

Abstract
Purpose: Farnesyltransferase (FTase) is a zinc-dependent enzyme that adds a farnesyl group to the Ras proteins. L778, 123 is a potent peptidomimetic imidazole-containing FTase inhibitor. Methods: L778123 was synthesized according to known methods and evaluated alone and in combination with doxorubicin against A549 (adenocarcinomic human alveolar basal epithelial cells) and HT29 (human colonic adenocarcinoma) cell lines by MTT assay. Results: L778123 showed weak cytotoxic activity with IC50 of 100 and 125 for A549 and HT-29 cell lines, respectively. The combination of doxorubicin and L778123 can decrease IC50 of doxorubicin in both cell lines significantly. Conclusion: It can be concluded that L778, 123 can be a good agent for combination therapy.
Keywords: Farnesyltransferase inhibitor, MTT assay, Combination therapy, L-778123
Introduction
The necessity for novel anticancer agents is obvious because of insufficient drugs. The anticancer agents must be effective for cancer therapy, increasing the lifetime and improving quality of life in cancer patients.1
Regulation of cellular growth is done by switching between the inactive and the active state of membrane-bound GTP binding proteins (G-proteins). Mutation of the GTP-binding protein Ras that has a key role in cell signaling pathways, can lead to uncontrolled proliferation.
Approximately, 30% of all human cancers are because of mutation of human ras proteins, reaching as high as 90% for pancreas cancer, 50% for colorectal cancer and and 40% for lung. A CAAX tetrapeptide motif exists at their C-terminal of these G-proteins (C: Cys, A: an aliphatic amino acid, X: Ser, Met, Gln, Ala typically Met).1-6 Ras proteins is undergone a series of modification for their biological functions. Their activation are started by the alkylation (i.e., farnesylation) of cysteine in CAAX motif. Farnesyltransferase (FTase) as a zinc-containing metalloenzyme identify the CAAX tetrapeptide sequence and add the 15 carbon isoprenoid, called a farnesyl group, from farnesyldiphosphate (FPP) to the thiol of cysteine. The farnesylation is an important step for the biological activity of Ras proteins and is a valuable target for chemotherapy.1,2,7,8 Peptidomimetic FTase inhibitors are a class of inhibitors which have a thiol moiety. Their thiol moiety could be bound to zinc ion of the FTase enzyme. Replacement of thiol group by another zinc binding moiety such as an imidazole or other heterocyclic rings made non-thiol, non-peptidic, imidazole or non-imidazole containing inhibitors.9 More than a decade later, researchers reported many groups of imidazole-containing FTase inhibitors which were tested in clinical trials, including L778123 and tipifarnib (Figure 1).
Figure 1 .
Chemical structures of farnesyltransferase inhibitors: (a) Tipifarnib, (b) L778123.
Doxorubicin is a potent chemotherapy drug which inhibits topoisomerase II. Doxorubicin-containing regimens are used in wide range of cancers include lung, colorectal, breast, esophageal, bladder, ovarian, and head and neck cancers, along with lymphomas and multiple myeloma.10,11
In this study, we report synthesis and comparison of cytotoxic activity of L778123 and doxorubicin alone and in combination with each other against A549 (adenocarcinomic human alveolar basal epithelial cells) and HT29 (human colonic adenocarcinoma) cell lines.
Materials and Methods
Chemistry
All reagents and solvents were prepared from Merck and Sigma Aldrich. L-778123 was synthesized was prepared according to known methods described previously.10 Melting points were measured with an Electrothermal-9100 melting point apparatus and are uncorrected. The IR spectra were recorded on a Shimadzu 4300 spectrophotometer (potassium bromide dicks). 1HNMR and 13CNMR spectra were recorded on a Varian unity 500 spectrometer and chemical shifts (δ) are reported in parts per million (ppm) using tetramethylsilane (TMS) as an internal standard. The mass spectra were run on an Agilent 6410 LC-MS at 70 eV. Merck silica gel 60. F254 plates were used for analytical TLC.
Synthesis of 4-{[5-(hydroxymethyl)-2-mercapto-1H-imidazol-1-yl]methyl-Benzonitrile (1)
A mixture of 4-(Aminomethyl)benzonitrile hydrochloride (4.21 g, 25 mmol), dihydroxyacetone dimmer (DHA) (2.47 g, 27.5 mmol), potassium thiocyanate (KSCN) (3.64 g, 37.5 mmol), acetic acid (3g, 50 mmol) in acetonitrile (24 mL) and water (1.0 mL) were stirred at 55 °C overnight. The mixture was cooled to room temperature and filtered. The precipitate was washed with acetonitrile (25 mL), water (50 mL) and ethylacetate (EtOAc) (25 mL) respectively. The solid was dried at 40 oC overnight to yield 1.
Yield: 73%; m.p = 159-161 oC; IR νmax (KBr)/cm-1: 3120(OH), 2570 (SH), 2230(nitrile), 1620(C=N). 1HNMR (250 MHz, d6-DMSO): δ 12.25 (s, 1H), 7.80 (d, J = 8.5, 2H), 7.37 (d, J = 8.5, 2H), 6.91 (s, 1H), 5.38 (s, 2H), 5.24 (s, 1H), 4.16 (s, 2H).
Synthesis of 4-{[5-(hydroxymethyl)-1H-imidazol-1-yl]methyl} benzonitrile (2)
A solution of sodium nitrite (1.1 g, 16.00 mmol) in water (1.2 mL) was added to a mixture of thioimidazole 1 (1 g, 4.00 mmol) in acetic acid (20 mL) over 30 min at room temperature. Ice (10 g) and then ammonia was added to bring the pH=11 at temperature between 20-30oC. The resulting mixture was stirred for 30 min and filtered. The solid was washed with 2:1 water-methanol (30 mL) respectively. The solid was dried to provide dethionated imidazole 2.
Yield: 85%; mp = 164-166 °C; IR νmax (KBr)/cm-1: 3130(OH), 2230 ( nitrile), 1680(C=N); 1HNMR (500 MHz, DMSO-d6): δ 7.83 (d, J = 8, 2H), 7.72 (s, 1H), 7.3 (d, J = 8, 2H), 6.86 (s, 1H), 5.35 (s, 2H), 5.12 (s, 1H), 4.29 (s, 2H).
Synthesis of 4-{[5-(chloromethyl)-1H-imidazol-1-yl] methyl}benzonitrile Hydrochloride (3)
Oxalyl chloride (1 mL, 11.5 mmol) was added over 30 min to a mixture of DMF (1.78 mL) and acetonitrile (25.6 mL) at temperature below 10 °C to give a white mixture. The mixture was added to a suspension of 2 (2.13 g, 10 mmol) in acetonitrile (17 mL) over 30 min while maintaining the temperature below 6 °C. The mixture was warmed to 25 °C, aged for 3 h and then cooled to 0 °C and aged for 60 min. The solid was filtrated, washed with ice-cold acetonitrile (15 mL) and dried to yield 3.
Yield: 90%; mp = 204-207 °C; IR νmax (KBr)/cm-101: 3100(NH+), 2230 ( nitrile), 1610(C=N); 1HNMR (500 MHz, DMSO-d6): δ 9.50 (d, J=1 Hz. 1H), 7.92 (s, 1H), 7.89 (d, J = 8 Hz, 2H), 7.56 (d, J = 8 Hz, 2H), 5.72 (s, 2H), 4.94 (s, 2H), 4.40 (br s, 1H).
Synthesis of N1-(3-chlorophenyl)-N2-(2-hydroxyethyl) glycinamide (4)
To a mixture of 3-chloroaniline (10 g, 78.25 mmol) in isopropyl acetate (90 mL) and potassium bicarbonate (15.62 g, 105.5 mmol) in water (75 mL) was added chloroacetyl chloride (12 g, 105.5 mmol) dropwise over 1 h maintained under 10 °C. The aqueous layer was removed the organic phase containing chloroacetamide was treated with ethanolamine (7.5 mL, 122.78 mmol). The mixture was warmed to 55 °C and aged for 1 h. Water (30 mL) and isopropyl acetate (7.5 mL) were added and the mixture was stirred vigorously at 55 °C for 30 minutes. The organic layer was separated and cooled to 0 °C over 1 h. The precipitate was filtrated, washed with cooled isopropyl acetate (2 × 15 mL) and dried to obtain 4.
Yield: 75%; mp = 103-105 °C; IR νmax (KBr)/cm-1 : 3260 (NH), 3200 (OH), 1690 (C=O); 1HNMR (400 MHz, DMSO-d6): δ 10.10 (br s, 1H), 7.86 (brt, 1H), 7.53 (d, J = 7.4, 1H), 7.33 (t, J = 8.0, 1H), 7.10 (d, J = 7.4, 1H), 4.66 (br s, 1H), 3.48 (t, J = 4.5, 2H), 3.31 (s, 2H), 2.62 (t, J =5.5, 2H); 13CNMR (75 MHz, DMSO-d6): δ 170.9, 140.1, 133.0, 130.3, 122.9, 118.6, 117.5, 60.3, 52.7, 51.5.
Synthesis of 1-(3-chlorophenyl) piperazine-2-one hydrochloride (5)
Diisopropylazodicarboxylate (DIAD) (5.1 g, 27 mmol) was added over 1.5 h to a mixture of tributylphosphine (5.5 g, 27 mmol) and EtOAc (15 mL) under the nitrogen atmosphere maintained under 0 °C. The mixture was aged at 0 °C for 30 min. The resulting solution was added over 1.5 h to a solution of amide alcohol 4 (4.52 g, 20 mmol) in EtOAc (30 mL) maintained under 0 °C and the mixture was warmed to room temperature over 1 h. Ethanolic HCl (4 M, 5.2 mL, 19.75 mmol) was added to the solution at 40 °C over 2 h. The resulting slurry was cooled to 0 °C and aged at 0 °C for 1 h. The hydrochloride salt was filtered, washed with chilled EtOAc (3 × 10 mL) and dried to afford 5.
Yield: 71%; mp = 232-235 °C; IR νmax (KBr)/cm-1: 3200-2400 (NH+), 1660 ( Amide C=O); 1HNMR (400 MHz, DMSO-d6): δ 10.24 (br s, 2H), 7.50-7.31 (m, 4H), 3.92 (t, J = 4.8, 2H), 3.85 (s, 2H), 3.52 (t, J = 4.8, 2H); 13CNMR (75 MHz, DMSO-d6): δ 162.1, 142.6, 132.9, 130.7, 127.0, 126.0, 124.5, 46.1, 44.9, 39.9.
Synthesis of 4[(5-{[4-(3-chlorophenyl)-3-oxopiperazin-1-yl] methyl}-1Himidazol-1 yl) methyl] benzonitrile (6)
A solution of 5 (4.11 g, 16.5 mmol) in acetonitrile (27 mL), i-Pr2NEt (9.30 mL, 52.9 mmol) and 3 (5.00 g, 17.2 mmol) was stirred at 0 °C for 48 h. Water (10 mL) and N,N-diisopropylethylamine (i-Pr2Net) (0.63 mL) were added. The solution was aged at 0 °C for 30 min and heated to 35 °C. Water (41 mL) was added over 5 min, and the solution was aged for 5- 10 min at 35 °C. Additional water (38 mL) was added over 30 min and the mixture was aged for 30 min at 35 °C. The mixture was cooled at 0 °C for 1 h. The crystals were filtrated and washed with ice-cold 1:5 acetonitrile/H2O (25 mL) and 1:9 acetonitrile/H2O at 5 °C (2 × 30 mL). The solid was dried to yield 6.
Yield: 80%; mp = 90-92 °C; IR νmax (KBr)/cm-1: 2240(C=N), 1640 ( Amide C=O),; 1HNMR (400 MHz, DMSO-d6): δ 10.24 (br s, 2H), 7.87-6.94 (m, 8H), 5.40 (s, 2H), 3.45 (s, 2H), 3.33 (s, 2H), 3.32 (d, J=5.5,2H), 3.03 (s, 2H), 2.6 (t, J = 5.1, 2H); 13CNMR (75 MHz, DMSO-d6): δ 165.6, 143.9, 143.2, 139.4, 132.8, 132.3, 130.2, 129.1, 127.4, 126.8, 126.1, 125.6, 124.0, 118.5, 109.9, 56.6, 49.1, 48.7, 48.0, 47.3. MS (ESI): 407.19 [M+H].
Growth inhibition assay
Cytotoxicity of L778, 123, doxorubicin and combination of them was examined with MTT assay at 1-200µM concentrations against two human cancer cell lines including A549 (adenocarcinomic human alveolar basal epithelial cells) and HT29 (human colonic adenocarcinoma) cells. Cell suspensions were seeded in 96-well plates with concentrations of 8000-10000 A549 cells and 15000-20000 HT29 cells per well and incubated for 24 h to allow cell attachment. The cells were treated for 72 h with four various concentrations of L778, 123, doxorubicin and combination of them. Culture medium were replaced with 150 μl fresh media plus 50 μl MTT (3-(4, 5-dimethylthiazol- 2-yl)-2, 5-diphenyl tetrazolium bromide) reagent (2mg/mL in PBS). An additional 4 h of incubation at 37 °C were done, and then the medium was discarded. 200 μl dimethyl sulfoxide plus 25μl sorenson buffer (0.1M NaCl, 0.1M glycine, pH: 10.5) was added to each well, and the solution was shacked for 15 min in 37 oC to dissolve the purple tetrazolium crystals. The absorbance of each well was measured by plate reader (SUNRISE TECAN, Austria) at a wavelength of 570 nm. The amount of produced purple formazan is proportional to the number of viable cells. Experiments were performed two times in triplicate for determination of sensitivity to each compound. The IC50 was calculated by linear regression analysis, expressed in mean±SD.12
Results and Discussion
The synthesis of L778, 123 (Figure 2) was started from 3-chloroaniline and 4-(Aminomethyl) benzonitrile hydrochloride. In one pot, 3-chloroaniline and chloroacetyl chloride in isopropyl acetate at 0 oC were mixed and then ethanolamine was added dropwise at 55°C to give amide alcohol 4 in quantitative yield. Ring closure of the piperazinone was performed with triphenylphosphine and DIAD in ethyl acetate via Mitsunobu reaction at -10 °C to afford 1-(3-Chlorophenyl) piperazine-2-one hydrochloride 5 in 75% overall yield.5,13,14
Figure 2 .

a) DHA, KSCN, CH3CN/H2O, 55 oC, 18h; b) NaNO2, AcOH, H2O, rt, 20 min; c) Oxalyl chloride, DMF/CH3CN, 0 oC to rt, 3h; d) Chloroacetyl chloride, iPrAc, 0 oC to rt, 1h; e) Ethanolamine, iPrAc, 55oC, 2h; f) DIAD, Tributylphosphine, EtOAc, -10 oC to rt, 1h; g) i-Pr2NEt, CH3CN/H2O, 0 oC, 39h.
In another pot ring closure of imidazole was performed by dihydroxyacetone, potassium thiocyanate and acetic acid in acetonotrile/H2O to yield thioimidazole 1. There have been many reports on the dethionation of thioimidazole compounds to their dethionated derivatives using hydrogen peroxide, sodium nitrite and etc.,5,10,13 but the sodium nitrite as a dethionation reagent has better yield than other reagents.10 So, the dethionation of thioimidazole was perfomed in water by sodium nitrite at 20-30 °C to afford the dethionated imidazole 2 in 85% yield. The chlorination of the dethionated imidazole 2 was achieved using oxalyl chloride in DMF and acetonitrile to obtain intermediate 3. Finally, the intermediates 3, 5 were reacted in presence of iPrNEt2 in acetonitrile for 48h at 0 oC to yield L778, 123 as free base with 80% yield. Synthesis of L778123 was done in 7 steps with overall yield of 24%.10
The results of cytotoxic activity were summarized as IC50 (µM) of L778, 123 and doxorubicin alone and in combination with each other by MTT assay in Table 1. The IC50 values of L778, 123 against A549 and HT29 cell lines showed that this compound had significant cytotoxic activity at concentrations lower than 100 µM. The IC50 values of doxorubicin alone and in combination with L778123 against both cell lines was 3.12, 2.75 µM and 1.72, 1.52 µM respectively. These results showed that however L778, 123 as a farnesyltransferase inhibitor can have a good cytotoxic activity but it is very weak in comparison with doxorubicin as a classic anti-cancer agent (Figure 3). On the other hand, combination of doxorubicin with L778123 can decrease IC50 of doxorubicin significantly. It means that combination of doxorubicin and L778123 may generate synergistic effects in both cell lines (Figure 4).
Table 1. Cytotoxic activity (IC50, µM) of L778123 and Doxorubicin (DOX) against HT-29 and A549 cell lines.
| A549 | HT29 | |
| L778123 | 100.72±2.16 | 125.78±2.45 |
| Doxorubicin | 3.13±0.11 | 2.75±0. 15 |
| Combined DOX and L778123 | 1.72±0.13 | 1.52±0.16 |
Figure 3 .
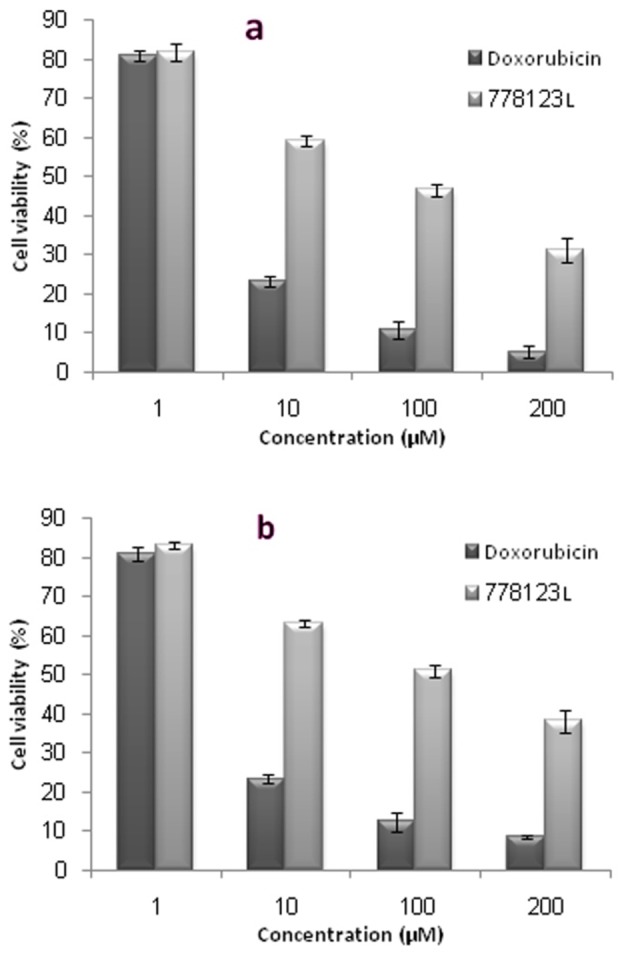
Cell viability of L778123, Doxorubicin and a combination of L778123 and Doxorubicin after 72 h treatment in: a) A549 and, b) HT-29 cell lines.
Figure 4 .
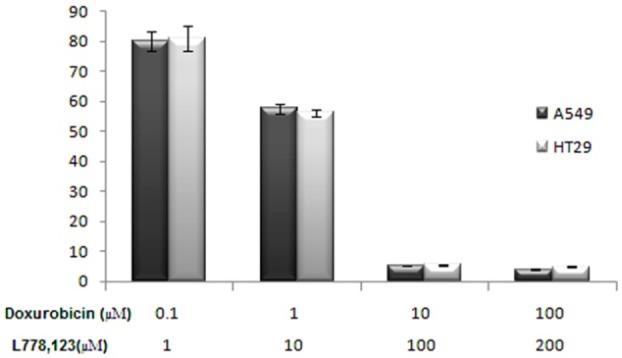
Cell viability of the combination of L778123 and Doxorubicin after 72 h treatment in A549 and HT-29.
Conclusion
In conclusion, L778, 123 was synthesized, characterized by IR, 1HNMR, 13CNMR, LC-Mass and identified as cytotoxic agents. Although this compound showed good potency with IC50 equal to 100 µM and 125 µM for A549 and HT29 cell lines respectively, it is very weak in comparison with potent anticancer doxorubicin. L778, 123 can be a good agent for combination therapy with doxorubicin because of synergetic effects.
Acknowledgement
The authors would like to thank the Tuberculosis and Lung Disease Research Center, the Faculty of Pharmacy, and research center for pharmaceutical nanotechnology Tabriz University of Medical Sciences for financial supports. This article was a part of a thesis submitted for PhD degree (No.45) in the Faculty of Pharmacy, Tabriz University of Medical Sciences, Tabriz, Iran.
Conflict of Interest
The authors report no conflicts of interest
References
- 1.Puntambekar DS, Giridhar R, Yadav MR. Insights into the structural requirements of farnesyltransferase inhibitors as potential anti-tumor agents based on 3D-QSAR CoMFA and CoMSIA models. Eur J Med Chem . 2008;43:142–54. doi: 10.1016/j.ejmech.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Asoh K, Kohchi M, Hyoudoh I, Ohtsuka T, Masubuchi M, Kenichi Kawasaki. et al. Synthesis and structure–activity relationships of novel benzofuran farnesyltransferase inhibitors. Bioorg Med Chem Lett . 2009;19:1753–7. doi: 10.1016/j.bmcl.2009.01.074. [DOI] [PubMed] [Google Scholar]
- 3.Angibaud P, Mevellec L, Meyer C, Bourdrez X, Lezouret P, Pilatte I. et al. Impact on farnesyltransferase inhibition of 4-chlorophenyl moiety replacement in the Zarnestra series. Eur J Med Chem . 2007;42(5):702–14. doi: 10.1016/j.ejmech.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 4.Gilleron P, Wlodarczyk N, Houssin R, Farce A, Laconde G, Goossens JF. et al. Design, synthesis and biological evaluation of substituted dioxodibenzothiazepines and dibenzocycloheptanes as farnesyltransferase inhibitors. Bioorg Med Chem Lett . 2007;17(19):5465–71. doi: 10.1016/j.bmcl.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka R, Rubio A, Harn NK, Gernert D, Grese TA, Eishima J. et al. Design and synthesis of piperidine farnesyltransferase inhibitors with reduced glucuronidation potential. Bioorg Med Chem . 2007;15(3):1363–82. doi: 10.1016/j.bmc.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Equbal T, Silakari O, Rambabu G, Ravikumar M. Pharmacophore mapping of diverse classes of farnesyltransferase inhibitors. Bioorg Med Chem Lett . 2007;17(6):1594–600. doi: 10.1016/j.bmcl.2006.12.087. [DOI] [PubMed] [Google Scholar]
- 7.Lu A, Zhang J, Yin X, Luo X, Jiang H. Farnesyltransferase pharmacophore model derived from diverse classes of inhibitors. Bioorg Med Chem Lett . 2007;17(1):243–9. doi: 10.1016/j.bmcl.2006.09.055. [DOI] [PubMed] [Google Scholar]
- 8.Bolchi C, Pallavicini M, Rusconi C, Diomede L, Ferri N, Corsini A. et al. Peptidomimetic inhibitors of farnesyltransferase with high in vitro activity and significant cellular potency. Bioorg Med Chem Lett . 2007;17:6192–6. doi: 10.1016/j.bmcl.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Xie A, Odde S, Prasanna S, Doerksen R. Imidazole-containing farnesyltransferase inhibitors: 3D quantitative structure–activity relationships and molecular docking. J Comput Aid Mol Des . 2009;23(7):431–48. doi: 10.1007/s10822-009-9278-z. [DOI] [PubMed] [Google Scholar]
- 10.Maligres PE, Waters MS, Weissman SA, McWilliams JC, Lewis S, Cowen J. et al. Preparation of a clinically investigated ras farnesyl transferase inhibitor. J Heterocycl Chem . 2003;40(2):229–41. [Google Scholar]
- 11.Chisholm-Burns Ma. Pharmacotherapy Principles and Practice. New York: The McGraw-Hill Companies; 2008. [Google Scholar]
- 12.Aliabadi A, Shamsa F, Ostad SN, Emami S, Shafiee A, Davoodi J. et al. Synthesis and biological evaluation of 2-phenylthiazole-4-carboxamide derivatives as anticancer agents. Eur J Med Chem . 2010;45(11):5384–9. doi: 10.1016/j.ejmech.2010.08.063. [DOI] [PubMed] [Google Scholar]
- 13.Askin D, Lewis S, Weissman SA, inventors; Merck & Co. Inc. assignee. Process for the synthesis of substituted piperazinones via Mitsunobu reaction. USA patent 6160118. 2000.
- 14.Weissman SA, Lewis S, Askin D, Volante R, Reider PJ. Efficient synthesis of N-arylpiperazinones via a selective intramolecular Mitsunobu cyclodehydration. Tetrahedron Lett . 1998;39(41):7459–62. [Google Scholar]