

Abstract
Therapeutic strategies controlling human cytomegalovirus (hCMV) infection are limited due to adverse side effects and emergence of antiviral resistance variants. A compound being evaluated for treating hCMV disease is maribavir (MBV) which disrupts replication by inhibiting the viral kinase pUL97. Previous studies have demonstrated that the antiviral activity of MBV is sensitive to the proliferation state of the infected cell. In these studies, we were interested in determining whether inhibition of the pro-proliferative transcription factor, signal transducer and activator of transcription-3 (STAT3), could influence the antiviral activity of MBV. The addition of the STAT3 inhibitor, S3i-201, during infection altered hCMV-mediated changes in cell cycle protein expression. Upon combining S3i-201 with MBV, our data suggest that STAT3 inhibition is acting synergistically with MBV to inhibit infection in vitro. Furthermore, specific concentrations of S3i-201 and MBV induced caspase-dependent death of infected but not uninfected cell. Our studies suggest that treating infection with both S3i-201 and MBV is a novel approach to inhibit hCMV replication.
Keywords: Human cytomegalovirus, HCMV, maribavir, STAT3, STAT3 inhibitor, UL97, viral kinase, ganciclovir, apoptosis, caspase, synergism
1. Introduction
Human cytomegalovirus (hCMV) is a beta-herpesvirus that infects a majority of the population and is typically asymptomatic in immunocompetent individuals (See review: Britt, 2008). Infection in immunocompromised individuals can result in severe disease (See review: Mercorelli et al., 2011). Several compounds have been developed and are currently used to treat infection. Although these antiviral agents have significantly improved the management of hCMV disease, several drawbacks limit their use including concerns of toxic side effects, poor oral bioavailability, and the emergence of drug resistant strains (See review: Hakki and Chou, 2011; Mercorelli et al., 2011).
The antiviral, maribavir (MBV), attenuates hCMV replication by inhibiting the activity of the hCMV kinase, pUL97 (See review: Prichard, 2009). pUL97 possesses activities similar to cyclin-dependent kinases (Hamirally et al., 2009; Hume et al., 2008) and contributes to several stages of viral replication. For example, pUL97 inhibits the retinoblastoma protein which contributes to altering the cell cycle of the infected cell (Hume et al., 2008; Kamil et al., 2009). This virally-induced cell cycle stage is required for productive infection (See review: Sanchez and Spector, 2008). Interestingly, treatment of infection with a pan-Cdk inhibitor enhanced the antiviral activity of MBV (Hertel et al., 2007). In addition, increased the levels of the cyclin-dependent kinase inhibitor p21CIP1 are associated with improved activity of MBV (Reitsma et al., 2011). These studies suggest that disruption of cellular proliferation enhances MBV activity.
We were interested in determining whether inhibition of the pro-proliferative transcription factor, signal transducer and activator of transcription-3 (STAT3), influences the antiviral activity of MBV. We demonstrate that inhibiting both STAT3 and the viral kinase results in an almost complete loss of virus production with limited impact on the viability of uninfected cells.
2. Material and Methods
2.1 Biological Reagents
MRC-5 fibroblasts, ARPE-19 epithelial and U-373 astrocytoma cells (ATCC) were propagated in DMEM with 7% FBS and 1% penicillin/streptomycin (Life Technologies). Unless otherwise stated, cells were grown until confluent, starved in 0.5% FBS for 2 days and infected at a multiplicity between 0.25-5 infectious units/cell (IU/cell). In several experiments, cells were treated 24 h prior to infection with 30-180 μM S3i-201 (ThermoFisher Scientific). The following inhibitors were added at the time of infection: 0.1-40 μM maribavir (ViroPharma) or 1-10 μM ganciclovir. Caspase inhibitors included 50 μM Z-VAD and Z-DEVD-FMK (EMD Millipore). Compounds were replaced every 24 h. As a control for STAT3-regulatable gene expression, cells were treated with 183 ng/ml of human interleukin-6 (IL6) (BioLegend) for 45 min. Viability and total cell numbers were determined with Viacount (EMD Millipore) and analyzed with a Gauva Flow Cytometer (Millipore). BAC-derived hCMV strain AD169 (ADwt) was propagated in fibroblasts. Strain TB40/E expressing mCherry was propagated in epithelial cells and harvested as previously described (O'Connor and Shenk, 2011). Cell-associated virus was isolated through sonicating infected cells and pelleting debris by centrifuging. Viral titers were determined by an infectious units assay (Mitchell et al., 2009) or standard plaque assay.
2.2 Analysis of Protein, DNA, and Gene Expression
Preparation of cell extracts, Western blot analysis, and immunofluorescence microscopy were completed as previously described (Terhune et al., 2010). Viral DNA and RNA were measured using quantitative real-time PCR as previously described (Reitsma et al., 2011) and oligonucleotides listed below.
2.3 Antibodies
The following antibodies with clone number were used: anti-GAPDH 0411 (Santa Cruz Biotechnology), anti-p21CIP1 CP74 (EMD Millipore), anti-STAT3 124H6 (Cell Signaling Technology), anti-cyclin B1 H-433 (Santa Cruz Biotechnology), anti-cyclin E C-19 (Santa Cruz Biotechnology) anti-cleaved caspase-3 D175 (Cell Signaling Technology), anti-cleaved PARP (Cell Signaling Technology). The antibodies against hCMV proteins were anti-pUL123 1B12, anti-pUL37 2A1D, and anti-pUL38 3D12 (generously provided by Dr. Tom Shenk), anti-pUL97 4-1 (Gill et al., 2012) (generously provided by Dr. Mark Prichard) and anti-pUL44 (Virusys). Secondary antibodies include anti-mouse and anti-rabbit HRP (Jackson ImmunoResearch). Cellular DNA was stained with DAPI (Life Technologies).
2.4 PCR Oligonucleotides
The following oligonucleotide pairs were used: cyclin B1 (5’-TGTGGATGCAGAAGATGGAGCTGA-3’ and 5’-TTGGTCTGACTGCTTGCTCTTCCT-3’), SOCS3 (5’- ATTCGCCTTAAATGCTCCCTGTCC-3’ and 5’-TGGCCAATACTTACTGGGCTGACA-3’), p21 (5’-GGCGTTTGGAGTGGTAGAAAT -3’ and 5’-TGGAGACTCTCAGGGTCGAAA-3’), GAPDH (5’-ACCCACTCCTCCACCTTTGAC -3’ and 5’-CTGTTGCTGTAGCCAAATTCGT-3’), UL123 (5’-GCCTTCCCTAAGACCACCAAT -3’ and 5’-ATTTTCTGGGCATAAGCCATAATC-3’), UL97 (5’-TCACGGTACATCGACGTTTCCACA -3’ and 5’-AGTGGGATACACGACACTGGTGAT-3’).
2.5 Cell Doubling
Cells were plated subconfluent in DMEM with 7% FBS. Following attachment to the plate, cells were treated with S3i-201. Cells were collected by dissociating into 500 μL of 1% trypsin (Life Technologies) and counted on a hemocytometer.
3. Results
3.1 Disruption of STAT3 alters proliferation and expression of cell cycle proteins during infection
We have recently observed that disruption of STAT3 inhibits hCMV DNA synthesis and virus production (Reitsma et al., 2013). In general, STAT3 influences the expression of genes involved in diverse activities including cell cycle regulation and proliferation (See review: Yu et al., 2009), and hCMV replication involves modulation of these activities (See review: Sanchez and Spector, 2008). Therefore, we hypothesized that inhibiting STAT3 might disrupt hCMV-mediated changes cell cycle regulation. To test our hypothesis, we utilized the compound S3i-201 which inhibits STAT3 dimerization and DNA binding (Siddiquee et al., 2007). To confirm inhibition, we pretreated U373 cells with S3i-201 and evaluated changes in expression of STAT3-regulated SOCS3 following IL6 stimulation (Reitsma et al., 2013). The addition of S3i-201 resulted in a 96% reduction in IL6-induced SOCS3 RNA levels (Figure 1A). MRC-5 cells are largely unresponsive to IL6 and we did not assess IL6-mediated STAT3 induction. Next, looking at proliferation, we quantified the number of U373 and MRC-5 cells over time treated with or without S3i-201. The addition of S3i-201 reduced cellular doubling for both cell types (Figure 1B). These data indicate that S3i-201 disrupts STAT3 signaling in U373 cells and proliferation of both U373 cells and MRC-5 primary fibroblasts.
Figure 1. Chemical inhibition of STAT3 activity decreases cellular proliferation.
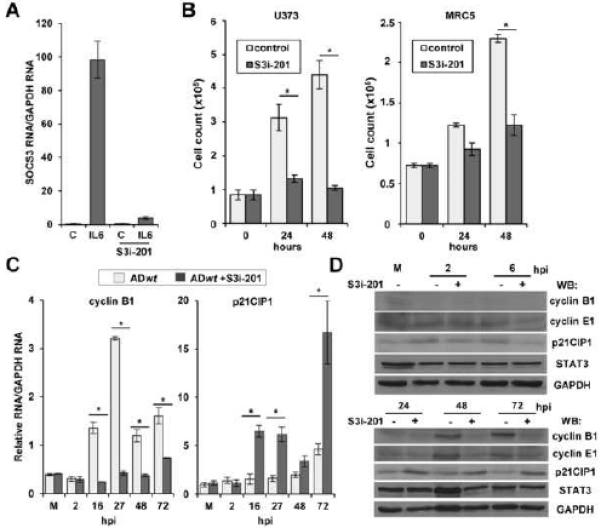
(A) U373 cells were pretreated with DMSO or 125 μM S3i-201. After 24 h, cells were treated with DMSO or recombinant human IL-6 for 45 min. SOCS3 RNA was quantified by qRT-PCR. Data represents the mean of two replicate experiments ±SEM. (B) U373 and MRC-5 cells were seeded subconfluent. After 24 h, U373 were treated with DMSO or 60 μM S3i-201 while fibroblasts were treated with 100 μM S3i-201. Total cell number was determined at the indicated times post drug treatment. (C) U373 cells were pretreated with DMSO or 125 μM S3i-201. After 24 h, cells were infected at 0.25 IU/cell using ADwt virus. Expression of RNAs was quantified by qRT-PCR. Data represents the mean of two replicate experiments ± SEM. (D) U373 cells were pretreated with drug and infected as described above. Western blot was completed using the indicated antibodies. (*p < 0.05)
Infection has been shown to alter the expression of several cell cycle regulators including cyclin B1 (Sanchez et al., 2003) and the CDK inhibitor p21CIP1 (Chen et al., 2001). To determine whether inhibition would alter hCMV-mediated changes in expression, we infected U373 cells treated with S3i-201using hCMV AD169 (ADwt). Addition of S3i-201 suppressed expression of cyclin B1 but increased p21CIP1 relative to the control (Figure 1C). We confirmed these changes using Western blot analysis and included an antibody against cyclin E1 (Figure 1D) which is also altered during infection (Bresnahan et al., 1998). These data demonstrate that inhibition of STAT3 alters hCMV-mediated changes in cell cycle genes.
3.2 Combining STAT3 inhibition and maribavir leads to a synergistic antiviral effect.
Previous studies suggest that manipulating the cell cycle can improve the antiviral activity of maribavir (MBV) (Hertel et al., 2007; Reitsma et al., 2011). To determine whether S3i-201 could influence MBV, we treated U373 cells with DMSO, MBV, S3i-201 or a combination of MBV and S3i-201. Viral DNA levels were determined. We observed that MBV or S3i-201 alone significantly decreased viral DNA levels by an average of 40% and 71%, respectively (Figure 2A). When infected cells were treated with both compounds, we quantified a 94% reduction in viral DNA levels. We observed similar changes using the hCMV clinically-derived isolate, TB40/E (Figure 2A).
Figure 2. Treatment with S3i-201 enhances the antiviral activity of maribavir (MBV).
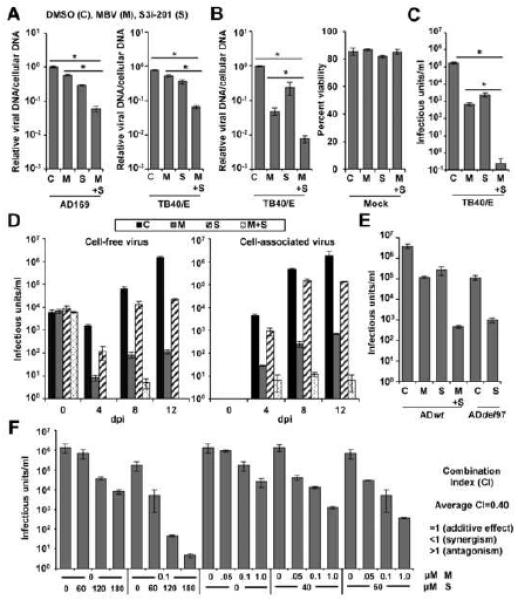
(A) U373 cells were pretreated with 100 μM S3i-201 (S) or DMSO (C). After 24 h, cells were infected at 3 IU/cell with either ADwt virus or TB40/E virus. Cells were treated with 40 μM MBV (M) or DMSO at the time of infection. At 72 hpi, viral genomes were quantified by qPCR and normalized to cellular DNA. Data represent the mean of two replicate experiments ±SEM. (B) MRC-5 fibroblasts were treated, infected, and analyzed as described above. Cell viability of uninfected cells was quantified using flow cytometry at 72 hpi. Data represent the mean of two replicate experiments ±SEM. (C) Fibroblasts were treated as above and infected at 3 IU/cell using TB40/E virus. Viral titers were determined from culture supernatants obtained at 96 hpi. Data represent the mean of four replicates ±SEM. (D) Fibroblasts were pretreated with 100 μM S3i-201 or DMSO. After 24 h, cells were infected at 0.25 IU/cell with TB40/E virus. Cells were treated with 40 μM MBV or DMSO at the time of infection. Cell free virus and cell associated virus were tittered at the indicated days post infection. Data represent the mean of two replicates ±SEM. (E) Fibroblasts were pretreated with 100 μM S3i-201 or DMSO. After 24 h, cells were infected at 3 IU/cell with either ADwt virus or ADdel97 virus. Cells were treated with 40 μM MBV or DMSO at the time of infection. Viral titers were determined from culture supernatants obtained at 96 hpi. Data represent the mean of four replicates ±SEM. (F) Fibroblasts were pretreated with different concentrations of S3i-201. After 24 h, cells were infected at 3 IU/cell with TB40/E virus. DMSO or MBV was added to cells at the time of infection. Viral titers were determined from culture supernatants obtained at 96 hpi. Data represent two replicates ± SEM. The average combination index (CI) as determined by CompuSyn software tool is indicated. (*p < 0.05)
We next investigated how the compounds influenced infection of primary fibroblasts. The addition of MBV resulted in an average 95.0% reduction in DNA accumulation while S3i-201 resulted in a 76.0% reduction (Figure 2B). The addition of both compounds resulted in an average 99.2% decrease. Drug treatments did not affect viability of uninfected growth-arrested cells (Figure 2B). We quantified the impact of these compounds on viral titers with MBV resulting in a 2.41 log reduction and S3i-201 resulting in a 1.86 log reduction (Figure 2C). When combined, we detected only one infected cell when titering virus from supernatants of four biological replicate experiments representing greater than a 5 log reduction. Similar results occurred for both cell-free and cell-associated virus during low MOI infection with TB40/E (Figure 2D), with lab-adapted strain AD169 at MOI 3 (Figure 2E) and upon addition of S3i-201 alone to a UL97-deficient viral infection (Figure 2E). Furthermore, the compounds reduced viral plaque formation over time (Figure S1). To provide evidence of synergism, we determined viral titers following treatment with varying concentrations of S3i-201 and MBV (Figure 2F). We evaluated the results using the CompuSyn software tool to determine the combination index (CI) based upon the theorem of Chou–Talalay (Chou, 2010). In general, drug combinations result in synergism (CI < 1; strong 0.1-0.3, moderate 0.7-0.85, slight 0.85-0.9), an additive effect (CI = 1), or antagonism (CI > 1) (Bijnsdorp et al., 2011). Using these non-constant ratio combinations of S3i-201 and MBV, we calculated the average CI value of 0.4. Examples of CI values determined upon disrupting multiple viral and/or cellular protein functions have been observed in studies with hepatitis C virus (Gottwein et al., 2013) and Kaposi's sarcoma-associated herpesvirus (Nayar et al., 2013). Our data suggest that STAT3 inhibition is acting synergistically with MBV to inhibit hCMV infection in vitro.
3.3 STAT3 inhibition has limited impact on the antiviral activity of ganciclovir.
The first line prophylaxis against hCMV is ganciclovir (GCV) (See review: Biron, 2006). The antiviral activity requires phosphorylation of GCV by the hCMV kinase pUL97 and functions by disrupting pUL54-dependent viral DNA synthesis (Littler et al., 1992; Sullivan et al., 1992). We next investigated the impact of inhibiting both STAT3 and viral DNA synthesis using GCV. Addition of GCV resulted in an average 89.8% reduction in DNA accumulation while S3i-201 resulted in an 83.0% reduction (Figure 3A). The addition of both compounds resulted in an average 95.3% decrease while not altering viability of uninfected cells (Figure 3A). Similar results also occurred when using 20 μM of GCV (Figure 3A). When evaluating titers, we observed 1.2, 1.6, and 1.8 log reductions using GCV, S3i-201, and GCV combined with S3i-201, respectively (Figure 3B). These data suggest that inhibition of STAT3 has limited impact on the activity of GCV. To further evaluate the inhibitors, we treated fibroblasts with increasing concentrations of S3i-201 in the presence or absence of two different concentrations of GCV. Again, we observed reduced viral yields upon increasing concentrations of S3i-201 (Figure 3C). The addition of 1 μM GCV resulted in an average 0.36 log reduction compared to control. In presence of 1 μM GCV, increasing concentrations of S3i-201 resulted in 0.8, 0.96, and 0.86 log reductions at 60, 120 and 180 μM S3i-201, respectively, when compared to GCV alone. We also evaluated 10 μM GCV which alone resulted in a substantial drop in viral yields compared to control. In the presence of 10 μM GCV, we quantified 0.43 log and 0.26 log reductions at 60 and 120 μM S3i-201, respectively, yet a small increase in viral yield using 180 μM. Our data indicate that S3i-201 does not improve the antiviral activity of GCV and is likely antagonistic to GCV.
Figure 3. S3i-201 treatment decreases UL97 expression.
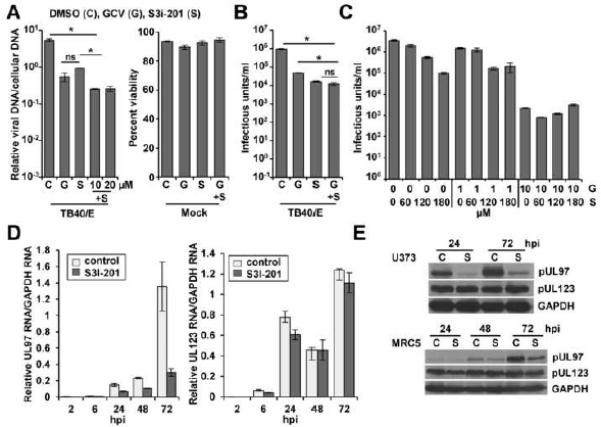
(A) Fibroblasts were pretreated with 100 μM S3i-201 (S) or DMSO (C). After 24 h, cells were infected at 3 IU/cell with either ADwt virus or TB40/E virus. Cells were treated with 10 or 20 μM GCV (G) and S3i-201 at the time of infection. At 72 hpi, viral genomes were quantified by qPCR and normalized to cellular DNA. Cell viability of uninfected cells was quantified using flow cytometry at 72 hpi. Data represent the mean of two replicate experiments ±SEM. (B) Fibroblasts treated as above using 10 μM GCV and infected at 3 IU/cell using TB40/E virus. Viral titers were determined from culture supernatants obtained at 96 hpi. Data represent the mean of four replicates ±SEM. (C) Fibroblasts were pretreated with different concentrations of S3i-201 with DMSO and then treated with 1 or 10 μM GCV upon infection. Cells were infected and analyzed as described above. Data represent the mean of two replicates ±SEM. (D) U373 cells were pretreated with DMSO or 100 μM S3i-201. After 24 h, cells were infected at 0.25 IU/cell with TB40/E virus. Expression of RNAs was quantified by qRT-PCR. Data represents the mean of two replicate experiments ±SEM. (E) U373 cells and MRC5 fibroblasts were pretreated with S3i-201, infected as described above, and analyzed by Western blot. (*p < 0.05)
Previous studies demonstrated that MBV is antagonistic to GCV (Chou and Marousek, 2006), which requires pUL97-mediated phosphorylation for its activity. To assess the impact of STAT3 inhibition on UL97 expression, we quantified changes in RNA levels during infection with and without S3i-201. Beginning at 24 hpi, we detected significant reductions in UL97 expression upon inhibition of STAT3 (Figure 3D). We observed similar levels of the hCMV immediate-early UL123 RNA between conditions. These observations were confirmed by Western blot analysis (Figure 3E). Unlike MBV, which inhibits kinase activity, our results indicate that inhibiting STAT3 disrupts UL97 expression. This response is likely indirect since S3i-201 inhibits hCMV DNA synthesis. Overall, these data suggest that S3i-201 is antagonistic to GCV activity.
3.4 Inhibition of STAT3 and pUL97 promotes caspase-dependent death of infected cells.
When primary fibroblasts were exposed to 40 μM MBV and 100 μM S3i-201, we observed a change in the morphology of infected but not uninfected cells (Figure 4B) as well as reduction in the total number of adherent cells (Figure 4C). This did not occur when using GCV or when using the tumor cells, U373 cells (data not shown). We hypothesized that inhibition of both STAT3 and kinase activity resulted in death of the infected cells. To test this hypothesis, we evaluated changes in the activation of caspase-3 and poly (ADP-ribose) polymerase (PARP) using Western blot. We detected cleaved caspase-3 and cleaved PARP using both compounds but not when using either compound alone or control (Figure 4A). A similar response occurred upon induction of apoptosis by using staurosporine with uninfected fibroblasts. We observed little difference in hCMV IE1 expression yet decreased levels of pUL37, pUL38, pUL44 and cyclin B1 when using both compounds. To evaluate the role of caspases, we completed experiments in the presence and absence of caspase inhibitors. Caspase inhibition reverted the morphology change (Figure 4B) seen when using MBV and S3i-201 together. Upon the addition of both MBV and S3i-201, we quantified an average 69.5% reduction in adherent cells which was fully rescued upon caspase inhibition (Figure 4C). No differences were detected when using either compound alone. Finally, we evaluated changes in viral DNA synthesis and viral titers. Again, MBV with S3i-201 resulted in a 98.3% decrease in viral DNA levels which was unaffected by the addition of caspase inhibitors (Figure 4D). However, addition of inhibitors resulted in a partial rescue of hCMV virus production going from a 5.2 log to a 4.1 log reduction (Figure 4D). Our data support the conclusion that inhibition of both STAT3 and hCMV kinase induces caspase-dependent loss of infected cells from the culture which contributes to the almost complete loss of viral yield.
Figure 4. Caspase-dependent loss of infected cells upon inhibition of both STAT3 and pUL97.
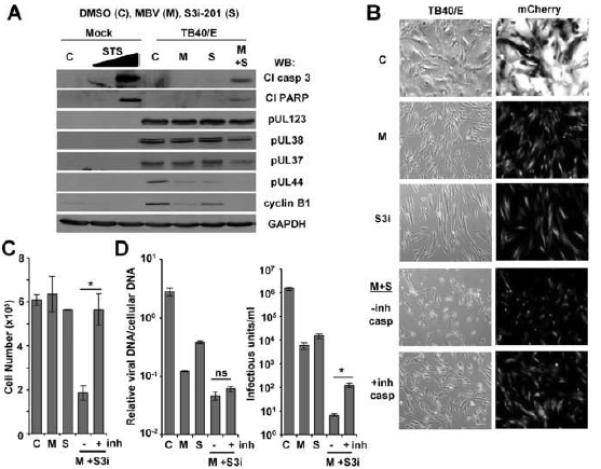
(A) MRC-5 fibroblasts were pretreated with 100 μM S3i-201 (S) or DMSO (C). After 24 h, cells were infected at 5 IU/cell using TB40/E virus. Cells were treated with 40 μM MBV (M) or MBV and S3i-201 at the time of infection and harvested at 48 hpi. Mock infected cells were treated with 0, 0.5, or 1.0 μM staurosporine (STS) for 3 h prior to Western blot analysis using the indicated antibodies. (B) MRC-5 fibroblasts were treated and infected as described above. At 48 hpi, cells treated with MBV and S3i-201 were treated with 50 μM of Z-VAD and Z-DEVD-FMK or DMSO control. At 72 hpi, live cells were imaged for viral infection (TB40/E expressing mCherry) and cell morphology (brightfield) using fluorescence microcopy. (C) MRC-5 fibroblasts were treated and infected as described above. At 48 hpi, cells treated with MBV and S3i-201 were treated with 50 μM of Z-VAD and Z-DEVD-FMK. At 72 hpi, adherent cells were harvested and total cell number was quantified using flow cytometry. Data represent the mean of three replicates ±SEM. (D) Fibroblasts were treated as described above. At 72 hpi, viral genomes were quantified by qPCR and normalized to cellular DNA (left panel). Viral titers were determined from culture supernatants obtained at 96 hpi (right panel). Data represent the mean of four replicates ±SEM. (*p < 0.05)
4. Discussion
The identification of pharmaceutical drug synergism has drastically improved the management of numerous diseases as well as treatment of bacterial and viral infections (See review: Chou, 2006). Our data indicate that inhibition of STAT3 synergizes with MBV to inhibit hCMV replication in vitro. When we treated infection with the STAT3 inhibitor, S3i-201, in the presence of MBV, we observed a substantial drop in DNA synthesis. A slight decrease occurred when combining S3i-201 with GCV. However, and to our surprise, combining S3i-201 with MBV but not GCV resulted in an almost complete loss of viral yield.
The difference in responses when combining compounds might be explained by the drop in UL97 expression observed upon S3i-201 treatment. Furthermore, past studies have shown that a reduction in UL97 expression or inhibition of kinase activity minimizes the antiviral efficacy of GCV (Chou and Marousek, 2006). Kinase-mediated phosphorylation of GCV is necessary for its antiviral activity (Littler et al., 1992; Sullivan et al., 1992). In addition, it is conceivable that S3i-201-mediated reduction in pUL97 levels could minimize the concentration of MBV needed to inhibit infection. Finally, previous studies suggest that manipulating the cell cycle could improve the antiviral activity of MBV (Hertel et al., 2007; Reitsma et al., 2011). We determined that inhibition of STAT3 influenced the expression of several cell cycle markers that are associated with hCMV infection. This suggests that inhibiting the pro-proliferative activities of STAT3 may also contribute to its synergistic effect with MBV.
Interestingly, at elevated concentrations of both MBV and S3i-201, infected cells, but not uninfected cells, exhibited activated caspase 3 and PARP cleavage as well as loss of the cells from the culture. The addition of caspase inhibitors prevented this loss and partially rescued viral titers without altering viral DNA levels. hCMV expresses diverse proteins that contribute to blocking cell death including pUL37 and pUL38 (See review: Brune, 2011), and expression of these proteins is lower in the presence of both compounds. Furthermore, STAT3 itself suppress expression of pro-apoptotic genes (Timofeeva et al., 2013). Taken together, death of the infected cell is likely a complex relationship between a reduction in anti-apoptotic viral proteins and inhibition of STAT3 activities.
Initial studies investigating MBV demonstrated that the compound exhibited promising antiviral activity against hCMV and low toxicity in patients. Unfortunately, in phase III clinical trials, MBV failed to prevent hCMV viremia after stem cell transplantation (See review: Marty et al., 2011). Studies investigating reasons for MBV’s poor efficacy suggested that the in vivo efficacy might not be as high as in cell culture conditions (Chou et al., 2006; Snydman, 2011). The compound has been returned to clinical trials to re-evaluate dosing in cases of resistance to first line antivirals. In general, advantages of drug synergism include increased inhibitory effect and decreased drug dosing while maintaining the same efficacy to avoid toxicity (See review: Chou, 2006). Our findings reveal that inhibiting STAT3 in the context of MBV displays both these properties of drug synergism. We have demonstrated that chemical antagonists of STAT3 alone significantly inhibit hCMV infection. Sen et al. (Sen et al., 2012) have also demonstrated that an inhibitor of STAT3 disrupts replication of the human herpesvirus, Varicella Zoster Virus. STAT3 inhibitors are being developed and are currently entering clinical trials as anti-cancer agents (See review: Johnston and Grandis, 2011). Several FDA-approved compounds have been shown to indirectly disrupt STAT3 signaling including celecoxib and sorafenib (Liu et al., 2012; Yang et al., 2008) and both compounds disrupt hCMV replication in vitro (Baryawno et al., 2011; Michaelis et al., 2011). Our studies suggest that treating infection with both S3i-201 and MBV may be a relevant strategy to inhibit hCMV infection.
Supplementary Material
Highlights.
S3i-201 synergizes with the hCMV kinase inhibitor maribavir to disrupt infection.
Specific concentrations of S3i-201 and maribavir induce death of only the infected cells.
STAT3 inhibitors including S3i-201 are a potential approach for anti-hCMV therapy.
Acknowledgements
We thank M. Nevels, C. Paulus, and M. Hakki for their comments and T. Bigley and A. Vallejos for calculating synergism. This work is supported by NIH R01AI083281 to S. Terhune.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baryawno N, Rahbar A, Wolmer-Solberg N, Taher C, Odeberg J, Darabi A, Khan Z, Sveinbjornsson B, FuskevAg OM, Segerstrom L, Nordenskjold M, Siesjo P, Kogner P, Johnsen JI, Soderberg-Naucler C. Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J Clin Invest. 2011;121:4043–4055. doi: 10.1172/JCI57147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijnsdorp IV, Giovannetti E, Peters GJ. Analysis of drug interactions. Methods Mol Biol. 2011;731:421–434. doi: 10.1007/978-1-61779-080-5_34. [DOI] [PubMed] [Google Scholar]
- Biron KK. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 2006;71:154–163. doi: 10.1016/j.antiviral.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Bresnahan WA, Albrecht T, Thompson EA. The cyclin E promoter is activated by human cytomegalovirus 86-kDa immediate early protein. J Biol Chem. 1998;273:22075–22082. doi: 10.1074/jbc.273.34.22075. [DOI] [PubMed] [Google Scholar]
- Britt W. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr Top Microbiol Immunol. 2008;325:417–470. doi: 10.1007/978-3-540-77349-8_23. [DOI] [PubMed] [Google Scholar]
- Brune W. Inhibition of programmed cell death by cytomegaloviruses. Virus Res. 2011;157:144–150. doi: 10.1016/j.virusres.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Chen Z, Knutson E, Kurosky A, Albrecht T. Degradation of p21cip1 in cells productively infected with human cytomegalovirus. J Virol. 2001;75:3613–3625. doi: 10.1128/JVI.75.8.3613-3625.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, Marousek GI. Maribavir antagonizes the antiviral action of ganciclovir on human cytomegalovirus. Antimicrob Agents Chemother. 2006;50:3470–3472. doi: 10.1128/AAC.00577-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S, Van Wechel LC, Marousek GI. Effect of cell culture conditions on the anticytomegalovirus activity of maribavir. Antimicrob Agents Chemother. 2006;50:2557–2559. doi: 10.1128/AAC.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- Gill RB, James SH, Prichard MN. Human cytomegalovirus UL97 kinase alters the accumulation of CDK1. J Gen Virol. 2012;93:1743–1755. doi: 10.1099/vir.0.039214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein JM, Jensen SB, Li YP, Ghanem L, Scheel TK, Serre SB, Mikkelsen L, Bukh J. Combination treatment with hepatitis C virus protease and NS5A inhibitors is effective against recombinant genotype 1a, 2a, and 3a viruses. Antimicrob Agents Chemother. 2013;57:1291–1303. doi: 10.1128/AAC.02164-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakki M, Chou S. The biology of cytomegalovirus drug resistance. Curr Opin Infect Dis. 2011;24:605–611. doi: 10.1097/QCO.0b013e32834cfb58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009;5:e1000275. doi: 10.1371/journal.ppat.1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel L, Chou S, Mocarski ES. Viral and cell cycle-regulated kinases in cytomegalovirus-induced pseudomitosis and replication. PLoS Pathog. 2007;3:e6. doi: 10.1371/journal.ppat.0030006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science. 2008;320:797–799. doi: 10.1126/science.1152095. [DOI] [PubMed] [Google Scholar]
- Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interv. 2011;11:18–26. doi: 10.1124/mi.11.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamil JP, Hume AJ, Jurak I, Munger K, Kalejta RF, Coen DM. Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proc Natl Acad Sci U S A. 2009;106:16823–16828. doi: 10.1073/pnas.0901521106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler E, Stuart AD, Chee MS. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature. 1992;358:160–162. doi: 10.1038/358160a0. [DOI] [PubMed] [Google Scholar]
- Liu DB, Hu GY, Long GX, Qiu H, Mei Q, Hu GQ. Celecoxib induces apoptosis and cell-cycle arrest in nasopharyngeal carcinoma cell lines via inhibition of STAT3 phosphorylation. Acta Pharmacol Sin. 2012;33:682–690. doi: 10.1038/aps.2012.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, Young JA, Rodriguez T, Maertens J, Schmitt M, Einsele H, Ferrant A, Lipton JH, Villano SA, Chen H, Boeckh M, Maribavir −300 Clinical Study, G. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial. The Lancet infectious diseases. 2011;11:284–292. doi: 10.1016/S1473-3099(11)70024-X. [DOI] [PubMed] [Google Scholar]
- Mercorelli B, Lembo D, Palu G, Loregian A. Early inhibitors of human cytomegalovirus: state-of-art and therapeutic perspectives. Pharmacology & therapeutics. 2011;131:309–329. doi: 10.1016/j.pharmthera.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis M, Paulus C, Loschmann N, Dauth S, Stange E, Doerr HW, Nevels M, Cinatl J., Jr. The multi-targeted kinase inhibitor sorafenib inhibits human cytomegalovirus replication. Cellular and molecular life sciences : CMLS. 2011;68:1079–1090. doi: 10.1007/s00018-010-0510-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DP, Savaryn JP, Moorman NJ, Shenk T, Terhune SS. Human cytomegalovirus UL28 and UL29 open reading frames encode a spliced mRNA and stimulate accumulation of immediate-early RNAs. J Virol. 2009;83:10187–10197. doi: 10.1128/JVI.00396-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayar U, Lu P, Goldstein RL, Vider J, Ballon G, Rodina A, Taldone T, Erdjument-Bromage H, Chomet M, Blasberg R, Melnick A, Cerchietti L, Chiosis G, Wang YL, Cesarman E. Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies. Blood. 2013 doi: 10.1182/blood-2013-01-479972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor CM, Shenk T. Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J Virol. 2011;85:3700–3707. doi: 10.1128/JVI.02442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prichard MN. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev Med Virol. 2009;19:215–229. doi: 10.1002/rmv.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma JM, Sato H, Nevels M, Terhune SS, Paulus C. Human Cytomegalovirus IE1 Protein Disrupts Interleukin-6 Signaling by Sequestering STAT3 in the Nucleus. J Virol. 2013 doi: 10.1128/JVI.01197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma JM, Savaryn JP, Faust K, Sato H, Halligan BD, Terhune SS. Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe. 2011;9:103–114. doi: 10.1016/j.chom.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Sanchez V, McElroy AK, Spector DH. Mechanisms governing maintenance of Cdk1/cyclin B1 kinase activity in cells infected with human cytomegalovirus. J Virol. 2003;77:13214–13224. doi: 10.1128/JVI.77.24.13214-13224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Spector DH. Subversion of cell cycle regulatory pathways. Curr Top Microbiol Immunol. 2008;325:243–262. doi: 10.1007/978-3-540-77349-8_14. [DOI] [PubMed] [Google Scholar]
- Sen N, Che X, Rajamani J, Zerboni L, Sung P, Ptacek J, Arvin AM. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis. Proc Natl Acad Sci U S A. 2012;109:600–605. doi: 10.1073/pnas.1114232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A. 2007;104:7391–7396. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snydman DR. Why did maribavir fail in stem-cell transplants? The Lancet infectious diseases. 2011;11:255–257. doi: 10.1016/S1473-3099(11)70033-0. [DOI] [PubMed] [Google Scholar]
- Sullivan V, Talarico CL, Stanat SC, Davis M, Coen DM, Biron KK. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature. 1992;359:85. doi: 10.1038/359085a0. [DOI] [PubMed] [Google Scholar]
- Terhune SS, Moorman NJ, Cristea IM, Savaryn JP, Cuevas-Bennett C, Rout MP, Chait BT, Shenk T. Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 2010;6:e1000965. doi: 10.1371/journal.ppat.1000965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeeva OA, Tarasova NI, Zhang X, Chasovskikh S, Cheema AK, Wang H, Brown ML, Dritschilo A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc Natl Acad Sci U S A. 2013;110:1267–1272. doi: 10.1073/pnas.1211805110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Van Meter TE, Buettner R, Hedvat M, Liang W, Kowolik CM, Mepani N, Mirosevich J, Nam S, Chen MY, Tye G, Kirschbaum M, Jove R. Sorafenib inhibits signal transducer and activator of transcription 3 signaling associated with growth arrest and apoptosis of medulloblastomas. Mol Cancer Ther. 2008;7:3519–3526. doi: 10.1158/1535-7163.MCT-08-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.