

Abstract
Treatment of allyl-1,1-dichlorovinyl ethers with n-BuLi at −78°C, followed by quenching with ketones, epoxides, and oxetanes, leads to highly substituted β-, γ-, and δ-lactones in good to excellent yields.
Electron-rich alkynes, such as ynamines and ynol ethers, are functional groups that possess significant potential in organic chemistry for the formation of carbon-carbon bonds.1 Due to their linear geometry, alkynyl ethers are relatively unhindered to approach by functional groups present in the same or different molecules; furthermore, alkynyl ethers can prospectively form up to three new bonds in a single reaction (Figure 1).
Figure 1.

Synthesis and reactivity of 1-alkynyl ethers.
Alkyllithium treatment of 1,1- and 1,2-dichlorovinyl ethers has been the traditional method for the synthesis of both substituted and unsubstituted 1-alkynyl ethers.2 In addition, we have shown that α–alkoxyketones are precursors of alkynyl ethers via treatment of the derived enol triflates or phosphates with potassium tert-butoxide or Schlosser’s base at −78°C.3 This process provides an additional protocol for the preparation of highly substituted 1-alkynyl ethers that cannot be prepared directly by electrophile trapping of lithioalkynyl ether intermediates available from 1,1- and 1,2-dichlorovinyl ethers.
We have previously shown that subjecting allyl-1,1-dichlorovinyl ethers to excess n-BuLi at −78°C in THF for 10 minutes, followed by reaction quench with alcohols, phenols, amines, or amides, leads to γ,δ–unsaturated carboxylic acid derivatives in high yields.4 These products likely arise from nucleophilic trapping of allyl ketene intermediates, which are formed from [3,3]-sigmatropic rearrangement of the in-situ-generated allyl-alkynyl ethers. The process is stereospecific and takes place through a cyclic transition state, as demonstrated by the failure of conformationally restricted substrates to undergo rearrangement. Intriguingly, a similar treatment of allyl-1,2-dichlorovinyl ethers with n-BuLi does not produce the expected rearrangement products after reaction quench but instead gives rise to the corresponding allylic alcohols (Figure 2). However, in situ generation of allyl-alkynyl ethers by treatment of the corresponding allyl enol triflates with KOt-Bu at −85°C, followed by quenching with alcohols, also produces the expected rearrangement products in good yields.3,5
Figure 2.

n-BuLi-induced [3,3]-sigmatropic rearrangement/ketene trapping of allyl-1,1-dichlorovinyl ethers.
Mechanistically, trapping of the intermediate lithioalkynyl ether or lithioketene species with alcohols, phenols, amines or amides likely proceeds through an initial protonation step followed by subsequent intermolecular nucleophilic attack on the neutral ketene species. We wished to assess if the organometallic intermediate in this process could be intercepted by a suitable carbon electrophile capable of subsequent intramolecular nucleophilic trap of the ketene intermediate, thus allowing the formation of products containing two new carbon-carbon bonds and a ring. In this communication we report our findings on the n-BuLi-induced reaction of allyl-1,1-dichlorovinyl ethers with ketones, epoxides, and oxetanes.
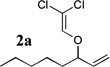
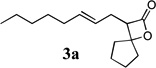
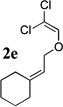
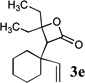
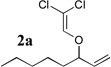
The allyl-1,1-dichlorovinyl ethers 2 used in this study were prepared in high yield (65–90%) from the corresponding allylic alcohols 1 by formylation (formic acetic anhydride, pyridine) and Wittig reaction (PPh3, CCl4, THF, 60°C, 7h).4 Subjecting 2a to n-BuLi (3 equiv) in THF at −78°C for 45 minutes, followed by addition of cyclopentanone (2 equiv) gave, after reaction quench at −78°C with saturated NaHCO3, spiro-β-lactone 3a in 70% isolated yield. Interestingly, a similar reaction of 2a with n-BuLi followed by acetaldehyde gave rise not to the expected β-lactone but rather β-hydroxy acid 4 as a mixture of diastereomers; elevating the reaction temperature before or after addition of acetaldehyde led to extensive side product formation due to decomposition of the ketene intermediate. Similar results were obtained with other aldehydes. These results suggest that the Thorpe-Ingold (gem-dialkyl) effect6 is important in facilitating the low-temperature 4-exo (or endo)16-dig ring closure to the strained β-lactone product for ketene intermediates arising from ketone quench.
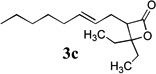
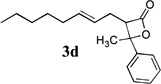
β-Lactones have been the subject of much synthetic effort due to their widespread occurrence in nature and intriguing biological activities,7 as well as their utility as synthetic intermediates8 and as monomers for the preparation of biodegradable polymers.9 Thus, the development of methods for their rapid preparation from simple precursors is of high importance to the fields of both pharmaceutical and polymer synthesis. A variety of symmetrical (Table 1, entries 1–3, 5–6, 8, 10) and unsymmetrical (Table 1, entries 4, 7, and 9) ketones may be used in the present reaction to produce the corresponding rearranged β-lactones. In addition, allyl-1,1-dichlorovinyl ethers derived from primary (entries 6–10)10–12 and secondary (entries 1–5) allylic alcohols could be employed successfully in the reaction, giving rise to complex β-lactone products, such as spiro 5-4 (entries 1 and 6) and 6-4 (entries 2 and 8) ring systems. Tetrasubstituted alkenes (entry 10) also proved to be excellent substrates for the rearrangement/ketone trapping process. However, quenching the rearrangement reaction with ketimines (such as N-phenylcyclohexyl imine, Scheme 1) instead of ketones did not provide β-lactams,13 even at elevated temperatures (rt) or in the presence of Lewis acids such as BF3•OEt2 (vide infra).
Table 1.
Scope of the n-BuLi-induced [3,3]sigmatropic rearrangement/ketone trapping reactiona

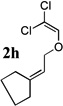
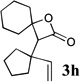
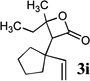
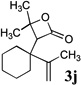
Compound 2 was treated with 3 equiv n-BuLi at −78°C for 45 minutes under argon atmosphere, and then the ketone (2 equiv) was added and the reaction was stirred for an additional 45 minutes before saturated NaHCO3 solution was added.
Isolated yield after silica gel chromatography.
Products obtained as a ~1:1 mixture of diastereomers.
Scheme 1.

Ketone vs. aldehyde/imine quench of the organometal intermediate arising from n-BuLi treatment of 2a.
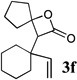
All of the β-lactones obtained were stable to silica gel chromatography, providing good to excellent isolated yields. However, low temperature (−78°C) treatment of the products with stoichiometric amounts of Lewis acids such as TMSOTf led to a rapid elimination reaction to afford the corresponding unconjugated carboxylic acids;14 for lactone 3f, the same carboxylic acid product (4f) was obtained quantitatively both in the presence and absence of the carbon nucleophile allyltrimethylsilane (Scheme 2).15
Scheme 2.

Lewis-acid mediated elimination reactions of β-lactones; attempted trapping of intermediate enolate A.
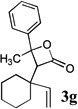
Intramolecular trapping of the ketene intermediate by the incipient alkoxide would be expected to provide a lactone enolate (e.g., species A, Scheme 2), which could be intercepted by yet another electrophile to provide α,α-disubstituted β-lactones. To test this hypothesis, ether 2h was subjected to rearrangement and trapping with cyclohexanone under standard reaction conditions, followed by quenching with iodomethane (2.5 equiv) and HMPA (20% by volume) and warming to room temperature for one hour. A 58% yield of β-lactone 3h was obtained, indicating that no enolate trapping had occurred. Although the high degree of steric hindrance around the enolate α-carbon in structure A may explain the reluctance of this species to undergo electrophilic trap, when rearrangements of the less hindered substrate 2a were quenched with excess ketone electrophiles, no α,α-disubstituted β-lactone products were recovered from the reaction mixtures. Thus, it appears that the added ketones may also be serving as a proton source for the quenching of the β-lactone enolate intermediate.
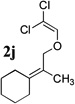
To extend the utility of this reaction to the preparation of γ– and δ-lactones, we envisioned utilizing epoxides and oxetanes for electrophilic trapping of the organometallic intermediate. It was also anticipated that the presence of a quaternary center in the alkoxyketene precursor would no longer be necessary in order to obtain high yields of lactone products due to the increased favorability of 5- or 6-exo-dig ring closures.16 Treatment of 2e with n-BuLi at −78°C for 40 minutes, followed by addition of 1-hexene oxide gave rise only to products arising from dimerization of the ketene intermediate. However, addition instead of equimolar amounts of 1-hexene oxide and BF3•OEt2 to the organolithium intermediate furnished an 89% yield of γ-butyrolactone 3o as a 1:1 mixture of diastereomers. Similarly, exposure of 2j to excess n-BuLi at −78 °C, followed by addition of trimethylene oxide and BF3•OEt2 gave rise to δ-lactone 3r in 68% yield (Scheme 3).
Scheme 3.

Synthesis of γ– and δ–lactones
The scope of γ- and δ-lactone synthesis available by this method is illustrated in Table 2. A variety of terminal and internal epoxides, as well as both substituted and unsubstituted oxetane, may be employed successfully in this protocol, allowing the preparation of highly complex lactone products. It was found that the yields of lactones available from ethers 2e, 2h and 2j were generally higher that those obtained from similar reactions of substrate 2a; indeed, products arising from dimerization of the ketene intermediate were also isolated from the rearrangement/trapping reactions of 2a. The decreased reactivity of epoxides and oxetanes as electrophiles, and the lower steric hindrance of the lithioketene intermediate derived from 2a compared to those arising from 2e, 2h, and 2j may explain these findings.
Table 2.
Synthesis of γ- and δ-lactones from allyl-1,1-dichlorovinyl ethers

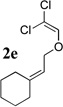
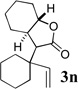
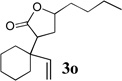
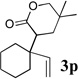
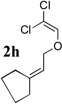

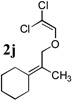
Compound 2 was treated with 3 equiv n-BuLi at −78°C for 45 minutes under argon atmosphere, and then the epoxide or oxetane (2 equiv) and BF3•OEt2 (2 equiv) was added and the reaction was stirred for an additional 45 minutes before saturated NaHCO3 solution was added.
Isolated yield after silica gel chromatography.
Product obtained as a ~1:1 mixture of regioisomers.
Products obtained as a ~1:1 mixture of diastereomers.
Conclusion
In summary, we have shown that allyl-1,1-dichlorovinyl ethers are precursors of β–, γ–, and δ–lactones via trapping of the organometallic intermediate arising from [3,3]-sigmatropic rearrangement reaction with ketones, epoxides and oxetanes. Continued attempts to trap the penultimate lactone enolate in reactions with electrophiles are under way and will be reported in due course.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health (SC3 GM 096899-01) and the Henry Dreyfus Teacher-Scholar program for their generous support of our research program.
Footnotes
Electronic Supplementary Information (ESI) available: full experimental details and charcterization data for all compounds reportd. See DOI: 10.1039/b000000x/
Notes and references
- 1.For reviews of the chemistry of ynol ethers and the methods for the synthesis of ynol ethers, see: Brandsma L, Bos HJ, Arens JF. In: The Chemistry of Acetylenes. Viehe HG, editor. New York: Marcel Dekker; 1969. pp. 751–860. Stang PJ, Zhdankin VV. In: The Chemistry of Triple-Bonded Functional Groups. Patai S, editor. chapter 19. New York: John Wiley & Sons; 1994.
- 2.(a) Smithers RH. Synthesis. 1985:556. [Google Scholar]; (b) Himbert G, Loffler A. Synthesis. 1992:495. [Google Scholar]; (c) Moyano A, Charbonnier F, Greene AE. J. Org. Chem. 1987;52(2):2919. [Google Scholar]
- 3.Sosa JR, Tudjarian AA, Minehan TG. Org. Lett. 2008;10:5091. doi: 10.1021/ol802147h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Christopher A, Brandes D, Kelly S, Minehan TG. Org. Lett. 2006;8:451. doi: 10.1021/ol052685j. (b) The allyl 1,1-dichlorovinyl ethers prepared were stable at room temperature and at 60 °C in THF under the conditions for their formation from the corresponding formate esters. The [3,3]-sigmatropic rearrangement of allyl 1,1-dichlorovinyl ethers at temperatures >100 °C has been described: Morimoto T, Sekiya M. Synthesis. 1981:308.
- 5.Tudjarian AA, Minehan TG. J. Org. Chem. 2011;76:3576. doi: 10.1021/jo200271s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beesley RM, Ingold CK, Thorpe F. J. Chem. Soc. 1915;107:1080. [Google Scholar]
- 7.(a) Wovkulich PM, Shankaran K, Kiegiel J, Uskokovic MR. J. Org. Chem. 1993;58:832. [Google Scholar]; (b) Pu Y, Lowe C, Sailer M, Vederas JC. J. Org. Chem. 1994;59:3642. [Google Scholar]; (c) Taunton J, Collins JL, Schreiber SL. J. Am. Chem. Soc. 1996;118:10412. [Google Scholar]; (d) Wan Z, Nelson SG. J. Am. Chem. Soc. 2000;122:10470. [Google Scholar]; (e) Nelson SG, Cheung WS, Kassick AJ, Hilfiker MA. J. Am. Chem. Soc. 2002;124:13654. doi: 10.1021/ja028019k. [DOI] [PubMed] [Google Scholar]; (f) Shen X, Wasmuth AS, Zhao J, Zhu C, Nelson SG. J. Am. Chem. Soc. 2006;128:7438. doi: 10.1021/ja061938g. [DOI] [PubMed] [Google Scholar]
- 8.(a) Pommier A, Pons J-M. Synthesis. 1993:441. [Google Scholar]; (b) Yang HW, Romo D. J. Org. Chem. 1997;62:4. doi: 10.1021/jo9619488. [DOI] [PubMed] [Google Scholar]; (c) Dymock BW, Kocienski PJ, Pons J-M. Synthesis. 1998:1655. [Google Scholar]; (d) Reddy LR, Saravanan P, Corey EJ. J. Am Chem Soc. 2004;126:6230. doi: 10.1021/ja048613p. [DOI] [PubMed] [Google Scholar]; (e) Purohit VC, Richardson RD, Smith JW, Romo D. J. Org. Chem. 2006;71:4549. doi: 10.1021/jo060392d. [DOI] [PubMed] [Google Scholar]
- 9.Albertsson A-C, Varma IK. Biomacromol. 2003;4:1466. doi: 10.1021/bm034247a. [DOI] [PubMed] [Google Scholar]
- 10.For a recent preparation of primary alcohol 1e, see: Koo J, Park H-S, Shin S. Tetrahedron Lett. 2013;54:834.
- 11.For a recent prepararion of primary alcohol 1h, see: Rigoli JW, Moyer SA, Pearce SD, Schomaker JM. Org. Biomol. Chem. 2012;10:1746. doi: 10.1039/c2ob06921k.
- 12.For a recent preparation of primary alcohol 1j, see: Kuroda C, Koshio H, Koito A, Sumiya H, Murase A, Hirono Y. Tetrahedron. 2000;56:6441.
- 13.Cossio FP, Arrieta A, Sierra MA. Acc. Chem. Res. 2008;41:925–936. doi: 10.1021/ar800033j. [DOI] [PubMed] [Google Scholar]
- 14.For Lewis acid-mediated rearrangement of β-lactones to α,β-unsaturated carboxylic acids, see: Black TH, Zhang Y, Huang J, Smth DC, Yates BE. Synth. Comm. 1995;25:15.
- 15.For a review of the utility of β-lactones as intermediates in natural product synthesis, see: Wang Y, Tennyson RL, Romo D. Heterocycles. 2004;64:605.
- 16.According to Baldwin’s rules: 4-exo dig ring closures are stereoelectronically disfavored, but 4-endo and 5- and 6-exo ring closures are allowed: Baldwin JE. J. Chem. Soc. Chem Commun. 1976;6:734.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.