mol Cancer. 2011;10:67. doi: 10.1186/1476-4598-10-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J, Tang X, Chen X. Comparative effects of TGF-beta2/Smad2 and TGF-beta2/Smad3 signaling pathways on proliferation, migration, and extracellular matrix production in a human lens cell line. Experimental eye research. 2011;92:173–9. doi: 10.1016/j.exer.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 53.Philippou A, Maridaki M, Theos A, Koutsilieris M. Cytokines in muscle damage. Advances in clinical chemistry. 2012;58:49–87. doi: 10.1016/b978-0-12-394383-5.00010-2. [DOI] [PubMed] [Google Scholar]
- 54.Philippou A, Halapas A, Maridaki M, Koutsilieris M. Type I insulin-like growth factor receptor signaling in skeletal muscle regeneration and hypertrophy. J Musculoskelet Neuronal Interact. 2007;7:208–18. [PubMed] [Google Scholar]
- 55.Schabort EJ, van der Merwe M, Niesler CU. TGF-beta isoforms inhibit IGF-1-induced migration and regulate terminal differentiation in a cell-specific manner. Journal of muscle research and cell motility. 2011;31:359–67. doi: 10.1007/s10974-011-9241-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.