

Abstract
Fifty-one chemicals including derivatives of sixteen flavonoids, three stilbenes, six pyrenes, seven naphthalenes, seven phenanthrenes, ten biphenyls, 17β-estradiol, and estrone were examined for their abilities to induce Reverse Type I binding spectra with human cytochrome P450 (P450) 1B1 and to inhibit 7-ethoxyresorufin O-deethylation (EROD) activities catalyzed by P450 1B1. Forty nine chemicals showed Reverse Type I spectra with P450 1B1 and we found that 3,5,7-trihydroxyflavone, 3′,4′-dimethoxy-5,7-dihydroxyflavone, 4′-methoxy-5,7-dihydroxyflavone, α- and β-naphthoflavones, 2,4,3′,5′-tetramethoxystilbene, pyrene, and several acetylenic pyrenes and phenanthrenes were strong inducers of the spectra and also potent inhibitors of EROD activities catalyzed by P450 1B1. Spectral dissociation constant (Ks) and the magnitude of the binding (ΔAmax/Ks) of 49 chemicals were correlated with the inhibition potencies of EROD activities by these chemicals (correlation coefficients (r) of 0.72 and 0.74, respectively). The Ks and ΔAmax/Ks values were more correlated with IC50 values when compared in a group of derivatives of flavonoids, stilbenes, and estrogens (r=0.81 and 0.88, respectively) or a group of derivatives of pyrenes, naphthalenes, phenanthrenes, and biphenyls (r=0.88 and 0.91, respectively). Among 14 flavonoids examined, 3,5,7-trihydroxyflavone and 4′-methoxy- and 3′,4′-dimethoxy-5,7-dihydroxyflavone were more active than flavone in interacting with P450 1B1, but the respective 7,8-dihydroxyflavones were less active. Pyrene itself was highly active in interacting with P450 1B1, but its binding was slightly decreased when substituted with acetylenic groups. In contrast, substitution of naphthalene with methyl- and ethyl propargyl ethers led to more interaction with P450 1B1 than with naphthalene itself. Similarly, substitution on phenanthrene or biphenyl with acetylenic groups and propargyl ethers increased affinities to P450 1B1. These results suggest that the Reverse Type I binding of chemicals to P450 1B1 may determine how they interact with and inhibit the catalytic activity of the enzyme. Substitutions on the compounds with various acetylenic groups and propargyl ethers cause an increase or decrease of their affinities to P450 1B1, depending on the parent compound used.
Introduction
P450 comprises a superfamily of enzymes that catalyze oxidative metabolism of a variety of xenobiotic chemicals including drugs, toxic chemicals, and chemical carcinogens as well as endogenous compounds such as steroids, fatty acids, prostaglandins, and lipid-soluble vitamins (1). P450 1B1 has been shown be expressed in extrahepatic organs and has important roles in the metabolic activation of polycyclic aromatic hydrocarbons (PAHs)1 and heterocyclic aromatic amines to active metabolites that attack DNA in the cell (2,3), as well as estrogens and possibly other endogenous compounds. Several lines of evidence have suggested that the levels of expression and catalytic roles of polymorphic P450 1B1 are related to the susceptibilities of individuals to chemical carcinogenesis in humans (4-6). Studies have also indicated that chemical inhibitors of P450 1B1 are able to decrease the incidence of cancers in experimental animals when potent carcinogens, such as PAHs, are simultaneously administered (7-10).
Chemical inhibitors of P450 1B1 have been identified in many laboratories; these include flavonoids, stilbenes, synthetic organoselenium compounds such as 1,2-, 1,3-, and 1,4-phenylenebis(methylene)selenocyanate, synthetic acetylenic PAHs such as 1-ethynylpyrene (1EP), 2-ethynylpyrene (2EP), 1-(1-propynyl)pyrene (1PP), 2-ethynylphenanthrene (2EPh), 2-(1-propynyl)phenanthrene (2PPh), and 4-(1-propynyl)biphenyl (4PB) (7,11-15). It has also been shown that many PAHs, e.g. benzo[a]pyrene, benz[a]anthracene, and 5-methylchrysene, are also potent inhibitors of human P450 1A1, 1A2, and 1B1 and that the IC50 values obtained with these PAHs in inhibiting P450 activities are similar to those of known potent inhibitors, such as several synthetic acetylenic PAHs and α-naphthoflavone (ANF) (16). These results are of interest because humans are exposed to mixtures of environmental PAHs through foods and the atmosphere, and these PAHs have activities both as carcinogens (through metabolism by P450s) and as chemoprevention agents by inhibiting P450s themselves (9, 10, 17, 18). Different mechanisms have been suggested to involve in inhibition of P450 1A1, 1A2, and 1B1 by various chemicals (19). For example, 1EP and 1PP inhibit P450 1A1 in a mechanism-based manner while they directly inhibit P450 1A2 and 1B1. Similarly, several carcinogenic PAHs (e.g. benzo[a]pyrene, benz[a]anthracene, and 5-methylchrysene) are converted by P450 1A2 to active metabolites that inhibit the enzyme, but these PAHs directly inhibit P450 1A1 and 1A2 (19). Our recent studies have also suggested that there are different binding sites in the P450 1B1 molecule for a variety of PAHs, acetylenic PAHs and biphenyls as judged by spectral interactions, and the quenching of P450 1B1-derived and inhibitor-based fluorescence (20). Molecular docking studies suggest differences in the interaction of selected PAH inhibitors (e.g. benzo[a]pyrene, 1PP, and 3-(1-propynyl)phenanthrene (3PPh) with the active sites of P450 1B1 and P450 1A2, and these results are consistent with these P450 enzymes forming different metabolites of these PAH inhibitors, thus yielding different mechanisms for inhibition of these P450 enzymes (20).
In this study, we further examined structure-function relationships of the interaction of human P450 1B1 with 51 chemicals including sixteen flavonoids, three stilbenes, six pyrenes, seven naphthalenes, seven phenanthrenes, ten biphenyls, 17β-estradiol (E2), and estrone (E1) that have been reported to be substrates or inhibitors of P450 1B1 (7, 11). We measured the spectral interaction of chemicals with P450 1B1 and the inhibition of P450 1B1-dependent 7-ethoxyresorufin O-deethylation (EROD) activity by these chemical inhibitors. Of the 51 chemicals examined, 49 were found to produce Reverse Type I binding spectra with human P450 1B1 and the spectral interaction was found to correlate significantly with inhibitory potencies of P450 1B1-dependent EROD activity. We conclude that the interaction of P450 1B1 with inhibitors can be ascribed in terms of a generally common process of interaction to change the spin state of the iron in the protein, presumably by favoring the liganding of water to the distal site of the heme.
Experimental Procedures
Chemicals
Chemical structures of 51 chemicals used in this study are presented in Figures 1 and 2. Flavone, stilbenes, estrone (E1), 17β-estradiol (E2), testosterone, pyrene, naphthalene, phenanthrene (Ph), biphenyl, 7-ethoxyresorufin, and resorufin were obtained from Sigma Chemical Co. (St. Louis, MO) or Kanto Kagaku Co. (Tokyo). Thirteen methoxylated flavonoids, with or without dihydroxy substitution at 5,7- and 7,8-positions, were synthesized and the methods for the synthesis of these chemicals have been described (22). Acetylenic pyrenes and phenanthrenes and derivatives of naphthalenes and biphenyls were also synthesized as previously described (21,23-26). Other chemicals and reagents used in this study were obtained from the sources described previously or were of the highest quality commercially available (16, 19, 20).
Figure 1.
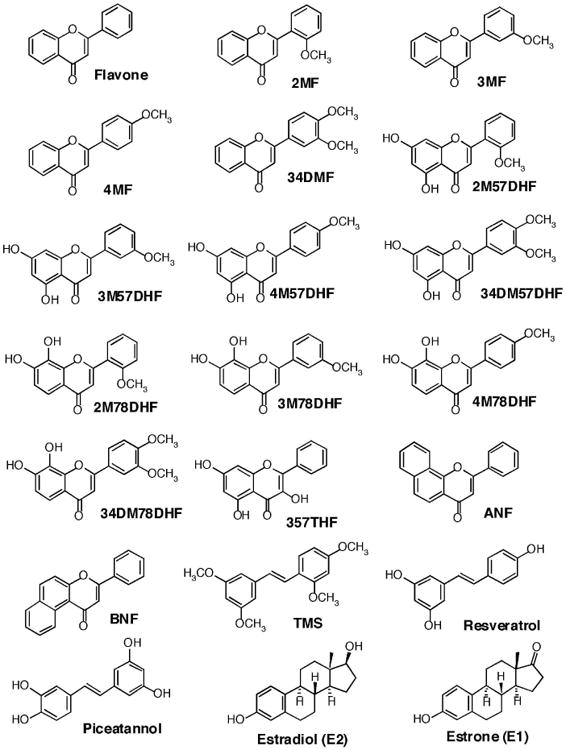
Structures of flavonoids, isoflavones, benzoflavones, and estrogens.
Figure 2.
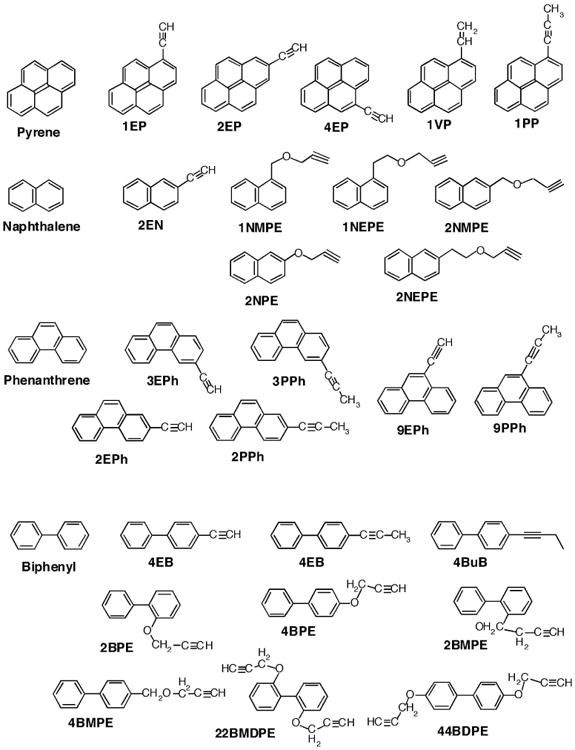
Structures of derivatives of pyrenes, naphthalenes, phenanthrenes, and biphenyls.
Enzymes
Bacterial “bicistronic” P450 1A1, 1A2, and 1B1 systems were prepared as described (16, 19). To facilitate expression and purification of P450s, six histidine residues were introduced at the position before the termination codon (C-terminual) (7). The plasmids for the expression of P450s 1A1, 1A2, or 1B1 plus human NADPH-P450 reductase were introduced into Escherichia coli DH5α cells by a heat shock procedure. Bacterial membranes were prepared and suspended in 10 mM Tris-HCl buffer (pH 7.4) containing 1.0 mM EDTA and 20% glycerol (v/v) (20).
For purification of P450 enzymes, the bacterial membranes were solubilized in 0.10 M potassium phosphate buffer (pH 7.4) containing 20% glycerol (v/v), 0.5 M NaCl, 10 mM β-mercaptoethanol, 0.5% sodium cholate (w/v), 1% Triton N-101 (w/v), and 30 μM ANF. The solubilized membranes were centrifuged (105 × g, 60 min), the supernatant was applied to a nickel-nitrilotriacetic acid column (Qiagen), and the P450 proteins were purified by the method described by Chun et al.(7).
Enzyme Assays
EROD assays were done at 37 °C in standard reaction mixtures (1.5 mL) containing recombinant P450 1B1 (3.3 nM), P450 1A1 (5.0 nM), or P450 1A2 (100 nM), NADPH (0.83 μM), and 7-ethoxyresorufin (4.0 μM). Reactions were started by adding NADPH, and product (resorufin) formation was monitored directly in a Hitachi F-4500 spectrofluorometer using an excitation wavelength of 571 nm and an emission wavelength of 585 nm (20). All of the PAH inhibitors and 7-ethoxyresorufin were dissolved in (CH3)2SO and added directly to the incubation mixtures; the final concentration of organic solvent in the mixture was <0.4% (v/v).
The 50% inhibition concentration (IC50) of EROD activities of P450 1B1, 1A1, and 1A2 were determined in a standard incubation mixture consisted of P450 enzymes, chemical inhibitors, and 7-ethoxyresorufin (2.5 μM) in a final volume of 0.5 mL of 100 mM potassium phosphate buffer (pH 7.4) and an NADPH-generating system (0.5 mM NADP+, 5 mM glucose 6-phosphate, and 0.5 unit of yeast glucose 6-phosphate dehydrogenase/mL) (16). Formation of resorufin from 7-ethoxyresorufin was determined fluorimetrically as described (20).
Spectral Binding Titrations
Purified P450 1B1 was diluted to 1.0 μM in 0.10 M potassium phosphate buffer (pH 7.4) containing 20% glycerol (v/v) and the binding spectra were recorded with subsequent additions of chemical inhibitors in a Jasco V-550 spectrophotometer (Figure 3). Chemical inhibitors were also added in a buffer solution and the spectra were determined as described above. The differences in absorbance between 350 nm and 500 nm were recorded as a function of substrate concentration (Figures 3A, 3D, and 3G). The P450 spectra (with or without inhibitors) were obtained by subtracting the blank spectra (in the absence of P450) from the P450 spectra (Figures 3B, 3E, and 3H). Difference spectra for the interaction of chemicals with P450 1B1 were obtained (Figures 3C, 3F, and 3I). Spectral dissociation constants (Ks) and the magnitude of spectral binding (ΔAmax/Ks) were estimated using nonlinear regression analysis with GraphPad Prism software (GraphPad Software, La Jolla, CA).
Figure 3.
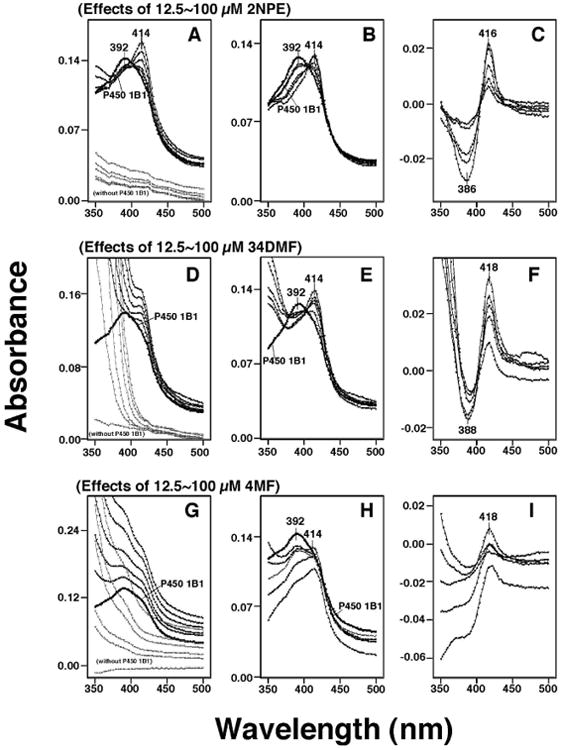
Spectral interactions of 2NPE (A, B, and C), 34DMF (D, E, and F), and 4MF (G, H, and I). Chemicals (at concentrations of 12.5, 25, 50, 75, and 100 μM) were added to the buffer, with or without 1 μM P450 1B1, and the spectra were recorded between 350 nm and 500 nm (A, D, and G). P450 spectra were obtained by subtracting the blank spectra (in the presence of chemicals and in the absence of P450 1B1) from the P450 spectra (containing 0-100 μM chemicals and P450 1B1) (B, E, and H). The difference spectra, showing the interaction of chemicals with P450 1B1 were also recorded (C, F, and I).
Other Assays
P450 and protein concentrations were estimated by the methods described previously (27, 28).
Results
Inhibition of P450 1A1-, 1A2-, and 1B1-Dependent EROD Activity by Flavonoids and other Inhibitors
We first determined the effects of flavonoids and other inhibitors on inhibition of EROD activities catalyzed by P450 1A1, 1A2, and 1B1 coexpressing NADPH-P450 reductase in membranes of E. coli (Figure 4). Among 10 flavonoids examined, 357THF was the most potent in inhibiting EROD activities by these three P450 enzymes and P450 1B1 was found to be more susceptible than P450 1A1 and 1A2. 3′,4′-Dimethoxy-5,7-dihydroxyflavone (34DM57DHF) and 4′-methoxy-7,8-dihydroxyflavone (4M57DHF) were also potent inhibitors of P450 enzymes, particularly for P450 1B1. Pyrene and phenanthrene inhibited P450 1A2 and 1B1 more strongly than P450 1A1. Naphthalene was weak in inhibiting P450 1A1 and 1A2 but inhibited P450 1B1 with an IC50 of ∼26 μM (Figure 4). As expected, ANF and β-naphthoflavone (BNF) were potent inhibitors of these P450s, particularly for P450 1A2 and 1B1. Because these results collectively indicated that P450 1B1 was more inhibited by these selected chemicals, we focused the study to P450 1B1 for the further analysis with 51 chemicals.
Figure 4.
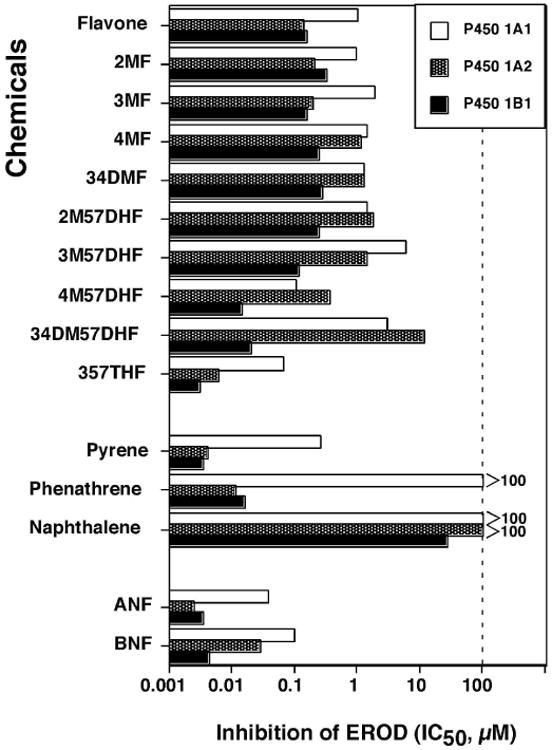
Effects of flavone and nine flavonoids, pyrene, phenanthrene, naphthalene, ANF and BNF on EROD activities catalyzed by P450 1A1 (
 ), P450 1A2 (shaded column,
), P450 1A2 (shaded column,
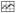 ), and P450 1B1 (■). The data presented are mean IC50 values (n=3) and the standard deviations in these cases were less than 15% of the values. Control activities for EROD by P450 1A1, 1A2, and 1B1 were 45 ± 5, 6.5 ± 0.8, and 26 ± 4 nmol/min/nmol P450, respectively.
), and P450 1B1 (■). The data presented are mean IC50 values (n=3) and the standard deviations in these cases were less than 15% of the values. Control activities for EROD by P450 1A1, 1A2, and 1B1 were 45 ± 5, 6.5 ± 0.8, and 26 ± 4 nmol/min/nmol P450, respectively.
Reverse Type I Spectral Changes of Human P450 1B1 Produced by Chemical Inhibitors
P450 1B1 had a wavelength maximum at ∼392 nm which changed to ∼414 nm upon the addition of 2NPE, 34DMF, or 4MF (Figs. 3A, 3D, and 3G). Because these chemicals had absorbance in the near-UV region, we calculated (subtracted) the absorbance due to the chemicals from the spectra of P450 1B1 interacted with the chemicals. 2-Naphthalene propargyl ether (2NPE) had weak spectral absorbance between 350 nm and 500 nm (Figure 3A) and did not cause significant interference in the absolute and difference spectra of P450 1B1 (Figure 3B and 3C, respectively). However, both 3′,4′-dimethoxyflavone (34DMF) and 4′-methoxyflavone (4MF) had significant absorbance in the near-UV region (Figures 3D and 3G) and interfered in the analysis of the spectral interaction of inhibitors with P450 1B1 (Figures 3E, 3F, 3H, and 3I). Particularly with 4MF (at high concentrations), the P450 difference spectra did not show a clear trough at ∼386-388 nm in the difference spectra (Figure 3I). We then compared the ratio of absorbance at 414 and 392 nm in the absolute spectra (cf. Figures 3B, 3E, and 3H) and the absorbance difference between 416 nm and 386 nm in the difference spectra (cf. Figures 3C, 3F, and 3I) for 12 chemicals (with P450 1B1) and found that there were good correlation between the data from the absolute and the difference spectra (Fig. 5). Therefore, we calculated the magnitude of spectral interaction of chemicals with P450 1B1 from either the absolute or difference spectral data.
Figure 5.
Correlation between the peak ratio of 414 nm vs 392 nm in the absolute spectra of P450 and the absorbance difference in 416 nm and 386 nm in the difference spectra for 12 chemical inhibitors. Methods for determination of the spectra are presented in Experimental Procedures. Concentrations of chemicals used were 12.5-100 μM except for TMS, 4M57DHF, and pyrene, for which concentrations of 1.25-10 μM were used.
Reverse Type I Binding Spectra and Inhibition of EROD Activity of P450 1B1 with Chemical Inhibitors
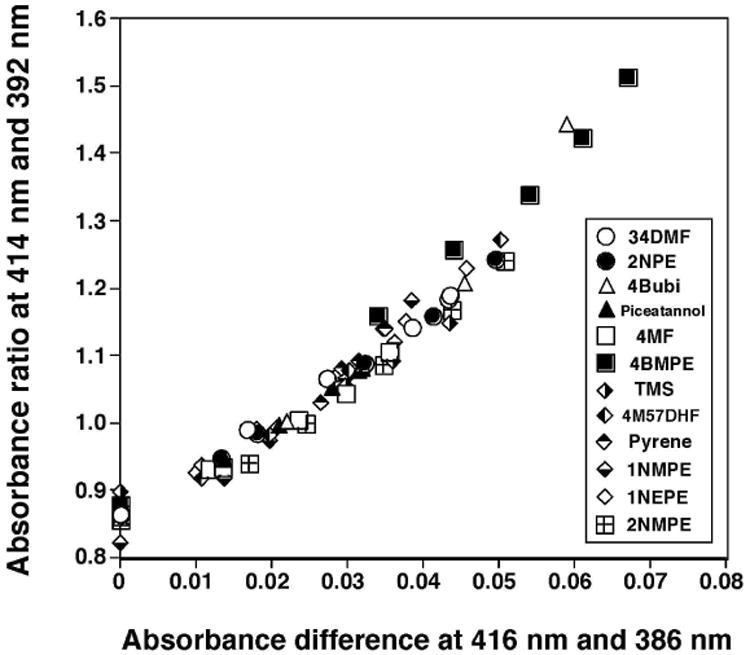
We analyzed sixteen flavonoids, three stilbenes, E1, and E2 for their abilities to induce Reverse Type I spectra and to inhibit EROD activities with P450 1B1 (Table 1 and Fig. 6). In our previous account we have obtained evidence that these chemicals directly inhibit EROD activities of P450 1B1, not in a mechanism-based manner (19,20). Among the 21 chemicals (Table 1), only 2M78DHF did not show any detectable spectral interaction with P450 1B1, even at 100 μM. 2M78DHF was also found to be less active in inhibiting EROD (IC50 of >100 μM). Flavone produced Reverse Type I spectra with P450 1B1 (Ks of 43 μM and ΔAmax 0.026) and inhibited P450 1B1-dependent EROD activity (IC50 0.15 μM) (Table 1). 357THF, 4M57DHF, 34DM57DHF, and 3M57DHF were more active than flavone in interacting with and inhibiting P450 1B1, but 3M78DHF, 4M78DHF, and 34DM78DHF were not as potent as flavone. Among the three stilbenes examined, TMS was the most potent in inducing a Reverse Type I binding spectrum and inhibiting EROD activity, as compared with resveratrol and picetannol. We compared the apparent Ks values or the ΔAmax/Ks values with IC50 values for EROD inhibition with these 20 chemicals (except for 2M78DHF) and found that these correlated well, r=0.81 and 0.88 respectively (Fig. 6).
Table 1. Reverse Type I spectral changes and inhibition of EROD activities of human P450 1B1 with derivatives of flavonoids, stilbenes, and estrogens.
| Reverse Type I spectra | inhibition of EROD | |||
|---|---|---|---|---|
| chemicals | Ks(μM) | ΔAmax | ΔAmax/Ks | IC50 (μM) |
| flavone | 43 ± 4.7 | 0.026 ± 0.002 | 0.0006 | 0.15 ± 0.02 |
| 2MF | 56 ± 8.0 | 0.070 ± 0.005 | 0.0013 | 0.32 ± 0.06 |
| 3MF | 51 ± 4.1 | 0.029 ± 0.001 | 0.0006 | 0.15 ± 0.02 |
| 4MF | 66 ± 11 | 0.048 ± 0.005 | 0.0007 | 0.23 ± 0.03 |
| 34DMF | 24 ± 3.2 | 0.051 ± 0.003 | 0.0021 | 0.26 ± 0.03 |
| 2M57DHF | 64 ± 10 | 0.100 ± 0.011 | 0.0016 | 0.24 ± 0.02 |
| 3M57DHF | 20 ± 3.0 | 0.075 ± 0.008 | 0.0037 | 0.11 ± 0.02 |
| 4M57DHF | 17 ± 0.7 | 0.130 ± 0.004 | 0.0078 | 0.014 ± 0.002 |
| 34DM57DHF | 2.0 ± 0.3 | 0.037 ± 0.002 | 0.0180 | 0.019 ± 0.003 |
| 2M78DHF | ND | ND | ND | >100 |
| 3M78DHF | 140 ± 15 | 0.044 ± 0.005 | 0.0003 | 3.7 ± 0.5 |
| 4M78DHF | 56 ± 15 | 0.073 ± 0.009 | 0.0013 | 1.1 ± 0.2 |
| 34DM78DHF | 190 ± 17 | 0.053 ± 0.003 | 0.0003 | 19 ± 2.3 |
| 357THF | 5.7 ± 0.9 | 0.090 ± 0.007 | 0.0160 | 0.0031 ± 0.0002 |
| ANF | 12 ± 1.6 | 0.073 ± 0.007 | 0.0061 | 0.0033 ± 0.0003 |
| BNF | 13 ± 1.2 | 0.079 ± 0.009 | 0.0061 | 0.0041 ± 0.0004 |
| TMS | 4.3 ± 0.7 | 0.047 ± 0.003 | 0.0110 | 0.023 ± 0.003 |
| resveratrol (Res) | 28 ± 3.3 | 0.034 ± 0.004 | 0.0012 | 1.2 ± 0.2 |
| Piceatannol (Pice) | 21 ± 1.1 | 0.034 ± 0.001 | 0.0016 | 0.83 ± 0.14 |
| E2 | 83 ± 18 | 0.039 ± 0.007 | 0.0005 | 1.5 ± 0.2 |
| E1 | 120 ± 13 | 0.014 ± 0.002 | 0.0001 | 16 ± 1.8 |
ND, not detected at the highest concentration of 100 μM. Ks, ΔAmax, and IC50 values presented are the means ± SEs obtained from the experiments with 5-8 different concentrations of the chemicals.
Figure 6.
Relationship between IC50 values and Ks values (A) or ΔAmax/Ks (B) in the interactions of flavone and flavonoids, isoflavones, E1, and E2 with P450 1B1.
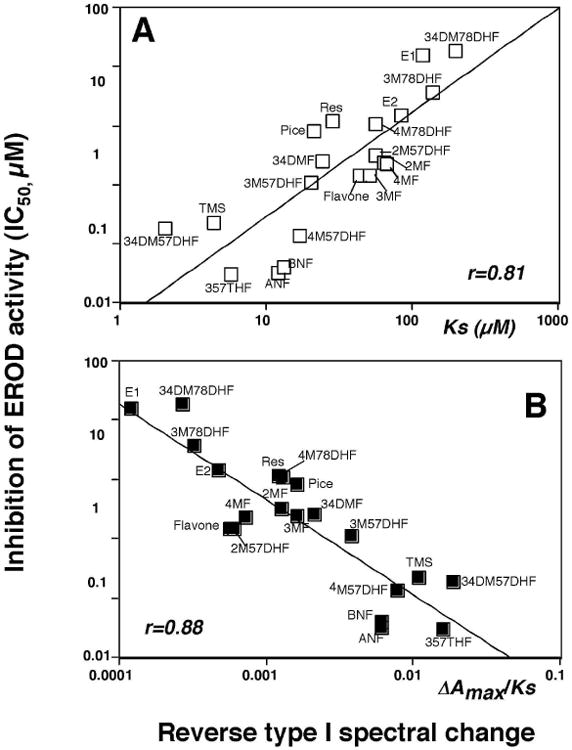
We also compared the spectral changes and inhibition of EROD activities with six pyrenes, seven naphthalenes, seven phenanthrenes, and ten biphenyls (Table 2 and Figure 7). It should be mentioned that these chemicals directly inhibited EROD activities of P450 1B1 except that 4PB inhibited P450 1B1 in a mechanism-based manner (results not presented). However, 4PB induced spectral changes with P450 1B1, indicating that this chemical inhibits by both a direct and in a mechanism-based manner. Among 30 chemicals examined, only naphthalene did not show any measurable spectral interaction with P450 1B1 (even at 100 μM). Pyrene induced Reverse Type I binding spectrum, with a Ks of 1.3 μM, and inhibited P450 1B1-EROD activity (IC50 0.025 μM); both of these values were the lowest among the pyrene derivatives examined. Naphthalene did not produce spectral changes with P450 1B1 and weakly inhibited P450 1B1-EROD activities (IC50 26 μM). The other six derivatives of naphthalene produced Reverse Type I spectra with Ks values of ∼100 μM (IC50 values of 1-5 μM), except that Ks value of 2EN was 15 μM. The Ks and IC50 values with phenanthrene were 14 μM and 0.51 μM, respectively, and other phenanthrene derivatives showed slightly lower Ks and IC50 values than phenanthrene. Biphenyl yielded a Ks value of 140 μM and IC50 of 19 μM, and the acetylenic biphenyls were found to be more active in interacting with P450 1B1 in both spectral changes and EROD inhibition. Three propargyl ethers of biphenyl—4BMPE, 22BDPE, and 44BDPE—interacted with P450 1B1 more strongly than biphenyl itself. The other propargyl ether derivatives of biphenyl—2BPE, 4BPE, 2BMPE—were not as potent in their interactions with P450 1B1.
Table 2. Reverse Type I spectral changes and inhibition of EROD activities of human P450 1B1 with pyrenes, naphthalenes, phenanthrenes, and biphenyls.
| Reverse Type I spectra | inhibition of EROD | |||
|---|---|---|---|---|
| chemicals | Ks (μM) | ΔAmax | ΔAmax/Ks | IC50 (μM) |
| pyrene | 1.3 ± 0.1 | 0.044 ± 0.001 | 0.0338 | 0.025 ± 0.003 |
| 1EP | 2.9 ± 0.3 | 0.049 ± 0.005 | 0.0169 | 0.036 ± 0.005 |
| 2EP | 3.4 ± 0.2 | 0.071 ± 0.001 | 0.0209 | 0.038 ± 0.004 |
| 4EP | 4.0 ± 0.5 | 0.052 ± 0.004 | 0.0130 | 0.17 ± 0.03 |
| 1VP | 4.4 ± 0.3 | 0.092 ± 0.003 | 0.0209 | 0.15 ± 0.02 |
| 1PP | 2.7 ± 0.3 | 0.042 ± 0.002 | 0.0156 | 0.045 ± 0.005 |
| naphthalene | ND | ND | ND | 26 ± 3.2 |
| 2EN | 15 ± 0.3 | 0.049 ± 0.002 | 0.0033 | 1.2 ± 0.2 |
| 1NMPE | 92 ± 10 | 0.074 ± 0.005 | 0.0008 | 1.2 ± 0.3 |
| 1NEPE | 88 ± 18 | 0.079 ± 0.009 | 0.0009 | 0.92 ± 0.15 |
| 2NPE | 97 ± 19 | 0.086 ± 0.008 | 0.0009 | 4.3 ± 0.6 |
| 2NMPE | 100 ± 17 | 0.090 ± 0.009 | 0.0009 | 2.1 ± 0.3 |
| 2NEPE | 110 ± 3.1 | 0.094 ± 0.016 | 0.0009 | 5.2 ± 0.7 |
| Phenanthrene | 14 ± 1.7 | 0.044 ± 0.002 | 0.0031 | 0.51 ± 0.10 |
| 2EPh | 4.2 ± 0.5 | 0.080 ± 0.004 | 0.0190 | 0.21 ± 0.03 |
| 3EPh | 3.0 ± 0.3 | 0.071 ± 0.001 | 0.0237 | 0.16 ± 0.02 |
| 9EPh | 3.6 ± 0.4 | 0.071 ± 0.004 | 0.0197 | 0.12 ± 0.01 |
| 2PPh | 2.2 ± 0.2 | 0.067 ± 0.005 | 0.0305 | 0.15 ± 0.02 |
| 3PPh | 3.1 ± 0.4 | 0.060 ± 0.006 | 0.0194 | 0.067 ± 0.008 |
| 9PPh | 3.2 ± 0.4 | 0.049 ± 0.002 | 0.0153 | 0.16 ± 0.03 |
| biphenyl | 140 ± 19 | 0.037 ± 0.008 | 0.0003 | 19 ± 2.3 |
| 4EB | 7.6 ± 1.2 | 0.023 ± 0.001 | 0.0030 | 2.2 ± 0.3 |
| 4PB | 12 ± 1.8 | 0.074 ± 0.008 | 0.0062 | 1.8 ± 0.2 |
| 4BuB | 54 ± 6.2 | 0.092 ± 0.007 | 0.0017 | 1.5 ± 0.3 |
| 2BPE | 114 ± 16 | 0.110 ± 0.009 | 0.0010 | 15 ± 0.2 |
| 4BPE | 83 ± 26 | 0.056 ± 0.010 | 0.0007 | 16 ± 0.3 |
| 2BMPE | 146 ± 15 | 0.130 ± 0.009 | 0.0009 | 12 ± 0.2 |
| 4BMPE | 13 ± 1.5 | 0.071 ± 0.002 | 0.0055 | 1.2 ± 0.1 |
| 22BDPE | 30 ± 4.5 | 0.018 ± 0.001 | 0.0006 | 12 ± 1.7 |
| 44BDPE | 23 ± 4.3 | 0.031 ± 0.002 | 0.0013 | 7.8 ± 1.0 |
ND, not detected at the highest concentration of 100 μM. Ks, ΔAmax, and IC50 values presented are the means ± SEs obtained from the experiments with 5-8 different concentrations of the chemicals.
Figure 7.
Relationship between IC50 values and Ks values (A) or ΔAmax/Ks (B) in the interaction of derivatives of pyrenes, naphthalenes, phenanthrenes, and biphenyls with P450 1B1.
Correlation between Ks or ΔAmax/Ks values (for Reverse Type I spectra) and IC50 values for P450 1B1-dependent EROD inhibition were compared for a combined set of 6 pyrenes, 6 naphthalenes (except naphthalene), 7 phenanthrenes, and 10 biphenyls and the correlation coefficients (r) were 0.88 and 0.91, respectively (Figure 7).
We also calculated the correlation coefficients between spectral data and IC50 values in several groups of chemical inhibitors determined in this study (Table 3). When the IC50 values for EROD activity were compared with Ks or ΔAmax/Ks values for Reverse Type I binding spectra with a total of 49 chemicals, the correlation coefficients (r) were 0.72 and 0.74, respectively. However, these correlations became higher when these values were calculated in a set of flavonoids and stilbenes (r=0.81 and 0.86, respectively in n=16), a set of biphenyls (r=0.75 and 0.89, respectively in n=10), or a group of phenanthrenes (r=0.80 and 0.77, respectively, n=7). The relationships in sets of pyrenes and naphthalenes were not so high, except that IC50 and Ks values were well correlated in a group of pyrenes (r=0.78). In all cases, the correlation coefficients between ΔAmax and IC50 values were not so high.
Table 3. Correlation between IC50 values for EROD inhibition and spectral parameters (Ks, ΔAmax, and ΔAmax/Ks) with various groups of chemical inhibitors in human P450 1B1 enzyme system.
| Chemicals | Comparison | correlation coefficient (r) |
|---|---|---|
| Total (n=49) | IC50 vs Ks | 0.72 |
| IC50 vs ΔAmax | 0.21 | |
| IC50 vs ΔAmax/Ks | 0.74 | |
| flavonoids, stilbenes, E1, and E2 (n=20) | IC50 vs Ks | 0.81 |
| IC50 vs ΔAmax | 0.29 | |
| IC50 vs ΔAmax/Ks | 0.86 | |
| biphenyls (n=10) | IC50 vs Ks | 0.75 |
| IC50 vs ΔAmax | 0.12 | |
| IC50 vs ΔAmax/Ks | 0.89 | |
| pyrenes (n=6) | IC50 vs Ks | 0.78 |
| IC50 vs ΔAmax | 0.52 | |
| IC50 vs ΔAmax/Ks | 0.55 | |
| naphthalene (n=6) | IC50 vs Ks | 0.42 |
| IC50 vs ΔAmax | 0.60 | |
| IC50 vs ΔAmax/Ks | 0.33 | |
| phenanthrenes (n=7) | IC50 vs Ks | 0.80 |
| IC50 vs ΔAmax | 0.43 | |
| IC50 vs ΔAmax/Ks | 0.77 |
Discussion
Chemical inhibitors (as well as substrates) for P450 enzymes have been shown to induce spectral changes with oxidized state of the enzymes and at least three types of spectral changes— Type I, Type II, and Reverse Type I—are found upon binding of different chemicals with various forms of P450s (29,30). Spectral dissociation constants (Ks) and magnitudes of spectral binding (ΔAmax/Ks) have been used as parameters for the affinity and potencies of chemicals to interact with P450 enzymes (31). Of 51 chemicals examined in this study, 49 were found to induce Reverse Type I binding spectra in interacting with human P450 1B1; only naphthalene and 2M78DHF did not show any spectral interaction, even at 100 μM. Our studies showed that all of the 49 chemicals caused almost the same spectral changes with P450 1B1, so-called Reverse Type I spectra or “Modified Type II” spectra (30). It should be noted that some of these compounds themselves had absorbance in the near-UV region, and these spectra sometimes caused interference in calculating the magnitude of the absorbance change in the difference spectra (Figure 3). Since good correlation was noted between the ratio of 392 nm versus 414 nm in the absolute spectra and the absorbance difference in 416 nm and 386 nm in the difference spectra, we determined the magnitude of interaction using either the results from absolute spectra or from difference spectra (Figure 4).
Previously, the chemicals that produce Reverse Type I spectra with P450 enzymes in several animal species have been suggested to have hydroxyl groups in the molecule and to ligand to the heme iron of the cytochrome (29-32). Our present results showed that several of the chemicals, such as hydroxylated flavones, stilbenes, and estrogens, contain hydroxyl groups in the molecule, however, there are many exceptions in that flavone and several flavonoids, pyrene, phenanthrene, biphenyl, and many other compounds do not have hydroxyl groups but have the ability to induce Reverse Type I binding spectra with P450 1B1. A more likely explanation for the ligand-induced conversion of high- to low-spin iron is that the ligands bind and alter the active site pocket of the P450 (1B1) to admit water as the distal ligand, in a similar way that the introduction of ligands into P450 3A4 active site causes the protein to release bound distal ligand waters (32).
Methoxylated flavones have been reported to be more resistant to metabolism by xenobiotic-metabolizing enzymes (e.g. UDP-glucuronyltransferases and sulfotransferases) than the unmethoxylated ones and thus accumulate in tissues and increase their biological potencies, including chemoprevention activities (15,34-37). Our results showed that (di)hydroxylation of flavones at the 5- and 7-positions of 3′-methoxy-, 4′-methoxy-, and 3′,4′-dimethoxy-flavones was found to stimulate both the magnitude of spectral binding and the potencies of EROD inhibition, but (di)hydroxylation at the 7- and 8-positions of the respective flavones decreased their affinities to P450 1B1 as compared with those of the parent flavone. Potent inhibition of human P450 1B1 by 5,7-dihydroxylated flavonoids has recently been reported by Kim et al. (38) using 5,7-dihydroxyflavones, 5,7-dihydroxyflavones, 4′-hydroxy-5,7-dihydroxyflavone, and 3′,4′-dihydroxy-5,7-dihydroxyflavone. These results suggest that hydroxylation position affects the interaction with P450 1B1, but that hydroxylation is not critical in determining the magnitude of the Reverse Type I binding spectra of chemicals with P450s.
Addition of any of several acetylenic groups to the pyrene molecule slightly increased both Ks (2.7∼-4.4 μM) and IC50 values (0.036-0.17 μM). It is not known at present whether or not acetylene moieties attached to pyrene affect affinities toward P450 1B1; however, our previous studies of molecular docking of human P450 1B1 with acetylenic PAH inhibitors have suggested that the aromatic positions of 1PP and 3PPh dock with the P450 1B1 heme, resulting in the formation of oxidative products at aromatic position that lose ability to inhibit P450 1B1 (20). In this regard, Sohl et al. (39) showed that pyrene binds to rabbit P450 1A2 and induces Type I binding spectra with a Ks value of 0.036 μM and that this value is increased to 0.2 μM for 1-hydroxypyrene.
2EN, 1NMPE, 1NEPE, 2NPE, 2NMPE, and 2NEPE were all found to induce Reverse Type I spectra with Ks values of 15-110 μM and inhibited EROD activities with IC50 values of 0.92-5.2 μM, although naphthalene itself did not show any spectra with P450 1B1, even at a 100 μM concentration, and weakly inhibited EROD activity catalyzed by P450 1B1. Similarly, substitution of phenanthrene with acetylenic groups increased the affinity towards P450 1B1, in terms of Reverse Type I binding spectra and inhibition of P450 1B1-dependent EROD activity. Biphenyl was also not very potent in producing Reverse Type I binding spectra or inhibiting catalytic activity, but substitution with either a 4-ethynyl-, 4-(1-propynyl)-, or 4-butynyl group caused an increase in affinity towards P450 1B1 (Ks values of 7.6, 12, and 54 μM, respectively). Substitution on biphenyl with mono- and di-propargyl ether also increased affinity toward P450 1B1. These results suggest that the substituted naphthalenes, phenanthrenes, and biphenyls—but not pyrenes— fit better into the pocket of P450 1B1 and thus significantly inhibit P450 1B1 catalytic activity.
Chun et al. (7) reported that TMS induces Reverse Type I binding spectra with P450 1B1 and inhibits P450 1B1-dependent EROD activity. In this work we found that resveratrol and piceatannol, as well as TMS, induced Reverse Type I binding spectra and inhibited EROD activity, although the former two polyphenols were less active than TMS in interacting with P450 1B1. These three polyphenols have been reported to have chemopreventive activity and several mechanisms for inhibition of cell growth have been presented (40,41). Interestingly, Potter et al. (42) have shown that resveratrol is converted by human P450 1B1 to piceatannol, which shows anti-leukemic activity and inhibits tyrosine kinase activity (43).
In conclusion, our current studies showed that 49 of 51 chemicals induced Reverse Type I binding spectra with human P450 1B1 and the magnitude of spectral binding (Ks and ΔAmax/Ks) were apparently related to the inhibition potencies (IC50 values) of these chemicals against P450 1B1. It was found that substitution of pyrene with acetylenic groups decreased the affinities to P450 1B1 while substitution of naphthalene and phenanthrene with acetylenic groups or propargyl ethers increased the affinity for P450 1B1. Substitution of biphenyl with these groups also increased affinity towards P450 1B1. In cases of flavonoids, 5,7-dihydroxy substituted flavonoids showed more interaction with P450 1B1 while 7,8-dihydroxylation decreased the affinity to P450 1B1. Collectively our current studies showed, for the first time, that interactions of chemicals with P450 1B1 (as determined by Reverse Type I binding spectra) may determine how these chemicals inhibit catalytic activity of P450 1B1 and that substitution on these particular chemical scaffolds sometimes affect their affinities to P450 1B1. The interaction of P450 1B1 with inhibitors can be ascribed in terms of a generally common process of interaction to change the spin state of the iron in the protein, presumably by favoring the liganding of water to the distal site of the heme. Our present results on structure-function relationships of these chemicals with P450 1B1 may be of use for further studies on molecular mechanisms of substrate and inhibitor binding at P450 1B1 active sites.
Acknowledgments
This work was supported in part by Grants from the Ministry of Education, Science, and Culture of Japan, the Ministry of Health and Welfare of Japan (T.S., M.K., H.Y.), NIH grants R37 CA090426 (F.P.G.), P30 ES000267 (F.P.G.), and S06 GM08008 (M.K.F.), and DOE grant DE-FC26-00NT40843 (M.K.F.).
Footnotes
Abbreviations: ANF, α-naphthoflavone; 22BDPE, 2,2-biphenyl dipropagyl ether; 44BDPE, 4,4-biphenyl dipropargyl ether; 2BMPE, 2-biphenyl methyl propargyl ether; 4BMPE, 4-biphenyl methyl propargyl ether; BNF; β–naphthoflavone; 2BPE, 2-biphenyl propargyl ether; 4BPE, 4-biphenyl propargyl ether; 4BuB, 4-butinylbiphenyl; 34DM57DHF, 3′,4′-dimethoxy-5,7-dihydroxyflavone; 34DM78DHF, 3′,4′-dimethoxy-7,8-dihydroxyflavone; 34DMF, 3′,4′-dimethoxyflavone; E1, estrone; E2, 17β-estradiol; 4EB, 4-ethynylbiphenyl; 2EN, 2-ethynylnaphthalene; 1EP, 1-ethynylpyrene; 2EP, 2-ethynylpyrene; 4EP, 4-ethynylpyrene; 2EPh, 2-ethynylphenanthrene; 3EPh, 3-ethynylphenanthrene; 9EPh, 9-ethynylphenanthrene; EROD, 7-ethoxyresorufin O-deethylation; 2M57DHF, 2′-methoxy-5,7-dihydroxyflavone; 3M57DHF, 3′-methoxy-5,7-dihydroxyflavone; 4M57DHF, 4′-methoxy-5,7-dihydroxyflavone; 2M78DHF, 2′-methoxy-7,8-dihydroxyflavone; 3M78DHF, 3′-methoxy-7,8-dihydroxyflavone; 4M78DHF, 4′-methoxy-7,8-dihydroxyflavone; 2MF, 2′-methoxyflavone; 3MF, 3′-methoxyflavone; 4MF, 4′-methoxyflavone; 1NEPE, 1-naphthalene ethyl propargyl ether; 2NEPE, 2-naphthalene ethyl propargyl ether; 1NMPE, 1-naphthalene methyl propargyl ether; 2NMPE, 2-naphthalene methyl propargyl ether; 2NPE, 2-naphthalene propargyl ether; PAHs, polycyclic aromatic hydrocarbons; 4PB, 4-(1-propynyl)biphenyl; Ph, phenanthrene; 1PP, 1-(1-propynyl)pyrene; 2PPh, 2-(1-propynyl)phenanthrene; 3PPh, 3-(1-propynyl)phenanthrene; 9PPh, 9-(1-propynyl)phenanthrene; 357THF, 3,5,7-trihydroxyflavone; TMS, 2,4,3′,5′-tetramethoxystilbene; 1VP, 1-vinylpyrene.
References
- 1.Guengerich FP. Human Cytochrome P450 Enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd. Kluwer Academic/Plenum Press; New York: 2005. pp. 377–530. [Google Scholar]
- 2.Shimada T, Hayes CL, Yamazaki H, Amin S, Hecht SS, Guengerich FP, Sutter TR. Activation of chemically diverse procarcinogens by human cytochrome P450 1B1. Cancer Res. 1996;56:2979–2984. [PubMed] [Google Scholar]
- 3.Shimada T. Xenobiotic-metabolizing enzymes involved in activation and inactivation of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab Pharmacokinet. 2006;21:257–276. doi: 10.2133/dmpk.21.257. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe J, Shimada T, Gillam EMJ, Ikuta T, Suemasu K, Higashi Y, Gotoh O, Kawajiri K. Association of CYP1B1 genetic polymorphism with incidence to breast and lung cancer. Pharmacogenetics. 2000;10:25–33. doi: 10.1097/00008571-200002000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Zheng W, Xie DW, Jin F, Cheng JR, Dai Q, Wen WQ, Shu XO, Gao YT. Genetic polymorphism of cytochrome P450 1B1 and risk of breast cancer. Cancer Epidem Biomarker Prev. 2000;9:147–150. [PubMed] [Google Scholar]
- 6.Rylander-Rudqvist T, Wedren S, Granath F, Humphreys K, Ahlberg S, Weiderpass E, Oscarson M, Ingelman-Sundberg M, Persson I. Cytochrome P450 1B1 gene polymorphisms and postmenopausal breast cancer risk. Carcinogenesis. 2003;24:1533–1539. doi: 10.1093/carcin/bgg114. [DOI] [PubMed] [Google Scholar]
- 7.Chun YJ, Kim S, Kim D, Lee SK, Guengerich FP. A new selective and potent inhibitor of human cytochrome P450 1B1 and its application to antimutagenesis. Cancer Res. 2001;61:8164–8170. [PubMed] [Google Scholar]
- 8.Slaga TJ, Jecker L, Bracken WM, Weeks CE. The effects of weak or non-carcinogenic polycyclic hydrocarbons on 7,12-dimethylbenz[a]anthracene and benzo[a]pyrene skin tumor-initiation. Cancer Lett. 1979;7:51–59. doi: 10.1016/s0304-3835(79)80076-2. [DOI] [PubMed] [Google Scholar]
- 9.Marston CP, Pereira C, Ferguson J, Fischer K, Hedstrom O, Dashwood WM, Baird WM. Effect of a complex environmental mixture from coal tar containing polycyclic aromatic hydrocarbons (PAH) on the tumor initiation, PAH-DNA binding and metabolic activation of carcinogenic PAH in mouse epidermis. Carcinogenesis. 2001;22:1077–1086. doi: 10.1093/carcin/22.7.1077. [DOI] [PubMed] [Google Scholar]
- 10.DiGiovanni J, Rymer J, Slaga TJ, Boutwell RK. Anticarcinogenic and cocarcinogenic effects of benzo[e]pyrene and dibenz[a,c]anthracene on skin tumor initiation by polycyclic hydrocarbons. Carcinogenesis. 1982;3:371–375. doi: 10.1093/carcin/3.4.371. [DOI] [PubMed] [Google Scholar]
- 11.Guengerich FP, Chun YJ, Kim D, Gillam EMJ, Shimada T. Cytochrome P450 1B1: A target for inhibition in anticarcinogenesis strategies. Mut Res. 2003;523-524:173–182. doi: 10.1016/s0027-5107(02)00333-0. [DOI] [PubMed] [Google Scholar]
- 12.Shimada T, Upadhyaya P, El-Bayoumy K, Strickland PT, Sutter TR, Guengerich FP, Yamazaki H. Inhibition of human cytochrome P450-catalyzed oxidations of xenobiotics and pro-carcinogens by synthetic organoselenium compounds. Cancer Res. 1997;57:4757–4764. [PubMed] [Google Scholar]
- 13.Doostdar H, Burke MD, Mayer RT. Bioflavonoids: selective substrates and inhibitors for cytochrome P450 CYP1A and CYP1B1. Toxicology. 2000;144:31–38. doi: 10.1016/s0300-483x(99)00215-2. [DOI] [PubMed] [Google Scholar]
- 14.Shimada T, Yamazaki H, Foroozesh M, Hopkins NE, Alworth WL, Guengerich FP. Selectivity of polycyclic inhibitors for human cytochromes P450 1A1, 1A2, and 1B1. Chem Res Toxicol. 1998;11:1048–1056. doi: 10.1021/tx980090+. [DOI] [PubMed] [Google Scholar]
- 15.Walle T, Ta N, Kawamori T, Wen X, Tsuji PA, Walle UK. Cancer chemopreventive properties of orally bioavailable flavonoids—methylated versus unmethylated flavones. Biochem Pharmacol. 2007;73:1288–1296. doi: 10.1016/j.bcp.2006.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimada T, Guengerich FP. Inhibition of human cytochrome P450 1A1-, 1A2-, and 1B1-mediated activation of procarcinogens to genotoxic metabolites by polycyclic aromatic hydrocarbons. Chem Res Toxicol. 2006;19:288–294. doi: 10.1021/tx050291v. [DOI] [PubMed] [Google Scholar]
- 17.Courter LA, Musafia-Jeknic T, Fischer K, Bildfell R, Giovanini J, Pereira C, Baird WM. Urban dust particulate matter alters PAH-induced carcinogenesis by inhibition of CYP1A1 and CYP1B1. Toxicol Sci. 2007;95:63–73. doi: 10.1093/toxsci/kfl137. [DOI] [PubMed] [Google Scholar]
- 18.Mahadevan B, Marston CP, Luch A, Dashwood WM, Brooks E, Pereira C, Doehmer J, Baird WM. Competitive inhibition of carcinogen-activating CYP1A1 and CYP1B1 enzymes by a standardized complex mixture of PAH extracted from coal tar. Int J Cancer. 2007;120:1161–1168. doi: 10.1002/ijc.22466. [DOI] [PubMed] [Google Scholar]
- 19.Shimada T, Murayama N, Okada K, Funae Y, Yamazaki H, Guengerich FP. Different mechanisms of inhibition for human cytochrome P450 1A1, 1A2, and 1B1 by polycyclic aromatic inhibitors. Chem Res Toxicol. 2007;20:489–496. doi: 10.1021/tx600299p. [DOI] [PubMed] [Google Scholar]
- 20.Shimada T, Murayama N, Tanaka K, Takenaka S, Imai Y, Hopkins NE, Foroozesh MK, Alworth WL, Yamazaki H, Guengerich FP, Komori M. Interaction of polycyclic aromatic hydrocarbons with human cytochrome P450 1B1 in inhibiting catalytic activity. Chem Res Toxicol. 2008;21:2313–2323. doi: 10.1021/tx8002998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hopkins NE, Foroozesh MK, Alworth WL. Suicide inhibitors of cytochrome P450 1A1 and P450 2B1. Biochem Pharmacol. 1992;44:787–96. doi: 10.1016/0006-2952(92)90417-h. [DOI] [PubMed] [Google Scholar]
- 22.McKendall ME, Smith TP, Anh K, Ellis J, McGee T, Foroozesh M, Zhu N, Klein Stevens C. Methoxyflavone inhibitors of cytochrome P450. J Chem Crystallogr. 2008;38:231–237. [Google Scholar]
- 23.Kelley AT, Mesbah JY, McKendall ME, Smith TP, Foroozesh M. Synthesis of a family of naphthyl propargyl ethers as potential cytochrome P450 inhibitors. J Undergrad Chem Res. 2004;3:103–106. [Google Scholar]
- 24.Zhu N, Lightsey K, Foroozesh M, Alworth W, Chaudhary A, Willett KL, Klein Stevens CLK. Naphthoflavone propargyl ether inhibitors of cytochrome P450. J Chem Crystallogr. 2006;36:289–296. [Google Scholar]
- 25.Bowman B, Lightsey D, McKendall M, Smith T, Zhu N, Stevens CLK, Foroozesh M. Synthesis of a family of biphenylpropagyl ethers as potential inhibitor of P450 enzymes. X-ray crystal structure of 2,2′-biphenyldipropargyl ether. J Undergrad Chem Res. 2005;2:57–61. [Google Scholar]
- 26.Foroozesh M, Primrose G, Guo Z, Bell LC, Guengerich FP, Alworth WL. Propynylaryl acetylenes as mechanism-based inhibitors of cytochrome P450 1A1, 1A2 and 2B1 enzymes. Chem Res Toxicol. 1997;10:91–102. doi: 10.1021/tx960064g. [DOI] [PubMed] [Google Scholar]
- 27.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 28.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 29.Yoshida Y, Kumaoka H. Studies on the substrate-induced spectral change of cytochrome P450 in liver microsomes. J Biochem (Tokyo) 1975;78:455–468. [PubMed] [Google Scholar]
- 30.Schenkman JB, Cinti DL, Orrenius S, Moldeus P, Kraschnitz R. The nature of the reverse type I (modified type II) spectral change in liver microsomes. Biochemistry. 1972;11:4243–4251. doi: 10.1021/bi00773a008. [DOI] [PubMed] [Google Scholar]
- 31.Jefcoate CR. In: Measurement of Substrate and Inhibitor Binding to Microsomal Cytochrome P-450 by Optical-Difference Spectroscopy. Colowick SP, Kaplan NO, editors. Academic Press; New York: 1978. pp. 258–279. [DOI] [PubMed] [Google Scholar]
- 32.Kumaki K, Sato M, Kon H, Nebert DW. Correlation of type I, type II, and reverse type I difference spectra with absolute changes in spin state of hepatic microsomal cytochrome P-450 iron from five mammalian species. J Biol Chem. 1978;253:1048–1058. [PubMed] [Google Scholar]
- 33.Isin EM, Guengerich FP. Kinetics and thermodynamics of ligand binding by cytochrome P450 3A4. J Biol Chem. 2006;281:9127–9136. doi: 10.1074/jbc.M511375200. [DOI] [PubMed] [Google Scholar]
- 34.Wen X, Walle T. Methylated flavonoids have greatly improved intestinal absorption and metabolic stability. Drug Metab Dispos. 2006;34:1786–1792. doi: 10.1124/dmd.106.011122. [DOI] [PubMed] [Google Scholar]
- 35.Walle UK, Walle T. Bioavailable flavonoids: cytochrome P450-mediated metabolism of methoxyflavones. Drug Metab Dispos. 2007;35:1985–1989. doi: 10.1124/dmd.107.016782. [DOI] [PubMed] [Google Scholar]
- 36.Walle T. Methoxylated flavones, a superior cancer chemopreventive flavonoid subclass? Semin Cancer Biol. 2007;17:354–362. doi: 10.1016/j.semcancer.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moon YJ, Wang X, Morris ME. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro. 2006;20:187–210. doi: 10.1016/j.tiv.2005.06.048. [DOI] [PubMed] [Google Scholar]
- 38.Kim HJ, Lee SB, Park SK, Kim HM, Park YI, Dong MS. Effects of hydroxy group numbers on the B-ring of 5,7-dihydroxyflavones on the differential inhibition of human CYP 1A and CYP1B1 enzymes. Arch Pharm Res. 2005;28:1114–1121. doi: 10.1007/BF02972971. [DOI] [PubMed] [Google Scholar]
- 39.Sohl CD, Isin EM, Eoff RL, Marsch GA, Stec DF, Guengerich FP. Cooperativity in oxidation reactions catalyzed by cytochrome P450 1A2: highly cooperative pyrene hydroxylation and multiphasic kinetics of ligand binding. J Biol Chem. 2008;283:7293–7308. doi: 10.1074/jbc.M709783200. [DOI] [PubMed] [Google Scholar]
- 40.Lu F, Zahid M, Wang C, Saeed M, Cavalieri EL, Rogan EG. Resveratrol prevents estrogen-DNA adduct formation and neoplastic transformation in MCF-10F cells. Cancer Prevent Res. 2008;1:135–145. doi: 10.1158/1940-6207.CAPR-08-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aziz MH, Kumar R, Ahmad N. Cancer chemoprevention by resveratrol: in vitro and in vivo studies and the underlying mechanisms. Int J Oncol. 2003;23:17–28. [PubMed] [Google Scholar]
- 42.Potter GA, Patterson LH, Wanogho E, Perry PJ, Butler PC, Ijaz T, Ruparelia KC, Lamb JH, Farmer PB, Stanley LA, Burke MD. The cancer preventative agent resveratrol is converted to the anticancer agent piceatannol by the cytochrome P450 enzyme CYP1B1. Br J Cancer. 2002;86:774–778. doi: 10.1038/sj.bjc.6600197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geahlen RL, McLaughlin JL. Piceatannol (3,4,3′,5′-tetrahydroxy-trans-stilbene) is a naturally occurring protein-tyrosine kinase inhibitor. Biochem Biophys Res Commun. 1989;165:241–245. doi: 10.1016/0006-291x(89)91060-7. [DOI] [PubMed] [Google Scholar]