

Abstract
Genetic polymorphisms in the IL-2Rα chain (CD25) locus are associated with several human autoimmune diseases, including multiple sclerosis (MS). Blockade of CD25 by the humanized monoclonal antibody (Ab) daclizumab decreases MS-associated inflammation, but has surprisingly limited direct inhibitory effects on activated T cells. The present study describes unexpected effects of daclizumab therapy on innate lymphoid cells (ILCs). The number of circulating RORγt+ ILCs, which include lymphoid tissue inducer (LTi) cells, was found to be elevated in untreated MS patients compared to healthy subjects. Daclizumab therapy not only decreased numbers of ILCs, but also modified their phenotype away from LTi cells and toward a natural killer (NK) cell lineage. Mechanistic studies indicated that daclizumab inhibited differentiation of LTi cells from CD34+ hematopoietic progenitor cells or c-kit+ ILCs indirectly, steering their differentiation towards immunoregulatory CD56bright NK cells through enhanced intermediate affinity IL-2 signaling. Because adult LTi cells may retain lymphoid tissue inducing capacity or stimulate adaptive immune responses, we indirectly measured intrathecal inflammation in daclizumab-treated MS patients by quantifying the cerebrospinal fluid CXCL13 and immunoglobulin G (IgG) index. Both of these inflammatory biomarkers were inhibited by daclizumab treatment. Our study indicates that innate lymphoid cells are involved in the regulation of adaptive immune responses, and their role in human autoimmunity should be investigated further, including their potential as therapeutic targets.
Introduction
Daclizumab, a humanized monoclonal antibody against the alpha chain of the IL-2 receptor (IL-2Rα; CD25), promotes development of tolerance in solid organ transplantation (1) and limits target-organ inflammation in inflammatory uveitis (2) and multiple sclerosis (MS) (3–5). Daclizumab selectively blocks the low affinity (Kd = 10 nM) IL-2-binding domain on CD25, a non-signaling chain of the IL-2R. The two remaining chains of IL-2R, the beta (CD122) and gamma (CD132) chains, both have intracellular signaling motifs, and together bind IL-2 with intermediate affinity (Kd = 1 nM). Association of CD25 with the intermediate affinity IL-2R enhances the affinity of IL-2 binding 10–100-fold, resulting in a high affinity IL-2R (Kd = 10 pM). (6)
Because T cells upregulate CD25 during activation, and activated T cells are the main consumers of IL-2 through the high-affinity IL-2R, daclizumab was designed as an immunotherapy with selective inhibitory action towards activated (effector) T cells. However, our previous studies demonstrated that daclizumab has limited direct effects on activated T cells; its immunomodulatory potency resides in unexpected effects on components of the innate immune system (7–10). Interestingly, some of these effects are an indirect result of daclizumab-driven inhibition of IL-2 consumption by activated T cells and FoxP3-expressing regulatory T cells (Tregs), resulting in greater availability of IL-2 for signaling by cells that express high levels of intermediate affinity IL-2R. (11)
Despite the prominent inhibition of Tregs (11, 12), MS-related inflammation is significantly inhibited by daclizumab (3, 4, 13). We have previously described two mechanisms that can explain this apparent paradox: 1) CD56bright NK cells, which are expanded (7) and activated (11) by daclizumab therapy, have overlapping immunoregulatory functions with FoxP3 Tregs (9) and 2) daclizumab also inhibits antigen-specific priming of effector T cells by blocking trans-presentation of IL-2 by mature dendritic cells (mDC) (10). The current paper describes a third mechanism of how daclizumab inhibits MS-related inflammation.
While investigating effects of CD25 blockade on mDCs (10), we observed a significantly decreased population of lineage negative lymphocytic cells lacking expression of typical DC markers in the daclizumab-treated cohort. A subsequent review of the literature indicated that these cells likely represent innate lymphoid cells (ILCs) (14, 15).
Three major categories of ILCs have been identified (14–16): 1. NK cells (also called ILC1 cells), which in humans are phenotypically subdivided into CD56bright and CD56dim subsets; 2. RORγt+ ILCs, which include fetal and adult lymphoid tissue inducer (LTi) cells, IL-22 producing ILCs (ILC22, which express NKp44) and IL-17 producing ILCs (ILC17; it is unclear whether these are distinct from LTi or ILC22 cells); and 3. Type 2 ILCs (ILC2 or nuocytes), which are independent of RORγt and secrete Th2-type cytokines, such as IL-5 and IL-13. Although this categorization is conceptually useful, it remains uncertain whether sub-groups of ILCs represent truly distinct lineages or developmentally related and plastic phenotypes.
Multiple ILC subsets originate from CD34+ hematopoietic precursors in an Id2-dependent manner (15, 16), because Id2-deficient mice have greatly diminished levels of NK cells and LTi cells (17). Despite the fact that multiple studies addressed developmental relationships between different ILC subsets (14, 18–23), a unifying concept is still missing. Although methodological differences, especially the origin and the exact phenotype of LTi cells may explain apparent discrepancies between published studies, we conclude that prevailing evidence implies existence of a common precursor at the hematopoetic stem cell level (24) and probably also of a less differentiated ILC that responds to different environmental cues to generate both LTi cells and NK cells. IL-2/IL-15 represents at least one environmental stimulus that drives differentiation toward NK cell lineage (18–20, 25).
Although fetal LTi cells play a vital role in the development of secondary lymphoid tissues (26), the role of adult LTi cells has been unclear. Although LTi cells retain lymphoid-tissue inducing capacity postnatally (at least in the gastrointestinal tract (26–30)), tertiary lymphoid follicles can form in RORγt-deficient animals, which lack LTi cells (31, 32). Both T cells (33, 34) and B cells (31) acquire lymphoid-tissue inducing capacity in different inflammatory animal models. Nevertheless, adult LTi cells may play an important role in the evolution of T cell memory and CD4-dependent high affinity Ab responses by providing OX40 and CD30 signals (35, 36), raising the possibility that these cells may participate in the development of autoimmunity. However, to our knowledge the role of LTi cells in human autoimmune diseases has not been investigated thus far. Our unforeseen observation that daclizumab therapy affects levels of ILCs prompted us to investigate the role of these cells in the MS disease process.
Results
Daclizumab decreases the number of circulating innate lymphoid cells
We used Influenza vaccination (Flulaval™) as a tool to assess daclizumab-induced changes in the activation of the immune system in vivo by standardized antigenic stimulation. Using ex vivo flow cytometry, we compared changes in the phenotype of immune cells induced by influenza vaccination in MS patients under long-term daclizumab therapy with age/gender matched controls. CD56bright NK cells were significantly expanded in daclizumab-treated patients. ILCs – lineage negative (CD3, -11c, -14, -19, -56, -123) lymphocytic cells were observed in significantly lower proportions in daclizumab-treated patients before and further decreased 7 days after Flu vaccination, whereas no vaccination-induced change was observed in controls (Fig. 1A). We confirmed that the decrease in circulating ILCs was daclizumab-driven in two additional cohorts: First, in matched cryopreserved samples from a published Phase II clinical trial of daclizumab in MS (Clinicaltrials.gov identifier NCT00071838 (13)) collected before- and 3- and 8-months after initiation of daclizumab treatment (Fig. 1B); and second, in the ongoing Phase I/II trial of daclizumab high-yield process (DAC-HYP; Clinicaltrials.gov identifier NCT01143441) that focuses on defining the full mechanism of action of daclizumab in MS. In this last cohort, fresh ex vivo samples were directly compared with cryopreserved samples (Fig. 1C). There were no significant differences in the numbers of ILCs, but cryopreservation significantly decreased RORγt and c-kit expression (fig. S1). Consequently, we used fresh blood samples from the ongoing trial to better characterize the effects of daclizumab therapy on ILCs (Fig. 1D).
Figure 1. Daclizumab therapy decreases numbers of pro-inflammatory ILCs.

(A) Fresh PBMCs were analyzed before (D0) and 7 days after (D7) vaccination with Influenza haemagglutinin (Flu-HA) in daclizumab treated MS patients (Dac) and age/gender matched controls by flow cytometry. Data were normalized so that the baseline (D0) of the control group represented 100%, to which the baseline of daclizumab-treated MS patients and Flu-HA induced changes in both cohorts (D7) were compared. (n=5–8)
(B) Cryopreserved PBMCs were analyzed similar to (A) and samples were from 17 MS patients at baseline (BL), after 3 months of treatment with daclizumab (Mo 3), and after 8 months of treatment with daclizumab (Mo 8).
(C) Cryopreserved PBMCs (n=6) and fresh PBMCs (n=6) in the DAC HYP clinical trial were analyzed similar to (A and B) at baseline (BL) and after 6 months of treatment with daclizumab (Mo 6). Gating on CD45+ cells prevented inclusion of unlysed erythrocytes into the lineage-negative ILC gate.
(D) Gating strategy for ILCs: ILCs were gated based on forward and side scatter, and subgated as CD45+ and lineage (CD3, CD14, CD19, CD20, CD56) negative, non-DCs (i.e. CD11c/CD123−). The proportion of ILCs that expressed surface markers CD161, CD7, CD122 and c-kit and intracellular LTα and RORγt are depicted in representative sample. Dark gray histograms represent appropriate isotype controls.
(E) Percentage of CD45+ c-kit+ ILCs, CD56dim and CD56bright NK cells which express OX40L, CD30L, CD25 and NKp44 were determined by flow cytometry. Fresh uncoagulated peripheral blood samples were used. (n=11–17)
(F) Expression of RORγt and c-kit of ILCs in fresh peripheral blood from healthy donors, untreated MS patients (UnTx MS) and daclizumab-treated MS patients (Dac MS) was determined by flow cytometry using gating strategy depicted in (D).
(G) Ratio of RORγt+/c-kit+ ILCs: CD3+ T cells was calculated from the peripheral blood samples of healthy donors, UnTx MS and Dac MS. The horizontal bars represent the mean of each group.
We confirmed that a large portion of ILCs represent LTi cells in the fresh blood of healthy donors (HD) as determined by constitutive expression of c-kit, RORγt and lymphotoxin-α (LTα; Fig. 1D). However, we also observed expression of NK lineage markers such as CD122, CD161 and CD7 on ILCs. In order to better assess what proportion of ILCs represent true LTi cells, as compared to ILC22 cells, we stained ILCs and NK cells for NKp44 (marker of ILC22), CD25 (expressed on LTi cells) and two TNF-super family receptor ligands, which have been previously shown to be expressed on virtually all (OX40L; (37)) or a proportion (CD30L; (35)) of human LTi cells (Fig. 1E). We observed that 52.1% of c-kit+ lineage negative lymphoid cells in fresh blood samples expressed OX40L (range 26.4–70.4%), whereas NK cells lacked expression of this marker. CD30L was expressed on 26.4% of ILCs (range 3.3–40.1%). CD25 was expressed on 42.2% of ILCs (range 13.8–83.7%) and as reported previously, was also expressed in a proportion of CD56bright NK cells (15.0%; range 1.9–28.9%). In contrast, NKp44 was expressed only on 13.4% of blood ILCs (range 0.0–31.0%) and 11.0% of CD56bright NK cells (range 0.1–23.2%).
When we analyzed the numbers of c-kit+/RORγt+ ILCs (normalized to 1000 T cells), we observed that untreated MS patients had significantly elevated numbers of these proinflammatory ILCs whereas daclizumab-treated MS patients had comparable levels of c-kit+/RORγt+ cells to those observed in HD (Fig. 1F&G).
Daclizumab treatment induces phenotype change of ILCs toward a NK cell lineage
Next we asked whether daclizumab therapy solely inhibits the absolute number of ILCs, or whether it also affects their phenotype. Daclizumab-treated MS patients compared to matched untreated MS controls had significantly decreased proportion of ILCs that expressed LTα, IL-22 and TNFα (Fig. 2A) and concomitantly increased proportions of ILCs that expressed NK cell markers CD122, CD161 and CD7 (Fig. 2B). In contrast, we observed no significant changes in expression of other surface markers, including OX40L and CD30L (fig. S2).
Figure 2. Daclizumab treatment modifies phenotype of ILCs away from LTi and toward NK cell lineage.

(A) Percentage of ILCs which express inflammatory the cytokines LTα, IL-22, and TNF upon PMA (20ng/ml) and Ionomycin (1μM) stimulation for 3 hours was determined by flow cytometry.
(B) Percentage of ILCs which express NK cell markers, CD161, CD7 and CD122 was determined. The horizontal bars represent the mean of each of the groups.
Furthermore, we observed a significant correlation between contraction of ILCs and expansion of CD56bright NK cells in all daclizumab-treated patients (r = 0.447, p<0.001) and between contraction of c-kit+/RORγt+ LTi cells and expansion of CD56bright NK cells in a smaller DAC-HYP patient cohort (r = 0.520, p =0.046).
IL-2 signaling drives differentiation of c-kit+ ILCs toward immunoregulatory CD56bright NK cells
Based on the observation that ILCs in daclizumab-treated patients expressed significantly higher levels of CD122, we tested the hypothesis that signaling through the shared IL-2/IL-15 intermediate affinity receptor on ILCs is also enhanced. In comparison to untreated controls, ILCs from daclizumab-treated MS patients had 2–3 fold higher Stat5 phosphorylation to IL-2 and IL-15, but not to IL-7 (Fig. 3A&B).
Figure 3. Enhanced intermediate IL-2/IL-15 signaling in daclizumab-treated patients promotes differentiation of ILCs toward functional NK cells.

(A) Representative pSTAT5 levels of untreated or daclizumab (Dac Th) treated MS patients. Purified ILCs were stimulated with IL-2 (100IU/ml), IL-7 (10ng/ml), IL-15 (10ng/ml) or no cytokine for 10 minutes (optimal concentrations determined in pilot experiments). ILCs were then immediately fixed and stained for phosphorylated Stat5 production. (B) Group signaling data analogous to (A).
(C) Purified c-kit+ ILCs cells were cultured for 7 days in media supplemented with SCF and Flt3L (both 10ng/ml) and in the presence of IL-2 (100IU/ml), IL-7 (10ng/ml), IL-15 (10ng/ml) or no cytokine control. At day 7, cultured cells were stained and analyzed by FACS for the presence of CD56+ NK cells (compared to ex vivo PBMCs of daclizumab-treated MS patients with significant expansion of CD56brightNK cells). (C) Raw data from a representative experiment and (D) represents group data of the number of CD56dim NK and CD56bright NK cells per 1,000 beads.
(E) Purified c-kit+ ILCs were cultured as in panel C. At day 7, cultured cells were FACS stained for functional NK markers (Perforin, Granzyme A and B; compared to ex vivo PBMC of daclizumab-treated MS patients with significant expansion of CD56bright NK cells). FACS plots are representative of 4 replications.
(F) Also on day 7, cultured cells were incubated for ~16hr with target GFP-tagged K562 cells at a 1:1 effector to target ratio (or K562 cell only control wells). K562 killing was then measured by flow cytometry. Killing was determined as the percentage of K562 cells that were lost relative to the target cells only condition. The horizontal bars represent the mean of each of the groups.
Though Cupedo et al. (23) used IL-7 as the common γ-chain (γc)-signaling cytokine to support differentiation of ILCs toward an NK cell lineage in vitro, based on our combined ex vivo/in vitro observations summarized above, we asked whether intermediate affinity IL-2/IL-15 signaling could also promote differentiation of lineage-negative, c-kit+ ILCs toward a NK cell lineage (e.g. into CD56bright NK cells). To examine this, we cultured >99% pure lineage negative ILCs with high expression of c-kit (fig. S3; purified from fresh aphaeresis samples by negative selection or cell-sorting), in the presence of stem cell factor (SCF) and FMS-like tyrosine kinase 3 ligand (Flt3L) with either no additional cytokine or with IL-2, IL-7 or IL-15 for 7 days. Both IL-2 and IL-15, but not IL-7, significantly enhanced differentiation of ILCs towards CD56bright NK cells (Fig. 3C&D).
We next sought to determine whether these NK cell populations were functional, as evidenced by their expression of cytolytic enzymes and their ability to kill MHC-I-deficient targets. We observed that IL-2/IL-15-expanded CD56bright NK cells from ILC cultures expressed comparable or higher levels of perforin and granzymes A and B as CD56bright NK cells in peripheral blood (Fig. 3E). Finally, we performed killing assays to determine the functional status of NK cells derived from ILCs and observed almost 3-fold higher cytotoxicity in IL-2- and IL-15-cultured NK cells as compared to IL-7-cultured NK cells (Fig. 3F).
IL-2 drives differentiation of common CD34+ HPC precursors toward CD56bright NK cells and away from LTi lineage
Previous reports by Freud et al. (24) demonstrated that the precursor for human CD56bright NK cells is a subpopulation of CD34+ hematopoietic progenitor cells (HPC) residing in the lymph nodes (or isolated from the blood, but not bone marrow) and that the differentiation process is dependent on IL-2 or IL-15 signaling. Therefore, we studied a unifying hypothesis that LTi cells and CD56bright NK cells share CD34+ HPCs and that intermediate affinity IL-2R signaling is the main driving force pushing differentiation away from LTi cells and toward CD56bright NK cells. To explore this, we isolated CD34+ HPC (>90 % purity; Fig. 4A) and cultured them with SCF and Flt3L in the presence or absence of IL-2. Without IL-2, CD34+ HPC differentiated into LTi cells (indicated by constitutive expression of LTα) but not to CD56bright NK cells (Fig. 4B, upper panels). In contrast, the presence of IL-2 inhibited differentiation to LTi lineage and instead produced high proportions of CD56bright NK cells (Fig. 4B, lower panels).
Figure 4. CD34+ HPCs differentiate in vitro to either LTi or CD56bright NK cells, depending on the presence of IL-2.

(A and B) Purified CD34+ HPC cells were cultured with SCF and Flt3L (both 10ng/ml) with either no cytokine or IL-2 (100IU/ml). After 10–14 days of culture the proportion of LTi cells (lineage-/c-kit+/LTα+ and CD56+ NK cell was determined by flow cytometry. (B) Representative raw data (C) Fold change calculated from the absolute numbers of LTi and CD56bright NK cells and (D) MFI of LTα and CD56 in CD34 HPC cultures after 10–14 days of differentiation. The horizontal bars represent the mean of each of the groups.
Daclizumab treatment decreases CSF levels of CXCL13 and intrathecal production of IgG
In order to determine whether inhibition of LTi cell development by daclizumab is clinically meaningful, we indirectly measured the functions of intrathecal inflammation by measuring CSF levels of CXCL13 (the chemokine linked to lymphoid neogenesis (38–41)) and IgG index (validated measure of intrathecal IgG production). After 6.5 months of daclizumab treatment, we observed that CXCL13 levels decreased by 50.4% (from an average of 35.92 pg/ml to 17.83 pg/ml; p = 0.008; Fig. 5A) and the IgG index decreased by 13.5% (from an average of 1.008 to 0.872; p = 0.003; Fig. 5B).
Figure 5. Daclizumab treatment decreases CSF levels of CXCL13 and intrathecal production of IgG.
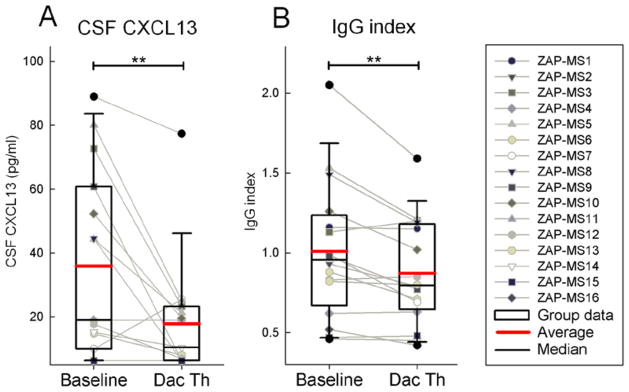
(A) CSF CXCL13 concentration was measured by ELISA.
(B) IgG index as measured by NIH clinical laboratory. Raw data for individual patients measured before (Baseline) and 6.5 months after initiation of daclizumab therapy (Dac Th) are depicted as gray dot and line blots and group data are depicted as box plots with mean highlighted as red and median as black horizontal line.
Discussion
The goal of our study was to investigate in detail effects of daclizumab therapy on ILC subtypes, the mechanisms behind these effects, as well as to explore potential role of ILC subsets in the MS disease process. We observed that although untreated MS patients have significantly elevated levels of pro-inflammatory ILCs, defined as c-kit+/RORγt+ lineage negative cells, which are known to contain LTi cells, daclizumab therapy restores ILC numbers to levels comparable to those observed in healthy subjects. Furthermore, daclizumab therapy skews the phenotype of ILCs away from the LTi lineage and toward the NK cell lineage. We were able to reproduce these in vivo effects by adding IL-2 (or IL-15) to in vitro differentiation assays of CD34+ HPC or c-kit+ ILCs: In both instances, addition of IL-2 drove differentiation of ILCs away from the LTi lineage and toward the NK cell lineage, in agreement with published animal studies (18–20, 25). We conclude that CD25 blockade alters differentiation of ILCs by paradoxically enhancing IL-2 signaling on common precursor(s) that can differentiate to either LTi cells or CD56bright NK cells, depending on environmental cues. By blocking high affinity IL-2 signaling on T cells, daclizumab increases the in vivo availability of IL-2 for cells that are capable of signaling via the intermediate affinity IL-2R (11), such as CD56bright NK cells (7) and subtypes of ILCs.
The crucial question is whether the phenotypic switch of ILCs from LTi to NK cell lineage induced by daclizumab inhibits aberrant adaptive immune responses associated with autoimmunity. Utilizing an open-label cross-over trial methodology, we previously demonstrated that daclizumab, either in combination with IFN-β (3) or as a monotherapy (8, 13) significantly inhibits formation of brain inflammatory lesions in MS and stabilizes neurological disability. These findings have now been fully reproduced in an independent double-blind, placebo-controlled clinical trial (4).
Although the current study is the first direct evidence linking adult LTi cells with autoimmunity in humans, by facilitating OX40 and CD30 signaling on T cells, LTi cells may play an important role in promoting the development of CD4 T cell memory (28, 35) and production of CD4 T cell-dependent high-affinity Ab (36). Both of these advanced functions of adaptive immunity are linked to secondary lymphoid tissues or organized lymphoid aggregates (such as “ectopic” or “tertiary” lymphoid follicles) that have been observed in many autoimmune diseases (26), including MS (42, 43). Indeed, the hallmark of MS is intrathecal production of IgG, measured clinically as IgG index, with characteristics of an antigen-driven affinity maturation process (44, 45). Formation of high affinity IgG against protein antigens is dependent on CD4 T cell help and occurs only in germinal centers of lymphoid organs. Persistent presence of oligoclonal bands (expanded IgG clonotypes detected only in the CSF but not in the serum), presence of all stages of B cell differentiation in the CSF of MS patients (46), and the recent discovery of CXCL13-rich tertiary lymphoid follicles in the meninges of some MS patients (42, 43) collectively imply that the intrathecal production of IgG in MS is facilitated and/or sustained within meningeal lymphoid aggregates.
Hence, we searched for in vivo evidence that would link our observation of daclizumab-driven inhibition of LTi cells to meningeal inflammation in MS. There is an abundance of literature linking ectopic lymphoid follicles in a variety of human autoimmune diseases with CXCL13 expression in the affected tissue (39) and elegant animal studies linked CXCL13 mechanistically to lymphoid neogenesis (40, 41). We observed that CSF CXCL13 levels decreased by 50.4% after 6.5 months of daclizumab therapy. Furthermore, this was associated with 13.5% decrease in the intrathecal production of IgG, measured as IgG index. This effect on IgG production was specific for the intrathecal compartment, as we have previously reported that daclizumab therapy has no effect on systemic production of immunoglobulins (3). To our knowledge, daclizumab is the first immunomodulatory drug that decreases levels of intrathecally produced IgG in MS, and it is doing so without depleting or limiting access of immune cells to the intrathecal compartment (13). Furthermore, because meningeal lymphoid follicles have been associated with neuronal loss in the underlying brain tissue (43, 47), inhibition of meningeal inflammation would be expected to slow down accelerated development of gray matter atrophy observed in MS. Indeed, our pilot data comparing 27 MS patients on long-term daclizumab therapy (total of 982 MRI scans) to a matched cohort of 44 MS patients treated with first line FDA-approved therapies (mainly interferons) indicate that daclizumab inhibits atrophy of brain structures that have direct contact with meninges/CSF by 38–60% in comparison to standard treatments (48). Although these encouraging observations need to be reproduced in large controlled studies, our data suggest that inhibition of LTi cells may be a highly efficacious therapeutic modality for MS.
There are limitations of the current study: Although our in vivo observations and in vitro mechanistic studies reinforce the evidence for a developmental link between LTi cells and NK cells through regulation of a common precursor, we have not performed detailed investigation and single cell cloning experiments that would define the precise phenotype of this precursor. Similarly, because the visualization of meningeal lymphoid follicles or even direct quantification of diffuse meningeal inflammation is currently not possible in living human subjects, our study provides only an indirect, albeit plausible, link between LTi cells and intrathecal inflammation in MS. Clearly, follow-up studies evaluating ILCs in autoimmune diseases amenable to biopsies of the targeted tissue, or clinical trials of novel therapies that target specifically pro-inflammatory ILCs will be necessary to establish a definite pathogenic role of LTi cells in human autoimmunity. We hope the current study will spark the necessary interest in these innate lymphoid effectors, which will lead to a better understanding of their role in human health and disease. Such understanding is the necessary prerequisite for development of targeted therapies.
Materials and Methods
Subjects
Daclizumab-treated MS patient data was obtained from patients participating in NINDS IRB-approved clinical protocols, and all patients signed informed consent. Five cohorts of patients and controls were utilized in the current study and described in Supplementary Materials.
Antibodies
The surface and intracellular flourochrome-conjugated antibodies from BD Bioscience, eBioscience or R&D systems were used and described in Supplementary Materials.
Flow Cytometry
Immunophenotyping of ILCs was performed on lysed whole blood within 30 minutes of ex vivo collection and stained according to a previously established protocol (7). Minimum of 1×106 cells were stained in order to acquire a minimum of 1000 gated ILCs. Gating for all intracellular and cell surface markers was based on isotype controls. Data were acquired on an LSR II with HTS delivery system and analyzed with FACSDiva 6.1(BD).
ILC purification and culture conditions
ILCs were isolated from fresh aphaeresis samples. PBMCs were isolated by Ficoll gradient and ILCs for each experiment were isolated from 2×108 PBMCs by negative selection using a combination of the lineage cell depletion kit (Miltenyi Biotec; 130-092-211) and anti-CD34 microbeads (Miltenyi Biotec; 130-046-702). Purity was verified by flow cytometry after isolation (fig. S2A). Purified ILCs were cultured in X-vivo-15 (Lonza) medium in the presence of stem cell factor (SCF) and Flt3L (both 10ng/ml; R&D Systems and Peprotech respectively) for 7 days with IL-2 (100 IU/ml), IL-7 (10ng/ml) or IL-15 (10ng/ml) as indicated (R&D Systems). Cultured ILCs were characterized by surface and intracellular flow cytometry for NK lineage markers after 0–7 days of cell culture.
STAT5 signaling assay
ILCs isolated from fresh PBMC samples were stimulated for 10 minutes at 37 °C with exogenous IL-2 (100 IU/ml), IL-15 (10ng/ml) or IL-7 (10ng/ml) and immediately formaldehyde fixed and stained cells for phosphorylated STAT5 or corresponding isotype according to the manufacturer protocol (BD Bioscience).
K562 cytotoxicity assay
The cytotoxicity of NK cells differentiated from ILC cultures toward MHC-I-deficient targets was assessed by a flow-cytometry based killing assay using GFP-tagged K562 cells. Targets were seeded with or without NK cells differentiated from ILCs at day 7 at a 1:1 effector:target ratio overnight. Cultures were then collected and the absolute number of live K562 cells was proportionally enumerated between conditions by flow cytometry, using equal amounts of fluorescent beads added to each condition before analysis as a reference. Percentage of K562 cell killing was calculated using the following formula: % killing = 100 − ((#K562 with NK/#K562control)*100).
In vitro differentiation of CD34+ HPC
CD34+ HPCs were purified from fresh PBMCs using magnetic-labeled CD34+ beads (Miltenyi Biotec;130-046-702) after lineage cell depletion and immediately cultured in X-vivo supplemented with FBS 20%, SCF and Flt3L (both 10ng/ml) +/− IL-2 (100IU/ml) for 14 days. CD34+ HPC were analyzed by flow cytometry at day 0, 7, 10 and 14 for differentiation into c-kit+ LTα+ LTi or CD56+ NK cells.
ELISA for CXCL13
CSF supernatant from 16 patients enrolled in the daclizumab clinical trial (NCT00071838) was collected and frozen at −80°C. Undiluted CSF was measured for CXCL13 by a DuoSet ELISA Development Kit (R&D Systems) according to the manufacturer’s instructions. The detection limit of the assay was determined to be 6.25 pg/ml.
IgG index measurement
IgG index was determined by immunonephelometric method using an automated analyzer (BN II, Siemens Healthcare Diagnostics).
Statistical analysis
Group differences were analyzed using one-way ordinary or repeated measures analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. For comparisons of two groups, student’s independent-samples t tests for analysis of independent groups or paired t test for analysis of repeated measurements within identical groups of patients. When data were not normally distributed, nonparametric tests were used. Data are presented as mean ± SEM using the GraphPad Prism 5.0 or SigmaPlot 11.0 program and p < 0.05 was considered significant. In figures, values of p are shown as follows: *p < 0.05; **p < 0.01;***p < 0.001.
Supplementary Material
Acknowledgments
We thank Azita Kashani for aphaeresis processing, the NIB nursing staff and clinicians for patient care and coordination of aphaeresis and blood collection, and Jayne Martin for technical assistance. Above all, we are grateful to all patients participating in NIB protocols whose samples contributed to this study.
Funding: This research was supported by the Intramural Research Program of the NINDS and NIH Clinical Center.
Footnotes
Author contributions: B.B. developed the concept of the study and supervised the project. J.S.A.P., S.H. and B.B. designed the experiments. J.S.A.P., S.H., Q.X., M.L.H., L.B.K., G.C., B.B. performed the experiments and analyzed the data. J.S.A.P., S.H. and B.B. wrote the paper.
Competing interests: B.B. is a co-inventor on NIH patents related to daclizumab therapy and as such has received royalty payments based upon license to US patents 7,575,742 and 7,258,859 (and patent applications claiming priority to these patents) and other National Stage patents and patent applications claiming priority to PCT/US2002/038290 or PCT/US2003/020428. The other authors declare no competing interests.
References and Notes
- 1.Waldmann TA, O’Shea J. The use of antibodies against the IL-2 receptor in transplantation. Curr Opin Immunol. 1998;10:507–512. doi: 10.1016/s0952-7915(98)80215-x. [DOI] [PubMed] [Google Scholar]
- 2.Nussenblatt RB, Thompson DJ, Li Z, Peterson JS, Robinson RR, Shames RS, Nagarajan S, Tang MT, Mailman M, Velez G, Roy C, Levy-Clarke GA, Suhler EB, Djalilian A, Sen HN, Al-Khatib S, Ursea R, Srivastava S, Bamji A, Mellow S, Sran P, Waldmann TA, Buggage RR. Humanized anti-interleukin-2 (IL-2) receptor alpha therapy: long-term results in uveitis patients and preliminary safety and activity data for establishing parameters for subcutaneous administration. J Autoimmun. 2003;21:283–293. doi: 10.1016/s0896-8411(03)00113-6. [DOI] [PubMed] [Google Scholar]
- 3.Bielekova B, Richert N, Howard T, Blevins G, Markovic-Plese S, McCartin J, Wurfel J, Ohayon J, Waldmann TA, McFarland HF, Martin R. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon-beta. Proc Natl Acad Sci U S A. 2004;101:8705–8708. doi: 10.1073/pnas.0402653101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wynn D, Kaufman M, Montalban X, Vollmer T, Simon J, Elkins J, O’Neill G, Neyer L, Sheridan J, Wang C, Fong A, Rose JW. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010;9:381–390. doi: 10.1016/S1474-4422(10)70033-8. [DOI] [PubMed] [Google Scholar]
- 5.Rose JW, Watt HE, White AT, Carlson NG. Treatment of multiple sclerosis with an anti-interleukin-2 receptor monoclonal antibody. Ann Neurol. 2004;56:864–867. doi: 10.1002/ana.20287. [DOI] [PubMed] [Google Scholar]
- 6.Rickert M, Wang X, Boulanger MJ, Goriatcheva N, Garcia KC. The structure of interleukin-2 complexed with its alpha receptor. Science. 2005;308:1477–1480. doi: 10.1126/science.1109745. [DOI] [PubMed] [Google Scholar]
- 7.Bielekova B, Catalfamo M, Reichert-Scrivner S, Packer A, Cerna M, Waldmann TA, McFarland H, Henkart PA, Martin R. Regulatory CD56bright natural killer cells mediate immunomodulatory effects of IL-2R-alpha-targeted therapy (daclizumab) in multiple sclerosis. PNAS. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bielekova B, Howard T, Packer AN, Richert N, Blevins G, Ohayon J, Waldmann TA, McFarland HF, Martin R. Effect of anti-CD25 antibody daclizumab in the inhibition of inflammation and stabilization of disease progression in multiple sclerosis. Arch Neurol. 2009;66:483–489. doi: 10.1001/archneurol.2009.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang W, Chai NR, Maric D, Bielekova B. Unexpected Role for Granzyme K in CD56bright NK Cell-Mediated Immunoregulation of Multiple Sclerosis. J Immunol. 2011;187:781–790. doi: 10.4049/jimmunol.1100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wuest SC, Edwan JH, Martin JF, Han S, Perry JS, Cartagena CM, Matsuura E, Maric D, Waldmann TA, Bielekova B. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med. 2011;17:604–609. doi: 10.1038/nm.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin JF, Perry JS, Jakhete NR, Wang X, Bielekova B. An IL-2 paradox: blocking CD25 on T cells induces IL-2-driven activation of CD56(bright) NK cells. J Immunol. 2010;185:1311–1320. doi: 10.4049/jimmunol.0902238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oh U, Blevins G, Griffith C, Richert N, Maric D, Lee CR, McFarland H, Jacobson S. Regulatory T cells are reduced during anti-CD25 antibody treatment of multiple sclerosis. Arch Neurol. 2009;66:471–479. doi: 10.1001/archneurol.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bielekova B, Richert N, Herman ML, Ohayon J, Waldmann TA, McFarland H, Martin R, Blevins G. Intrathecal effects of daclizumab treatment of multiple sclerosis. Neurology. 2011;77:1877–1886. doi: 10.1212/WNL.0b013e318239f7ef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, Eberl G. Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science. 2010;330:665–669. doi: 10.1126/science.1194597. [DOI] [PubMed] [Google Scholar]
- 15.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 16.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annual review of immunology. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 17.Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, Gruss P. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 1999;397:702–706. doi: 10.1038/17812. [DOI] [PubMed] [Google Scholar]
- 18.Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Holscher C, Honig M, Pannicke U, Schwarz K, Ware CF, Finke D, Diefenbach A. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 2010;33:736–751. doi: 10.1016/j.immuni.2010.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3− LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity. 1997;7:493–504. doi: 10.1016/s1074-7613(00)80371-4. [DOI] [PubMed] [Google Scholar]
- 20.Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci U S A. 2010;107:10961–10966. doi: 10.1073/pnas.1005641107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colonna M. Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity. 2009;31:15–23. doi: 10.1016/j.immuni.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 22.Crellin NK, Trifari S, Kaplan CD, Cupedo T, Spits H. Human NKp44+IL-22+ cells and LTi-like cells constitute a stable RORC+ lineage distinct from conventional natural killer cells. The Journal of experimental medicine. 2010;207:281–290. doi: 10.1084/jem.20091509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10:66–74. doi: 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- 24.Freud AG, Becknell B, Roychowdhury S, Mao HC, Ferketich AK, Nuovo GJ, Hughes TL, Marburger TB, Sung J, Baiocchi RA, Guimond M, Caligiuri MA. A human CD34(+) subset resides in lymph nodes and differentiates into CD56bright natural killer cells. Immunity. 2005;22:295–304. doi: 10.1016/j.immuni.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Satoh-Takayama N, Lesjean-Pottier S, Vieira P, Sawa S, Eberl G, Vosshenrich CA, Di Santo JP. IL-7 and IL-15 independently program the differentiation of intestinal CD3-NKp46+ cell subsets from Id2-dependent precursors. The Journal of experimental medicine. 2010;207:273–280. doi: 10.1084/jem.20092029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nature reviews Immunology. 2006;6:205–217. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 27.Finke D. Fate and function of lymphoid tissue inducer cells. Curr Opin Immunol. 2005;17:144–150. doi: 10.1016/j.coi.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Lane P, Kim MY, Withers D, Gaspal F, Bekiaris V, Desanti G, Khan M, McConnell F, Anderson G. Lymphoid tissue inducer cells in adaptive CD4 T cell dependent responses. Semin Immunol. 2008;20:159–163. doi: 10.1016/j.smim.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Schmutz S, Bosco N, Chappaz S, Boyman O, Acha-Orbea H, Ceredig R, Rolink AG, Finke D. Cutting edge: IL-7 regulates the peripheral pool of adult ROR gamma+ lymphoid tissue inducer cells. J Immunol. 2009;183:2217–2221. doi: 10.4049/jimmunol.0802911. [DOI] [PubMed] [Google Scholar]
- 30.Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 31.Lochner M, Ohnmacht C, Presley L, Bruhns P, Si-Tahar M, Sawa S, Eberl G. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. The Journal of experimental medicine. 2011;208:125–134. doi: 10.1084/jem.20100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, Woodland DL, Lund FE, Randall TD. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med. 2004;10:927–934. doi: 10.1038/nm1091. [DOI] [PubMed] [Google Scholar]
- 33.Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, Wucherpfennig K, Turley S, Carroll MC, Sobel RA, Bettelli E, Kuchroo VK. Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity. 2011;35:986–996. doi: 10.1016/j.immuni.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, Chervonsky AV, Fu YX. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 35.Withers DR, Gaspal FM, Bekiaris V, McConnell FM, Kim M, Anderson G, Lane PJ. OX40 and CD30 signals in CD4(+) T-cell effector and memory function: a distinct role for lymphoid tissue inducer cells in maintaining CD4(+) T-cell memory but not effector function. Immunol Rev. 2011;244:134–148. doi: 10.1111/j.1600-065X.2011.01057.x. [DOI] [PubMed] [Google Scholar]
- 36.Lane PJ, McConnell FM, Withers D, Gaspal F, Saini M, Anderson G. Lymphoid tissue inducer cells and the evolution of CD4 dependent high-affinity antibody responses. Prog Mol Biol Transl Sci. 2010;92:159–174. doi: 10.1016/S1877-1173(10)92007-3. [DOI] [PubMed] [Google Scholar]
- 37.Kim S, Han S, Withers DR, Gaspal F, Bae J, Baik S, Shin HC, Kim KS, Bekiaris V, Anderson G, Lane P, Kim MY. CD117 CD3 CD56 OX40Lhigh cells express IL-22 and display an LTi phenotype in human secondary lymphoid tissues. European journal of immunology. 2011;41:1563–1572. doi: 10.1002/eji.201040915. [DOI] [PubMed] [Google Scholar]
- 38.Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, Hamburger J, Ainsworth J, Mathews J, Salmon M, Bowman SJ, Buckley CD. Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjogren’s syndrome. Arthritis and rheumatism. 2001;44:2633–2641. doi: 10.1002/1529-0131(200111)44:11<2633::aid-art443>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 39.Carlsen HS, Baekkevold ES, Morton HC, Haraldsen G, Brandtzaeg P. Monocyte-like and mature macrophages produce CXCL13 (B cell-attracting chemokine 1) in inflammatory lesions with lymphoid neogenesis. Blood. 2004;104:3021–3027. doi: 10.1182/blood-2004-02-0701. [DOI] [PubMed] [Google Scholar]
- 40.Luther SA, Ansel KM, Cyster JG. Overlapping roles of CXCL13, interleukin 7 receptor alpha, and CCR7 ligands in lymph node development. The Journal of experimental medicine. 2003;197:1191–1198. doi: 10.1084/jem.20021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, Kusser K, Hopken UE, Lipp M, Niederreither K, Blomhoff R, Sitnik K, Agace WW, Randall TD, de Jonge WJ, Mebius RE. Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol. 2009;10:1193–1199. doi: 10.1038/ni.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14:164–174. doi: 10.1111/j.1750-3639.2004.tb00049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magliozzi R, Howell OW, Reeves C, Roncaroli F, Nicholas R, Serafini B, Aloisi F, Reynolds R. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol. 2010;68:477–493. doi: 10.1002/ana.22230. [DOI] [PubMed] [Google Scholar]
- 44.Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH. Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol. 2003;171:2725–2733. doi: 10.4049/jimmunol.171.5.2725. [DOI] [PubMed] [Google Scholar]
- 45.Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, Reynolds R, O’Connor KC, Hafler DA. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain. 2011;134:534–541. doi: 10.1093/brain/awq350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corcione A, Casazza S, Ferretti E, Giunti D, Zappia E, Pistorio A, Gambini C, Mancardi GL, Uccelli A, Pistoia V. Recapitulation of B cell differentiation in the central nervous system of patients with multiple sclerosis. Proc Natl Acad Sci U S A. 2004;101:11064–11069. doi: 10.1073/pnas.0402455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Howell OW, Reeves CA, Nicholas R, Carassiti D, Radotra B, Gentleman SM, Serafini B, Aloisi F, Roncaroli F, Magliozzi R, Reynolds R. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134:2755–2771. doi: 10.1093/brain/awr182. [DOI] [PubMed] [Google Scholar]
- 48.Borges I, Shea C, Ohayon J, Bielekova B, Reich DS. Daclizumab reduces the rate of brain atrophy in multiple sclerosis. Mult Scler. 2011;17:540. doi: 10.1016/j.msard.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.