

Abstract
Purpose: Induction of oxidative stress by Organophosphate compounds (OPs) has been previously reported. In the present work, the mechanism of protective effects of N-acetylcysteine as a glutathion (GSH) prodrug against malathion–induced cell toxicity was investigated. In this work, freshly isolated rat hepatocytes were used to determine the effect of NAC on malathion-induced cytotoxicity, formation of reactive oxygen species (ROS) and mitochondrial dysfunction. Methods: Rat hepatocytes were isolated using collagenase perfusion and then cell viability, mitchondrial membrane potential (MMP) and ROS formation were determined using trypan blue exclusion, Rhodamine 123 fluorescence and fluorogenic probe, 2', 7' -dichlorofluorescin diacetate (DCFH-DA), respectively. Results: Despite the protective effect of NAC on malathion-induced cell toxicity and MMP dysfunction, its efficacy against ROS formation was not adequate to completely protect the cells. Conclusion: Cytotoxic effects of malathion regardless of its cholinergic feature, is started with gradual free radical production but, the main factor that causes cell death, is mitochondrial dysfunction, so that reduction of ROS formation alone is not sufficient for cell survival, and the maintenance of mitochondrial integrity through different mechanisms is the most ameliorative factor specially at high levels of cell damage, as NAC seemed to protect cells with various fashions apart from ROS scavenging in concentrations higher than malathion’s LC50.
Keywords: Organophosphate Pesticides, Cytotoxicity, Oxidative Stress, Mitochondria, N-actylcycteine, Hepatocytes
Introduction
Malathion is one of the most widely used organophosphate compounds (OPs) applied in different fields like agriculture (as a pesticide), veterinary practice (as an ectoparasiticide), treatment of body lice on humans, food preparation and processing areas.1 OPs are cholinesterase inhibiting chemicals and the main cause of pesticide poisonings.2 Besides inhibiting cholinesterase (ChE), oxidative stress has been recently proposed as a main toxicity mechanism for OPs both in acute and chronic poisoning cases.3-5 Increased lipid peroxidation and diminished antioxidant defense capacity particularly glutathione are important findings accounted for OPs toxicity.6 Generation of oxidative intermediates induced by various agents has been predicated in cell death processes mainly mediated by the intracellular organelle mitochondriaMitochondria.7 are important intracellular source of ROS because of being the oxidative phosphorylation station in the cells and a susceptive target for damaging effects of ROS under certain phathological situations owing to abundance of tetraacyl phospholipid cardiolipin in their membrane.8-10 Mitochodrial membrane integrity is important for efficient oxidative phosphorylation.11 While increased ROS production is a harmful outcome of mitochondrial dysfunction12, there is lots of evidence underlining the responsibility of mitochondrial impairment for ROS signaling via apoptotic pathways. 13-14 Despite this, mitochondria have some antioxidant defense systems that help protect it against deleterious effects of ROS. The most important agents for mitochondrial antioxidant protection are the tripeptide glutathione (GSH) and multiple GSH-linked antioxidant enzymes.15
Considering oxidative stress induction by OPs pesticides, several studies proposed that a drug that could act as a multiplier of GSH content and a reductant would improve the cell tolerance towards OP-toxicity. In this regard, N-acetyl-L-cysteine (NAC) as a cell-permeable GSH prodrug that acts through direct scavenging of free radicals and revival of glutathione and cysteine can be a good candidate. Recently, our team reported that NAC can prevent toxicity of OPs in rats.16
The liver that is of extraordinary importance in detoxification of toxic metabolites acts as a potential site for OPs- induced damages.17,18 Since bio- transformation of thiono-OP as an imperative mechanism of their activation is mostly occurred in the liver, this organ is considered as one of the preliminary targets for malathion- induced toxicities.19
In this study, the role of mitochondrial dysfunction and enhanced ROS formation in malathion- induced cytotoxicity as well as the protective effect of NAC against it has been examined.
Materials and Methods
Chemicals
Bovine serum albumin, collagenase A from clostridium histolyticum and HEPES from Roche Diagnostics (Indianapolis, IN), 2',7'-dichlorofluorescin diacetate (DCFH-DA) and rhodamine 123 from Fluka (Italy), N-acetyl-cysteine (NAC) and 1-bromoheptane from Acros Pharmaceuticals, heparin sodium salt grade 1-A, trypan blue, methanol, and malathion 90% from Merck (Germany) were used in this study.
Animals
Male Sprague–Dawley rats (200–250 g) were obtained from the Laboratory of Animal Research Center of Tabriz University (Medical Sciences). The rats were housed in an air-conditioned room, under controlled temperature of 23 ± 1°C, relative humidity of 36 ± 6% and 12 h light/12 h dark conditions for 1 week before starting the experiments. They were allowed to feed with standard laboratory chaw and tap water ad libitum. Procedures involving animals and their care were conducted in conformity with the NIH guidelines for the care and use of laboratory animals.
Preparation of hepatocytes
Hepatocytes were isolated from adult male Sprague–Dawley rats (220–250 g), by a two-step collagenase perfusion of the liver as described previously.20 After isolation, the cells were suspended (106cells/ml) in Krebs-Henseleit buffer (NaCl 118 mM; KCl 4.7 mM; KH2PO4 1.17 mM; MgSO4.7H2O 1.19 mM; CaCl2.2H2O 2.58 mM; NaHCO3 25mM) containing 12.5 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) and incubated under a stream of 95% O2 and 5% CO2 in continuously rotating round-bottomed 50 ml flasks at 37 °C for 30 minutes before the addition of chemicals. Stock solutions of chemicals were made either in incubation buffer or in methanol and added to the hepatocyte suspensions at the indicated time points. Cell viability was measured by Trypan blue exclusion method.20 The hepatocytes used in this study were at least 85-90% viable immediately after isolation.
Exposure of hepatocytes to malathion and NAC
According to a previous report on the toxicity and metabolism of malathion in isolated rat hepatocytes 21 as well as our pilot studies, concentrations in the range of 0.5-1.5 mM were chosen to be used in this experimental set up. malathion was dissolved in methanol and NAC was dissolved in deionized double distilled water and added to the flasks containing the hepatocytes (106 cells /ml) at mentioned time intervals. 1- bromoheptane was dissolved in methanol and was added to the flasks 30 minutes before adding malathion. The utilized concentration of both NAC and 1- bromoheptane was 200 μM in cell suspensions.
Measurement of ROS formation
ROS formation was measured according to Eghbal et al., 2004b.22 Briefly, DCFH-DA was dissolved in methanol. The final concentration of DCFH-DA in cell suspensions (106 cells per ml) was 1 μM. DCFH-DA was added to the cell suspensions at the same time as malathion and a 100 μl aliquot was taken and diluted in 2ml of the incubation buffer. The samples were taken and the fluorescence intensity was measured at the excitation wavelength 485 nm and the emission wavelength 530 nm.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential in hepatocytes was assessed by monitoring the uptake of the cationic dye, rhodamine 123 as described.23-24 Isolated cells were extracted then resuspended in original media containing 1 μM rhodamine 123. After 10 minutes of incubation, the cells were centrifuged and the supernatant was measured with a Shimadzu RF-5000U spectrofluorimeter. The amount of dye remaining in the supernatant was inversely proportional to the membrane potential of the cells.
Statistical analysis
Statistical comparisons were carried out using a one-way analysis of variance (ANOVA) followed by the Bonferroni t-test (post-hoc) for multiple comparisons to determine statistical significance (P < 0.05) between treatments and control groups.
Results
Cytotoxic effect of malathion on freshly isolated rat hepatocytes
Figure 1 indicates the absolute toxicity of malathion in concentrations above 0.5 mM whereas the 1 and/or 1.5 mM caused significant cell death. Malathion at 2 mM caused 100% mortality 3 hours after incubation.
Figure 1.
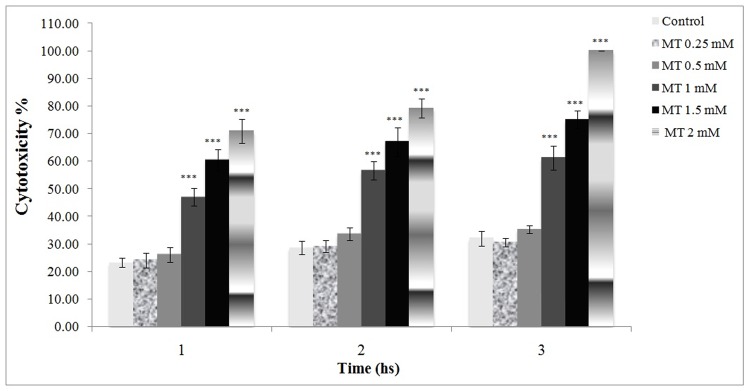
Cytotoxic effect of malathion on freshly isolated rat hepatocytes.
Malathion was added to the flasks containing the hepatocytes to make the concentrations (1 and 1.5 mM) and cell viability was measured with uptake of trypane blue. All results were shown in mean ±S.E.Mand are at least from 3 independent experiments. MT (malathion)
***Shows significant difference (p<0.001) with control
Effect of malathion on MMP in freshly isolated rat hepatocytes
Figure 2 illustrates the relative percentages of MMP in test groups in comparison with the control, where the 100 percent of Rhodamine 123 trapping occurred in the intact mitochondria. As shown, there was a significant difference in MMP of the malathion-treated cells (1 &1.5 mM) in comparison with the control.
Figure 2.

Effect of malathion on mitochondrial membrane potential in freshly isolated rat hepatocytes.
Rhodamine was added to the aliquots of cell suspensions and the fluorescence amounts of the samples were measured with a Shimadzu RF-5000U spectrofluorimeter. The amount of dye remaining in the supernatant was inversely proportional to the membrane potential of the cells. All results were shown in mean ±S.E.Mand are at least from 3 independent experiments. MT (malathion), MMP (mitochondrial membrane potential)
**Shows significant difference (p<0.01) with control
***Shows significant difference (p<0.001) with control
Effect of malathion on ROS formation in freshly isolated rat hepatocytes
As shown in Figure 3, ROS formation was significantly increased in 1 and 1.5 mM of malathion-treated cells in comparison with the control while 0.5 mM did not show a significant effect.
Figure 3.

Effect of malathion on ROS formation in freshly isolated rat hepatocytes.
DCF was added to the flasks containing hepatocytes at the same time as malathion and the fluorescence intensity was measured at the excitation wavelength 485 nm and the emission wavelength at 530 nm. All results were shown in mean ±S.E.Mand are at least from 3 independent experiments. MT (malathion), ROS (reactive oxygen species)
***Shows significant difference (p<0.001) with control
Effect of 1-bromoheptane on malathion toxicity
Table 1 compares the different rates of malathion- induced cytotoxicity in the cells pre-incubated with 1- bromoheptane to that in the cells treated with different concentrations of malathion only. In the presence of 1- bromoheptane, the cytotoxicity of malathion was intensified and the rate of cell mortality was increased even with nontoxic concentrations of malathion (0.25, 0.5 mM).
Table 1. Effect of 1-bromoheptane on malathion cytotoxicity .
| Treatments \ Time Intervals | Cytotoxicity (%) | ||
| 1 Hs | 2 Hs | 3Hs | |
| Normal Cells (without treatment) | 23.±1.7 | 28±2.3 | 32±2.7 |
| “ + MT 0.25mM | 24±2.7 | 29±2.1 | 30±1.4 |
| “ + MT 0.5mM | 26±2.6 | 33±2.3 | 35±1.4 |
| “ + MT 1mM | 47±3.2 c | 56±3.3 c | 61±4.4 c |
| “ + MT 1. 5mM | 60±3.8 c | 67±5.2 c | 75±3.1 c |
| 1-BH 200µM treated Cells | 24±3.1 | 31±3.4 | 32±2.3 |
| “ + MT 0.25mM | 43±2.8 b | 46±3.4 a | 49±3.5 a |
| “ + MT 0. 5mM | 48±4.1 b | 55±2.9 b | 57±4.3 b |
| “ + MT 1mM | 54±2.3 c | 81±2.6 c | 83±4.6 c |
| “ + MT 1. 5mM | 72±3.4 c | 86±3.2 c | 100±0.0 c |
Hepatocytes were incubated in Krebs–Henseleit solution, pH 7.4 at 37 °C under the atmosphere of 95%O2/5%CO2.The samples were taken at mentioned time intervals and cell death was assessed by trypan blue exclusion method.All data were shown in mean ±S.E.Mand are at least from 3 independent experiments.
a: Significant difference (p<0.05) with control
b: Significant difference (p<0.01) with control
c: Significant difference (p<0.001) with control
Protective effect of NAC against malathion-induced cytotoxicity
Figure 4 shows that NAC protected hepatocytes against malathion-induced cytotoxicity at different time intervals.
Figure 4.
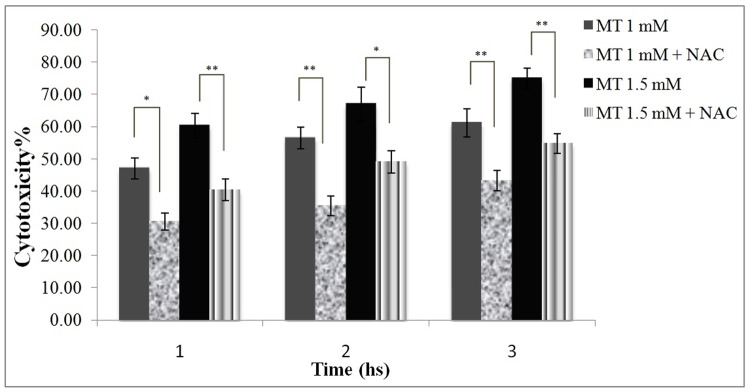
Protective effect of NAC against malathion toxicity in freshly isolated rat hapatocytes.
Malathion (1 and 1.5 mM) and NAC (200μM) were added to the flasks containing the hepatocytes and cell viability was measured with uptake of trypane blue. All results were shown in mean ±S.E.Mand are at least from 3 independent experiments. MT (malathion), NAC (N-acetylcysteine)
* Shows significant difference (p<0.05)
**Shows significant difference (p<0.01)
Protection by NAC of MMP against malathion toxicity
As shown in Figure 5, NAC was able to prevent the reduction of MMP in malathion-treated cells in all 3 time intervals.
Figure 5.
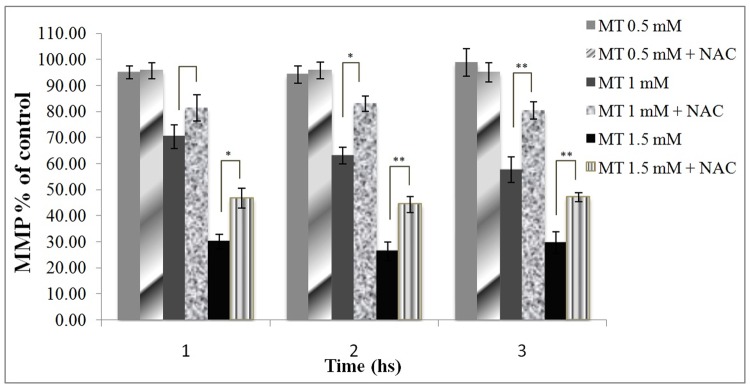
Protection of mitochondrial membrane potential against malathion toxicity by NAC.
Rhodamine was added to the aliquots of cell suspensions and the fluorescence amounts of the samples were measured with a Shimadzu RF-5000U spectrofluorimeter. The amount of dye remaining in the supernatant was inversely proportional to the membrane potential of the cells. All results were shown in mean ±S.E.Mand are at least from 3 independent experiments. MT (malathion), NAC (N-acetylcysteine), MMP (mitochondrial membrane potential)
* Shows significant difference (p<0.05)
**Shows significant difference (p<0.01)
Effect of NAC on malathion-induced ROS formation
As shown in Figure 6, NAC was able to prevent malathion- induced ROS formation only in the cells treated with 1 mM malathion.
Figure 6.

Effect of NAC on malathion – induced ROS formation in freshly isolated rat hepatocytes.
DCF was added to the flasks containing hepatocytes at the same time as Malathion and the fluorescence intensity was measured at the excitation wavelength 485 nm and the emission wavelength at 530 nm. All results were shown in mean ±S.E.Mand are at least from 3 independent experiments. MT (malathion), NAC (N-acetylcysteine), ROS (reactive oxygen species)
* Shows significant difference (p<0.05)
**Shows significant difference (p<0.01)
Discussion
This study evaluated the cytotoxic effects of malathion as well as the protective effect of NAC against it in freshly isolated rat hepatocytes. The results indicate that NAC has beneficial effects on some parameters of malathion- induced cytotoxicity so that the decrease in cell mortality, modification of altered MMP and reduction of ROS formation are obvious. The results of the present study indicate that hepatocytes were able to tolerate malathion in concentrations less than 0.5 mM, while the cell viability was significantly decreased at higher concentrations so that 100% cell mortality was seen in the group receiving 2 mM malathion after 3 hours incubation and the LC50 was estimated to be around 1mM in this experimental set-up.
Malathion in its toxic concentrations in isolated rat hepatocytes (1, 1.5 mM) significantly increased ROS formation which is in parallel with the result of the previous studies showing the oxidative effects of various OPs in vitro24, and also in vivo.26-28
The basis of OP toxicity in induction of oxidative stress has been speculated in two prospects; (a) redox-cycling activity recounted as the ability to accept an electron and produce free radicals that is subsequently followed by superoxide anions and hydrogen peroxide formation through transmission of an electron to oxygen and dismutation reactions, respectively; and (b) antioxidant depletion because of disruption of antioxidant homeostasis that results in increased ROS formation.29-32
This study not only confirmed the previous reports on the proxidative activity of OPs, but also examined if the toxicity of malathion could be an outcome of mitochondrial permeability transition (MPT). According to the results obtained from the rhodamine 123 uptake experiment, the loss of MMP significantly occurred in isolated rat hepatocytes incubated with 1 and/or 1.5 mM malathion. MPT is an important mechanism in various hepatotoxicities and defined as an abrupt increase in the inner mitochondrial membrane permeability to solutes of size less than 1500 Da. As a lethal event for the cells, MPT occurs with uncontrolled proton influx into matrix and is accompanied with dissipation of MMP, syncope of ATP synthesis, efflux of intramitochondrial ions specially calcium as well as mitochondrial swelling because of osmotic influx of water. It can be mediated by oxidative stress and causes increased oxidative stress.24,33
Two main functions of mitochondria are maintenance of cellular energy metabolism and regulation of cell death showing the unrivaled capacity of this organelle in ROS production. Indeed, mitochondrial respiratory chain is not only the main source of ROS production but also a sensitive target for the harmful effects of ROS. Ranjbar et al (2010)34 have previously reported that malathion-induced mitochondrial dysfunction in rat brain is mostly mediated through oxidative stress. Since, release of cytochrom c toward triggering apoptosis can be largely mediated by mitochondria-generated ROS35 as well as close correlation between apoptosis and collapse of MMP.36 it can be ideated that hepatotoxic effect of malathion is mediated through occurrence of MPT toward programmed cell death. In addition, mitochondrial antioxidant defense system has a crucial role in the survival of cells.37 But the most important factors for mitochondrial antioxidant protection are the tripeptide GSH and multiple GSH-linked antioxidant enzymes.38,39 So we evaluated the effect of GSH-depleted condition induced by 1-bromoheptane on malathion cytotoxicity. 1-Bromoheptane and other 1-Halogenoalkanes are GSH-S-transferase substrates and produce a substantial depletion of cellular GSH with minimal effects on phase I biotransformation and minimal cytotoxicity. 1-bromoheptane is the most effective and least cytotoxic GSH-S-transferase substrate tested, and therefore has been used as a tool to modulate cellular GSH levels in isolated hepatocytes.40 It is also clear that GSH depleted cells have adequate antioxidant and enzymatic systems to counteract physiological oxidative stress. It has been reported that treatment of cells with 1-bromoheptane depleted GSH by 87% within 30 min and the cells remained GSH-depleted for 4 hrs without significant cytotoxicity. A 30 fold higher concentration of 1-bromoheptane was required before cytotoxicity ensued. Therefore, 1-bromoheptane is particularly useful for studying the role of GSH in modulating xenobiotic cytotoxicity.40,41Paying attention to noticeable increase in mortality of hepatocytes incubated with 1-bromoheptane in this study, GSH depletion is considered as a main mechanism of malathion-induced toxicity. In other words, GSH plays a critical role in detoxification of malathion. As shown, in the presence of 1- bromoheptane, cytotoxicity of malathion was intensified and the rate of cell mortality was increased even with nontoxic concentrations of malathion (0.25, 0.5 mM)(Table 1 ).
NAC, as a precursor of the sulfur amino acid, cysteine, exerts antioxidative effect by reacting directly with electrophiles or by facilitating generation of GSH, after incorporation into cells. It is a small molecule that passes freely through cell membranes into the intracellular compartments and is subsequently deacylated to deliver cysteine. As an antioxidant, NAC directly scavenges hydrogen peroxide, hydroxyl free radicals and hypochloric acid in vitro.42-44 Furthermore, Saito et al 45 showed that administration of NAC or GSH results in a rapid and complete recovery of hepatic GSH level within 1 hour in mice and that the efficacy in supplying cysteine for recovery of GSH level and ROS scavenging in liver was similar between NAC and GSH treatment.
In this experimental set-up, NAC decreased malathion-induced cytotoxicity and ROS formation. Likewise, malathion-induced loss of MMP in freshly isolated rat hepatocytes was markedly terminated following NAC treatment. But the point is that NAC in malathion 1 mM-treated group declined cytotoxicity in parallel with both reduction of ROS formation and MMP obviation, whereas its cytoprotective effect in the next group (malathion 1.5 mM) is more similar to the pattern of MMP than the ROS one (Figures 5 & 6). Hence, it seems that maintenance of the cell survival is more related to the mitochondrial function integrity than the cellular ROS level, as the recent works on cell death mechanisms has focused on MPT. The mitochondrion has two redox-sensitive sites; one of them under the effect of mitochondrial GSH and the other influenced by NAD(P)H that can be modulated by ROS.46 So, depletion of mitochondrial GSH may be considerably responsible for sensitization of cells to oxidative injury. 47 In hepatocytes, mitochondria include ~ 10-15 % of the total cellular GSH content. Lack of the enzymatic machinery for de novo GSH synthesis in mitochondria makes it dependent on GSH uptake from the cytosol by carrier-mediated transport systems located in the inner membrane. It means that inconvenience of the mitochondrial membrane can violate the function of these carriers towards the mitochondrial dysfunction.48-50 NAC reduced toxicity in malathion 1.5 mM-treated hepatocytes in parallel with MMP upgrade but not distinctly ROS reduction. Thus, other mechanisms in addition to the ROS scavenging are responsible for the mitochondrial protective effects of NAC. In this regard, improvement of cellular energy supply is one of these proposed possible mechanisms, as the increasing effect of NAC on cytochrom c oxidase activity in mice synaptic mitochondria has been reported by Banaclocha & Martinez (1999).51 Furthermore, Saito et al (2010)45explained that ameliorative effect of NAC on cellular ATP level in the liver may be the result of its capacity to supply substrates needed for Krebs cycle. Thereupon, improvement of energy supply to mitochondria may be responsible for the accelerated uptake of cytosolic GSH into mitochondria as was observed after GSH treatment previously.45
Conclusion
Based on the results of the current study, we cold conclude that the cytotoxic effects of malathion as a common OP, regardless of its cholinergic effect, is started with gradual free radical production but, the main factor that causes cell death, is mitochondrial dysfunction, so that reduction of ROS formation alone is not sufficient for cell survival, and the maintenance of mitochondrial integrity through different mechanisms is the most ameliorative factor specially at high levels of cell damage, as NAC seemed to protect cells with various fashions apart from ROS scavenging in concentrations higher than malathion’s LC50.
Acknowledgments
This study was supported by a grant from Drug Applied Research Center, Tabriz University of Medical Sciences to M.A. Eghbal. This research received no specific grant from any other funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest
Authors declare no conflict of interest.
Abbreviations
OPs (Organophosphate Compounds), ROS (Reactive Oxygen Species), MMP (Mitochondrial Membrane Potential), GSH (Gluthatione), NAC (N-Acetylcysteine), DCFH-DA (2', 7' -Dichlorofluorescin Diacetate), LC50 (Lethal Concentration 50)
References
- 1.Gupta RC. Toxicology of Organophosphate and Carbamate Compounds. Elsevier Academic Press: 2006
- 2.Rahimi R, Nikfar S, Abdollahi M. Increased morbidity and mortality in acute human organophosphate-poisoned patients treated by oximes: a meta-analysis of clinical trials. Human and Experimental Toxicology. 2006;25(3):157–162. doi: 10.1191/0960327106ht602oa. [DOI] [PubMed] [Google Scholar]
- 3.Ranjbar A, Pasalar P, Abdollahi M. Induction of oxidative stress and acetylcholinesterase inhibition in organophosphorus pesticide manufacturing workers. Human and Experimental Toxicology. 2002;21(4):179–182. doi: 10.1191/0960327102ht238oa. [DOI] [PubMed] [Google Scholar]
- 4.Ranjbar A, Solhi H, Mashayekhi FJ, Susanabdi A, Rezaie A, Abdollahi M. Oxidative stress in acute human poisoning with organophosphorus insecticides; a case control study. Environ Toxicol Pharmacol. 2005;20(1):88–91. doi: 10.1016/j.etap.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 5.Soltaninejad K, Abdollahi M. Current opinion on the science of organophosphate pesticides and toxic stress: a systematic review. Med Sci Monit. 2009;15(3):75–90. [PubMed] [Google Scholar]
- 6.Abdollahi M, Mostafalou S, Pournourmohammadi S, Shadnia S. Oxidative stress and cholinesterase inhibition in saliva and plasma of rats following subchronic exposure to malathion. Comparative Biochemistry and Physiology Part C Toxicology & Pharmacology. 2004;137(1):29–34. doi: 10.1016/j.cca.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Syng-ai C, Kumari AL, Khar A. Effect of curcumin on normal and tumor cells: role of glutathione and bcl-2. Molecular Cancer Therapy. 2004;3:1101–1108. [PubMed] [Google Scholar]
- 8.Kirkland RA, Franklin JL. Bax, reactive oxygen, and cytochrome c release in neuronal apoptosis. Antioxidant & Redox Signaling. 2003;5(5):589–596. doi: 10.1089/152308603770310257. [DOI] [PubMed] [Google Scholar]
- 9.Roue G, Bitton N, Yuste VJ, Montange T, Rubio M, Dessauge F, Delettre C, Merle-beral H, Sarfati M, Susin SA. Mitochondrial dysfunction in cd47- mediated caspase-independent cell death: ros production in the absence of cytochrome c and aif release. Biochimie. 2003;85(8):741–746. doi: 10.1016/s0300-9084(03)00129-9. [DOI] [PubMed] [Google Scholar]
- 10.Ahmed T, Tripathi AK, Suke SG, Kumar V, Ahmed RS, Das S, Banerjee BD. Role of hsp27 and reduced glutathione in modulating malathion-induced apoptosis of human peripheral blood mononuclear cells: ameliorating effect of n-acetylcysteine and curcumin. Toxicology in Vitro. 2009;23:1319–1325. doi: 10.1016/j.tiv.2009.07.016. [DOI] [PubMed] [Google Scholar]
- 11.Shafiee H, Mohammadi H, Rezayat SM, Hosseini A, Baeeri M, Hassani S, Mohammadirad A, Bayrami Z, Abdollahi M. Prevention of malathion-induced depletion of cardiac cells mitochondrial energy and free radical damage by a magnetic magnesium-carrying nanoparticle. Toxicology Mechanisms and Methods. 2010;20(9):538–543. doi: 10.3109/15376516.2010.518173. [DOI] [PubMed] [Google Scholar]
- 12.Eghbal MA, Pennefather PS, O'brien PJ. H2s cytotoxicity mechanism involves reactive oxygen species formation and mitochondrial depolarisation. Toxicology. 2004;203:69–76. doi: 10.1016/j.tox.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 13.Lambert C, Apel K, Biesalski HK, Frank J. 2-methoxyestradiol induces caspase-independent, mitochondria-centered apoptosis in ds-sarcoma cells. International Journal of Cancer. 2004;108(4):493–501. doi: 10.1002/ijc.11579. [DOI] [PubMed] [Google Scholar]
- 14.Karami-mohajeri S, Abdollahi M. Toxic effects of organophosphate, carbamate, and organochlorine pesticides on cellular metabolism of lipids, proteins, and carbohydrates: a comprehensive review. Hum Exp Toxicol. 2011;30(9):1119–1140. doi: 10.1177/0960327110388959. [DOI] [PubMed] [Google Scholar]
- 15.Czarna M, Jarmuszkiewicz W. Role of mitochondria in reactive oxygen species generation and removal, relevance to signaling and programmed cell death. Postepy Biochemii. 2006;52(2):145–156. [PubMed] [Google Scholar]
- 16.Shadnia S, Dasgar M, Taghikhani S, Mohammadirad A, Khorasani R, Abdollahi M. Protective effects of α-tocopherol and n-acetyl-cysteine on diazinon-induced oxidative stress and acetylcholinesterase inhibition in rats. Toxicology Mechanisms and Methods. 2007;17(2):109–115. doi: 10.1080/15376510600860318. [DOI] [PubMed] [Google Scholar]
- 17.Teimouri F, Amirkabirian N, Esmaily H, Mohammadirad A, Aliahmadi A, Abdollahi M. Alteration of hepatic cells glucose metabolism as a non-cholinergic detoxication mechanism in counteracting diazinon-induced oxidative stress. Hum Exp Toxicol. 2006;25(12):697–703. doi: 10.1177/0960327106075064. [DOI] [PubMed] [Google Scholar]
- 18.Amirkabirian N, Teimouri F, Esmaily H, Mohammadirad A, Aliahmadi A, Abdollahi M. Protection by pentoxifylline of diazinon-induced toxic stress in rat liver and muscle. Toxicol Mech Methods. 2007;17(4):215–221. doi: 10.1080/15376510600943783. [DOI] [PubMed] [Google Scholar]
- 19.Yang MC, Mclean AJ, Rivory LP, Le COUTEUR DG. Hepatic disposition of neurotoxins and pesticides. Pharmacol Toxicol. 2000;87:286–291. doi: 10.1034/j.1600-0773.2000.pto870608.x. [DOI] [PubMed] [Google Scholar]
- 20.Moldeus P, Mogberg J, Orrenius S. Isolation and use of liver cells. Methods in Enzymology. 1978;52:60–71. doi: 10.1016/s0076-6879(78)52006-5. [DOI] [PubMed] [Google Scholar]
- 21.Malik JK, Summer HK. Toxicity and metabolism of malathion and its impurities in isolated rat hepatocytes: role of glutathione. Toxicology and applied pharmacology. 1982;66:69–76. doi: 10.1016/0041-008x(82)90061-8. [DOI] [PubMed] [Google Scholar]
- 22.Eghbal MA, Tafazoli S, Pennefather P, O’brien PJ. Peroxidase catalysed formation of cytotoxic prooxidant phenothiazine free radicals at physiological ph. Chemico-biological Interactions. 2004;151:43–51. doi: 10.1016/j.cbi.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Mingatto FE, Rodrigues T, Pigoso AA, Uyemura SA, Curti C, Santos AC. The critical role of mitochondrial energetic impairment in the toxicity of nimesulide to hepatocytes. The Journal of Pharmacology and Experimental Therapeutics. 2002;303(2):601–607. doi: 10.1124/jpet.102.038620. [DOI] [PubMed] [Google Scholar]
- 24.Anoush M, Eghbal MA, Fathiazad F, Hamzeiy H, Kouzehkonani NS. The protective effect of garlic extract against acetaminophen-induced loss of mitochondrial membrane potential in freshly isolated rat hepatocytes. Iranian J pharm sci. 2009;5(3):141–150. [Google Scholar]
- 25.Altuntas I, Kilinc I, Orhan H, Demirel R, Koylu H, Delibas N. The effects of diazinon on lipid peroxidation and antioxidant enzymes in erythrocites in vitro. Human and Experimental Toxicology. 2004;23(1):9–13. doi: 10.1191/0960327104ht408oa. [DOI] [PubMed] [Google Scholar]
- 26.John S, Kale M, Rathore N, Bhatnagar D.. Protective effect ofvitamin e in dimethoate and malathion induced oxidative stress in rat erythrocytes. The Journal of Nutritional Biochemistry. 2001;12(9):500–504. doi: 10.1016/s0955-2863(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 27.Akhgari M, Abdollahi M, Kebryaeezadeh A, Hosseini R, Sabzevari SABZEVARI, O O. Biochemical evidence for free radicalinduced lipid peroxidation as a mechanism for subchronic toxicity of malathion in blood and liver of rats. Human & Experimental Toxicology. 2003;22:205–211. doi: 10.1191/0960327103ht346oa. [DOI] [PubMed] [Google Scholar]
- 28.Shadnia S, Azizi E, Hosseini R, Khoei S, Fouladdel S, Pajoumand A, Jalali N, Abdollahi M. Evaluation of oxidative stress and genotoxicity in organophosphorus insecticide formulators. Human and Experimental Toxicology. 2005;24(9):439–445. doi: 10.1191/0960327105ht549oa. [DOI] [PubMed] [Google Scholar]
- 29.Banerjee BD, Seth V, Bhattacharya A, Pasha ST, Chakraborty AK. Biochemical effects of some pesticides on lipid peroxidation and free-radical scavengers. Toxicol Lett. 1999;107(13):33–47. doi: 10.1016/s0378-4274(99)00029-6. [DOI] [PubMed] [Google Scholar]
- 30.Kovacic P. Mechanism of organophosphates (nerve gases and pesticides) and antidotes: electron transfer and oxidative stress. Current Medicinal Chemistry. 2003;10(24):2705–2709. doi: 10.2174/0929867033456314. [DOI] [PubMed] [Google Scholar]
- 31.Vidyasagar J, Karunakar N, Reddy MS, Rajnarayana K, Surender T, Krishna DR. Oxidative stress and antioxidant status in acute organophosphorous insecticide poisoning. Indian J Pharmacol. 2004;36(2):76–79. [Google Scholar]
- 32.Possamai FP, Fortunato JJ, Feier G, Agostinho FR, Quevedo J, Wilhelm FILHO D, Dal-pizzol F. Oxidative stress after acute and sub-chronic malathion intoxication in wistar rats. Environ Toxicol Pharmacol. 2007;23(2):198–204. doi: 10.1016/j.etap.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 33.Reid AB, Kurten RC, Mccullough SS, Brock RW, Hinson JA. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. The Journal of Pharmacology and Experimental Therapeutics. 2005;312(2):509–516. doi: 10.1124/jpet.104.075945. [DOI] [PubMed] [Google Scholar]
- 34.Ranjbar A, Ghahremani MH, Sharifzadeh M, Golestani A, Ghazi-khansari M, Baeeri M, Abdollahi M. Protection by pentoxifylline of malathion-induced toxic stress and mitochondrial damage in rat brain. Human and Experimental Toxicology. 2010;29(10):851–864. doi: 10.1177/0960327110363836. [DOI] [PubMed] [Google Scholar]
- 35.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 36.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 37.Han D, Antunes F, Canali R, Rettori D. Cadenas e. voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytoso. The Journal of Biological Chemistry. 2003;278(8):5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 38.Soderdahl T, Enoksson M, Lundberg M. et al. Visualization of the compartmentalization of glutathione and protein-glutathione mixed disulfides in cultured cells. The FASEB Journal. 2003;17(1):124–126. doi: 10.1096/fj.02-0259fje. [DOI] [PubMed] [Google Scholar]
- 39.Marí M, Morales A, Colell A, García-ruiz C, Fernández-checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal. 2009;11(11):2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan S, O’brien P J. 1-bromoalkanes as new potent nontoxic glutathione depletors in isolated rat hepatocytes. Biochemical and Biophysical Research Communications. 1991;179(1):436–441. doi: 10.1016/0006-291x(91)91389-t. [DOI] [PubMed] [Google Scholar]
- 41.Kanz M F, Dugas T R, Liu H AND. Glutathione depletion exacerbates methylenedianiline toxicity to biliary epithelial cells and hepatocytes in rats. Toxicological Sciences. 2003;74:447–456. doi: 10.1093/toxsci/kfg125. [DOI] [PubMed] [Google Scholar]
- 42.Custodio JBA, Cardoso CMP, Almeida LM. Thiol protecting agents and antioxidants inhibit the mitochondrial permeability transition promoted by etoposide: implications in the prevention of etoposide-induced apoptosis. Chemico-biological Interactions. 2002;140(2):169–184. doi: 10.1016/s0009-2797(02)00020-0. [DOI] [PubMed] [Google Scholar]
- 43.Soltan-sharifi MS, Mojtahedzadeh M, Najafi A, Reza KHAJAVI M, Reza ROUINI M, Moradi M, Mohammadirad A, Abdollahi M. Improvement by n-acetylcysteine of acute respiratory distress syndrome through increasing intracellular glutathione, and extracellular thiol molecules and anti-oxidant power: evidence for underlying toxicological mechanisms. Human and Experimental Toxicology. 2007;26(9):697–703. doi: 10.1177/0960327107083452. [DOI] [PubMed] [Google Scholar]
- 44.Kamboj S S, Chopra K, Sandhir R. Neuroprotective effect of n-acetylcysteine in the development of diabetic encephalopathy in streptozotocin-induced diabetes. Metab Brain Dis. 2008;23(4):427–443. doi: 10.1007/s11011-008-9104-7. [DOI] [PubMed] [Google Scholar]
- 45.Saito C, Zwingmann C, Jaeschke H. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and n-acetylcysteine. Hepatology. 2010;51(1):246–254. doi: 10.1002/hep.23267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Constantini P, Chernyak BV, Petronilli V, Bernardi P. Modulation of the mitochondrial permeability transition pore by pyridine nucleotides and dithiol oxidation at two separate sites. The Journal of Biological Chemistry. 1996;271(12):6746–6751. doi: 10.1074/jbc.271.12.6746. [DOI] [PubMed] [Google Scholar]
- 47.Circu ML, Rodriguez C, Maloney R, Moyer MP, Aw TY. Contribution of mitochondrial gsh transport to matrix gsh status and colonic epithelial cell apoptosis. Free Radical Biology & Medicine. 2008;44(2):768–778. doi: 10.1016/j.freeradbiomed.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meister A, Anderson M E. Glutathione. Annual Review of Biochemistry. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 49.Martensson J, Lai JC, Meister A. High-affinity transport of glutathione is part of a multi-component system essential for mitochondrial function. Proc Natl Acad Sci U S A. 1990;87(18):7185–7189. doi: 10.1073/pnas.87.18.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han D, Canali R, Rettori D, Kaplowitz N. Effect of glutathione depletion on site and topology of superoxide and hydrogen peroxide production in mitochondria. Molecular Pharmacology. 2003;64(5):1136–1144. doi: 10.1124/mol.64.5.1136. [DOI] [PubMed] [Google Scholar]
- 51.Banaclocha MM, Martinez N. N-acetylcysteine elicited increase in cytochrome c oxidase activity in mice synaptic mitochondria. Brain Research. 1999;842:249–251. doi: 10.1016/s0006-8993(99)01819-3. [DOI] [PubMed] [Google Scholar]