

Abstract
Development of SAR in an aryl ether series of mGlu5 NAMs leading to the identification of tool compound VU0409106 is described in this Letter. VU0409106 is a potent and selective negative allosteric modulator of mGlu5 that binds at the known allosteric binding site and demonstrates good CNS exposure following intraperitoneal dosing in mice. VU0409106 also proved efficacious in a mouse marble burying model of anxiety, an assay known to be sensitive to mGlu5 antagonists as well as clinically efficacious anxiolytics.
The metabotropic glutamate receptors (mGlus) are a family of eight related G-protein-coupled receptors (GPCRs) that act through binding glutamate (L-glutamic acid), the major excitatory transmitter in the mammalian central nervous system (CNS). The orthosteric binding sites of these seven transmembrane (7TM) receptors are located in the extracellular domain while allosteric binding sites identified to date are located in the transmembrane domain.1 Due to a highly conserved orthosteric binding site across the members of the mGlu family, the design of selective orthosteric ligands has been challenging. A solution to this problem that has garnered much attention and proven effective in many instances has been the development of allosteric modulators of mGlus.2 One of the more developed areas within the mGlu allosteric modulator field has been the design of small molecule negative allosteric modulators (NAMs), also known as non-competitive antagonists, of mGlu5.3
Extensive preclinical in vivo work has been published with two structurally related disubstituted alkyne tool compounds, MPEP4 and MTEP5 (Fig. 1). Efficacy in numerous animal models has been noted with these compounds, including pain,6 anxiety,7 gastroesophageal reflux disease (GERD),8 Parkinson’s disease levodopa induced dyskinesia (PD-LID),9 and fragile X syndrome (FXS).10 Furthermore, another alkyne tool compound known as CTEP11 was recently shown to reverse an already established FXS phenotype in adult Fmr1 knockout mice following chronic dosing.12 Recent studies in mice with MPEP and yet another alkyne tool compound known as GRN-529 point to a potential role for mGlu5 NAMs in the treatment of other autism spectrum disorders.13 GRN-529 has also recently proven efficacious in rodent models of treatment resistant depression (TRD).14 Finally, both MPEP and MTEP have produced encouraging results in animal models of addiction with various drugs of abuse, including cocaine,15 nicotine,15g,16 methamphetamine,17 morphine,18 and ethanol.19
Figure 1.
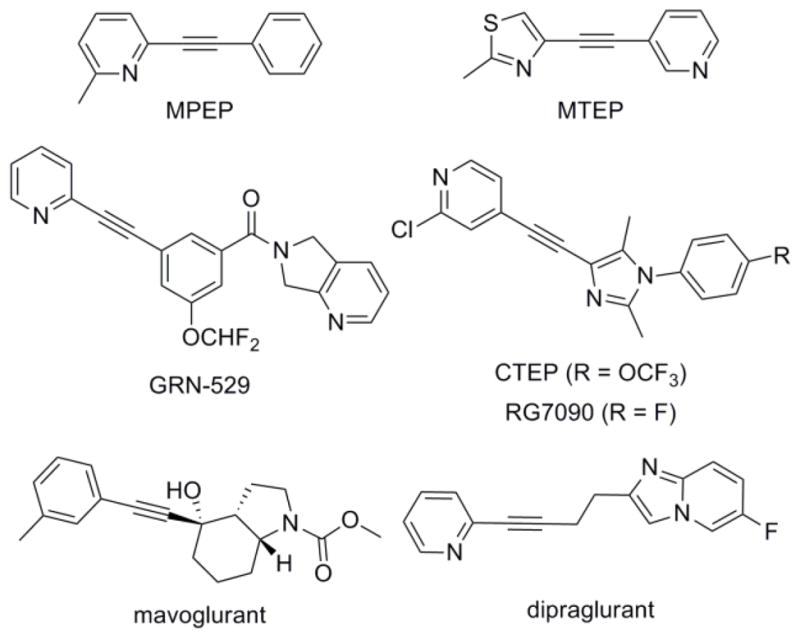
mGlu5 NAM tool and clinical compounds
Multiple mGlu5 NAMs have advanced to clinical trials, with the most encouraging results thus far observed in GERD,20 FXS,21 and PD-LID.22 The majority of clinical compounds are from within the disubstituted alkyne structure class, including each of the three molecules with confirmed ongoing clinical activity: dipraglurant (ADX48621), mavoglurant (AFQ056), and RG7090 (RO4917523) (Fig. 1).3a We have been interested in the identification and optimization of mGlu5 NAMs within chemotypes that do not contain a disubstituted alkyne motif. One approach that we have successfully employed in this endeavor was based on the development of hits identified using a functional cell-based high-throughput screen (HTS) of a collection of 160,000 compounds.23 We have also used both rational design approaches24 as well as virtual screening methods to identify new mGlu5 NAM tool compounds.25
Among the confirmed hits from our functional HTS was aryl ether benzamide 1 (Fig. 2), which demonstrated good potency in our functional assay. This assay also serves as our primary assay for lead optimization by measuring the ability of the compound to block the mobilization of calcium induced by an EC80 concentration of glutamate in HEK293A cells expressing rat mGlu5.26 As part of our initial hit evaluation process, the primary amine functional group was removed to afford analog 2, which was approximately 4-fold more potent than hit 1. Another early structural modification involved preparation of the compound with the alternative orientation of the amide bond of 2 to produce analog 3. Though compound 3 was more than 20-fold less potent than 2, our anticipation was that optimization within this series might restore lost potency and yield interesting analogs. Indeed, such an effort was fruitful and is described in detail herein.
Figure 2.
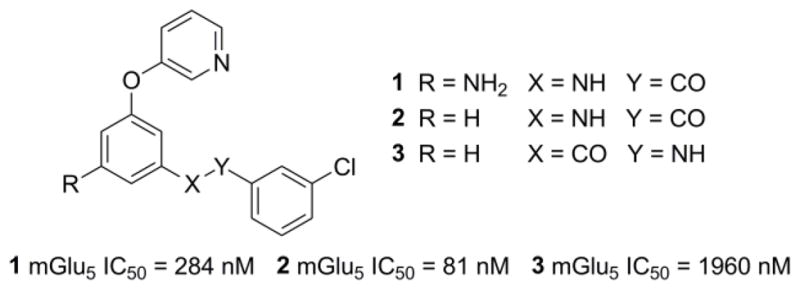
mGlu5 NAM HTS hit and early analogs
Preparation of aryl ether analogs of 3 was straightforward and followed the general methods outlined here (Scheme 1).27 Though certain reactions proceeded in poor to moderate yield, these reactions were not optimized and were sufficient for analog generation. For selected compounds of interest, scalable routes with improved yields were developed.28 A nucleophilic aromatic substitution reaction between pyridine or pyrimidine alcohols 4 and suitable 3-halobenzonitrile compounds 5 afforded ether intermediates 6. Basic hydrolysis of the nitrile functional group provided the carboxylic acid intermediates 7. In certain cases acids 7 were coupled with primary amines under standard conditions to give the desired amide compounds directly. Alternatively, acids 7 were first converted to the corresponding methyl esters 8. Subsequent treatment of 8 with primary amines in the presence of potassium bis(trimethylsilyl)amide yielded the desired amide compounds. This alternative route was especially useful for the incorporation of nitrile groups into the scaffold at a late stage.
Scheme 1.
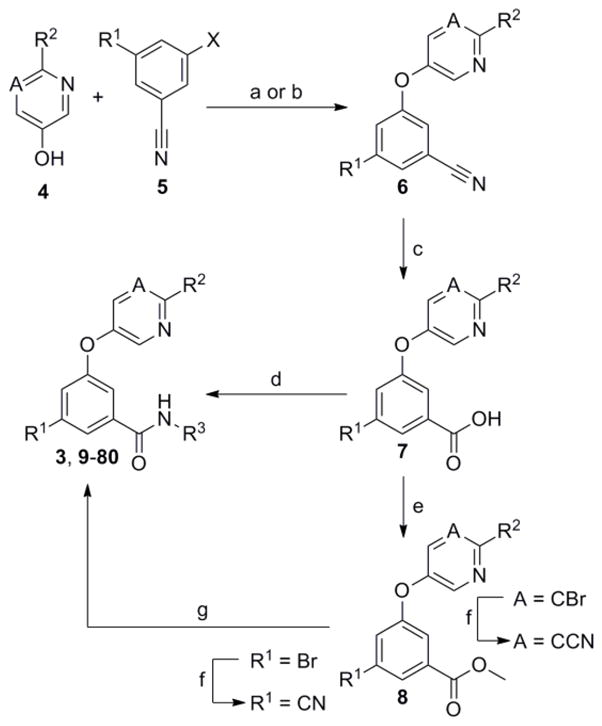
Reagents and conditions: (a) For X = Br; CuO, K2CO3, pyridine, 80 °C (50–73%) or CuI, KOtBu, DMG, DMF, μw, 190 °C (35–50%); (b) For X = F; K2CO3, DMF, μw, 150–180 °C (48–72%); (c) aq. NaOH, EtOH or dioxane, sealed tube, 100 °C (77–99%); (d) R3NH2, DIEA, HATU, DMF, CH2Cl2 (15–58%) or R3NH2, POCl3, pyridine, −15 °C (36–65%); (e) H2SO4, MeOH, reflux (84–92%); (f) Pd(OAc)2, PS-PPh3 or Pd(PPh3)4, Zn(CN)2, DMF, μw, 140 °C (32–81%); (g) R3NH2, KN(SiMe3)2, THF (10–65%).
The first area of the chemotype that was targeted for SAR exploration was the eastern secondary amide group (Table 1). Potency data in the primary assay is presented here as both pIC50 and IC50 values for convenient evaluation of SAR. Not surprisingly, the 3-chlorophenyl group (3) can be replaced with a 3-methylphenyl group (9) with little effect on potency. More interesting was the enhanced potency observed with 2-pyridyl derivative 10. While compound 3 was considered relatively lipophilic (cLogP = 3.86), the more polar analog 10 was considerably less lipophilic (cLogP = 2.70).29 Lipophilicity can be an important parameter to monitor during the course of a CNS drug discovery program.30 For this reason, the flexibility to install a heteroaryl ring at this position of the chemotype was considered attractive, and SAR development continued along those lines. Further modification of this ring in the form of pyrimidine 11 was not well tolerated. Efforts to evaluate substituted analogs of 10 produced 6-methylpyridine 12, which was equipotent to the original hit 1. Other substituted analogs of 10 (14–16) were 2 to 5-fold less potent than 10. 4-Pyridyl analog 13 was only weakly active and more than 25-fold less active than 12, highlighting the importance of the location of the nitrogen atom in the pyridine ring.
Table 1.
Initial Amide SAR
| ||||
|---|---|---|---|---|
| cpd | R | mGlu5 pIC50 (± SEM)a | mGlu5 IC50 (nM) | % Glu Max (± SEM)a,b |
| 1 |
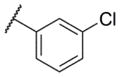
|
6.55 ± 0.19 | 284 | 1.3 ± 0.2 |
| 2 |
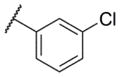
|
7.09 ± 0.07 | 81 | 1.0 ± 0.2 |
| 3 |
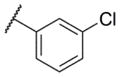
|
5.71 ± 0.14 | 1960 | 1.6 ± 0.4 |
| 9 |
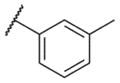
|
5.66 ± 0.11 | 2190 | 1.4 ± 0.1 |
| 10 |
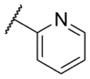
|
6.07 ± 0.13 | 844 | 1.6 ± 0.6 |
| 11 |
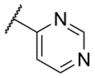
|
< 5.0c | > 10,000 | 49.9 ± 16.4 |
| 12 |
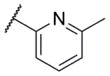
|
6.45 ± 0.16 | 354 | 1.3 ± 0.4 |
| 13 |
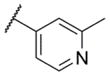
|
< 5.0c | > 10,000 | 27.8 ± 7.3 |
| 14 |
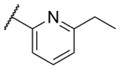
|
5.73 ± 0.13 | 1860 | 1.9 ± 0.2 |
| 15 |
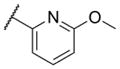
|
5.44 ± 0.11 | 3610 | 3.2 ± 0.9 |
| 16 |
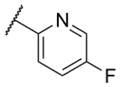
|
5.17 ± 0.05 | 3780 | 6.8 ± 0.9 |
Calcium mobilization mGlu5 assay; values are average of n ≥ 3
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3
Concentration-response curve (CRC) does not plateau
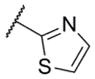
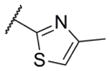
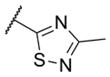
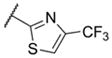
In addition to these six-membered ring heteroaryl analogs, several five-membered ring heteroaryl analogs were also prepared (Table 2). Although unsubstituted thiazole 17 was only weakly active, simple modification of the thiazole ring by installation of a 4-methly group (18) resulted in substantially enhanced potency. As can often be the case in allosteric modulator chemotypes, large changes in potency were observed with quite minor structural modifications. For example, both thiadiazole 19 and 4-trifluoromethylthiazole 20 were inactive up to the highest concentration tested (30 μM) in spite of the fact that each compound was quite closely related to 18 from a structural standpoint. Triazole derivatives 21 and 22 were also both inactive up to the highest concentration tested. One of the early modifications made within this chemotype was evaluation of the pyrimidine ether (23) as an alternative to the pyridine ether in the northern portion of the chemotype (Table 3). This modification not only reduced lipophilicity (cLogP 23 = 1.27; cLogP 18 = 2.32)29 but also enhanced potency. As such, substantial SAR was developed in the context of the pyrimidine ether group.
Table 2.
Initial Amide SAR
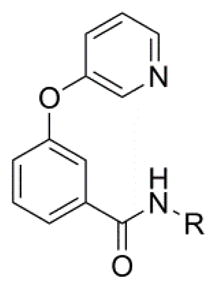
| ||||
|---|---|---|---|---|
| cpd | R | mGlu5 pIC50 (± SEM)a | mGlu5 IC50 (nM) | % Glu Max (± SEM)a,b |
| 17 |
|
< 5.0c | > 10,000 | 14.4 ± 6.7 |
| 18 |
|
6.11 ± 0.12 | 772 | 1.7 ± 0.2 |
| 19 |
|
< 4.5 | > 30,000 | 3 |
| 20 |
|
< 4.5 | > 30,000 | 3 |
| 21 |
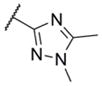
|
< 4.5 | > 30,000 | 3 |
| 22 |
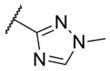
|
< 4.5 | > 30,000 | 3 |
Calcium mobilization mGlu5 assay; values are average of n ≥ 3
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3
CRC does not plateau
Table 3.
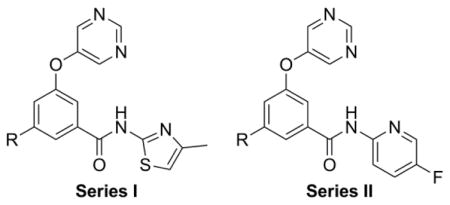
Phenyl Ring SAR
| |||||
|---|---|---|---|---|---|
| cpd | Series | R | mGlu5 pIC50 (± SEM)a | mGlu5 IC50 (nM) | % Glu Max (± SEM)a,b |
| 23 | I | H | 6.69 ± 0.25 | 205 | 1.7 ± 0.3 |
| 24 | I | F | 7.62 ± 0.03 | 24 | 1.2 ± 0.1 |
| 25 | I | Cl | 7.94 ± 0.19 | 11 | 1.4 ± 0.2 |
| 26 | I | CF3 | 6.55 ± 0.10 | 283 | 1.7 ± 0.2 |
| 27 | II | F | 6.52 ± 0.07 | 305 | 1.3 ± 0.3 |
| 28 | II | Cl | 7.49 ± 0.02 | 33 | 1.2 ± 0.2 |
| 29 | II | CH3 | 7.72 ± 0.06 | 19 | 0.8 ± 0.2 |
| 30 | II | CF3 | 5.26 ± 0.01 | 5530 | 3.2 ± 0.3 |
| 31 | II | CN | 6.50 ± 0.08 | 318 | 1.1 ± 0.4 |
Calcium mobilization mGlu5 assay; values are average of n ≥ 3
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3
One area of interest that was investigated was substitution of the phenyl core. A number of substituents were tolerated at the position meta to both the ether oxygen and the benzamide group, and representative examples are pictured here (Table 3). Installation of fluoro (24) and chloro (25) groups on the phenyl core provided analogs with enhanced potency relative to 23, while trifluoromethyl derivative 26 was essentially equipotent to 23. Additional texture in the SAR can be observed by moving to the 5-fluoropyridyl amide, which was only moderately potent in an earlier analog (16). In this case, the chlorophenyl analog 28 was approximately 9-fold more potent than its fluorophenyl counterpart (27). Interestingly, methyl analog 29 was more than 250-fold more potent than trifluoromethyl analog 29. The cyano derivative 31 was similarly potent to fluorophenyl analog 27.
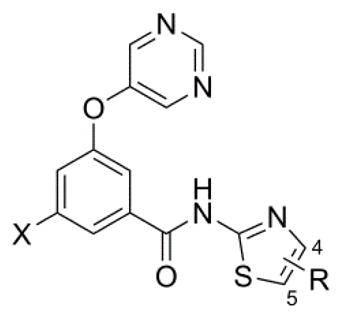
A second generation library of thiazole amides was prepared in the context of both fluorophenyl and chlorophenyl cores (Table 4). The importance of the optimized functional group modifications presented thus far is evidenced by moderately potent unsubstituted thiazole analog 32 (compare to 17). Unfortunately, efforts to further enhance potency by modification or replacement of the 4-methyl group on the thiazole ring proved unsuccessful. For example, the cyclopropyl derivates 33 and 38 reduced potency by 10 and 7-fold, respectively (compare to 24 and 25). Likewise progressive fluorination of the methyl group (35–37 and 40–42) was deleterious for mGlu5 activity, though monofluorinated derivatives 35 and 40 were more potent than their difluoromethyl (36 and 41) and trifluoromethyl (37 and 42) counterparts. A more successful modification was fluorination at the 5-position of thiazole ring, where only a 2 (34 vs. 24) to 3-fold (39 vs. 25) loss of potency was observed.
Table 4.
Second Generation Amide SAR – Thiazoles
| |||||
|---|---|---|---|---|---|
| cpd | X | R | mGlu5 pIC50 (± SEM)a | mGlu5 IC50 (nM) | % Glu Max (± SEM)a,b |
| 32 | F | H | 5.75 ± 0.25 | 1790 | 3.2 ± 1.2 |
| 33 | F | 4-(C3H5) | 6.62 ± 0.08 | 238 | 1.7 ± 0.3 |
| 34 | F | 4-CH3, 5-F | 7.27 ± 0.03 | 54 | 1.5 ± 0.1 |
| 35 | F | 4-CH2F | 6.66 ± 0.03 | 217 | 1.6 ± 0.4 |
| 36 | F | 4-CHF2 | 5.66 ± 0.06 | 2200 | 1.7 ± 0.4 |
| 37 | F | 4-CF3 | 5.49 ± 0.05 | 3230 | 1.5 ± 0.5 |
| 38 | Cl | 4-(C3H5) | 7.09 ± 0.19 | 82 | 1.7 ± 0.2 |
| 39 | Cl | 4-CH3, 5-F | 7.44 ± 0.09 | 37 | 1.4 ± 0.2 |
| 40 | Cl | 4-CH2F | 7.01 ± 0.08 | 99 | 1.3 ± 0.2 |
| 41 | Cl | 4-CHF2 | 6.51 ± 0.07 | 310 | 1.2 ± 0.2 |
| 42 | Cl | 4-CF3 | 6.68 ± 0.04 | 210 | 1.2 ± 0.4 |
Calcium mobilization mGlu5 assay; values are average of n ≥ 3
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3
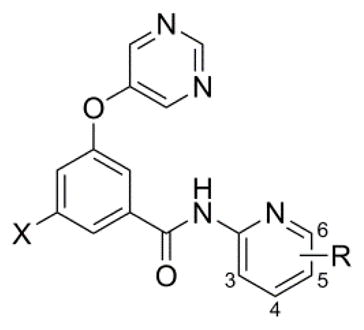
A parallel second generation library of 2-pyridyl amides was also prepared in the context of both fluorophenyl and chlorophenyl cores (Table 5). The unsubstituted 2-pyridyl analogs 43 and 55 were prepared in order to have a baseline for comparison, and both were potent with 55 demonstrating excellent potency. Substitution at the 3-position was not favorable as evidenced by weak antagonism seen with 3-fluoropyridine analogs 44 and 56. SAR at the 4-position was more nuanced. In the context of the fluorophenyl core, the 4-chloropyridine 45 demonstrated enhanced potency relative to 43, while the 4-methylpyridine 46 and 4-trifluoromethylpyridine 47 were less potent than 43. In the case of the chlorophenyl core, each of the substituted analogs (57–61) was less potent than unsubstituted comparator 55. 5-Fluoropyridines 27 and 28 were discussed previously and proved to be the most potent among the 5-substituted analogs (48–49 and 62–63). Fluorophenyl core analogs with substituents at the 6-position (50–54) generally demonstrated good to moderate potency, and 6-chloropyridine 51 exhibited enhanced potency relative to 43. The most potent compounds were obtained through the preparation of 6-substituted pyridine analogs in the context of the chlorophenyl core (64–70). In this case, a number of substituents (64–67) were well tolerated and demonstrated potency on par with 55. Difluoromethylpyridine 68 and methoxypyridine 70 were only 2 and 3-fold less potent than 55, respectively. Finally, a limited number of disubstituted pyridines (71–74) were prepared and tested. Among these analogs, only 72 demonstrated potency within 3-fold of comparator 55. Interestingly, compound 72 was slightly less potent than both 5-fluoropyridine 28 and 6-methylpyridine 66 indicating that there was no additive effect on potency with these substituents.
Table 5.
Second Generation Amide SAR – Pyridines
| |||||
|---|---|---|---|---|---|
| cpd | X | R | mGlu5 pIC50 (± SEM)a | mGlu5 IC50 (nM) | % Glu Max (± SEM)a,b |
| 43 | F | H | 7.02 ± 0.09 | 95 | 1.4 ± 0.2 |
| 44 | F | 3-F | < 5.0c | > 10,000 | 70.5 ± 3.2 |
| 45 | F | 4-Cl | 7.38 ± 0.03 | 42 | 1.5 ± 0.1 |
| 46 | F | 4-CH3 | 6.67 ± 0.01 | 215 | 1.4 ± 0.2 |
| 47 | F | 4-CF3 | 5.98 ± 0.06 | 1050 | 1.5 ± 0.3 |
| 27 | F | 5-F | 6.52 ± 0.07 | 305 | 1.3 ± 0.3 |
| 48 | F | 5-Cl | 5.97 ± 0.14 | 1080 | 2.0 ± 0.4 |
| 49 | F | 5-CN | < 4.5 | > 30,000 | — |
| 50 | F | 6-F | 6.59 ± 0.13 | 258 | 1.6 ± 0.2 |
| 51 | F | 6-Cl | 7.35 ± 0.09 | 45 | 1.5 ± 0.5 |
| 52 | F | 6-OCH3 | 6.18 ± 0.11 | 668 | 1.6 ± 0.3 |
| 53 | F | 6-CHF2 | 6.83 ± 0.08 | 149 | 1.2 ± 0.4 |
| 54 | F | 6-CF3 | 5.64 ± 0.13 | 2320 | 3.7 ± 1.3 |
| 55 | Cl | H | 7.77 ± 0.03 | 17 | 1.2 ± 0.4 |
| 56 | Cl | 3-F | < 5.0c | > 10,000 | 52.6 ± 5.4 |
| 57 | Cl | 4-F | 6.47 ± 0.08 | 341 | 1.4 ± 0.2 |
| 58 | Cl | 4-Cl | 6.77 ± 0.02 | 169 | 1.2 ± 0.2 |
| 59 | Cl | 4-CH3 | 6.85 ± 0.05 | 140 | 1.7 ± 0.2 |
| 60 | Cl | 4-CH2CH3 | 5.35 ± 0.02 | 4450 | 2.0 ± 0.7 |
| 61 | Cl | 4-CF3 | 5.36 ± 0.12 | 4340 | 2.0 ± 0.0 |
| 28 | Cl | 5-F | 7.49 ± 0.02 | 33 | 1.2 ± 0.2 |
| 62 | Cl | 5-Cl | 6.99 ± 0.11 | 103 | 1.3 ± 0.4 |
| 63 | Cl | 5-CF3 | < 4.5 | > 30,000 | — |
| 64 | Cl | 6-F | 8.00d | 10d | 0.8d |
| 65 | Cl | 6-Cl | 7.86 ± 0.06 | 14 | 1.2 ± 0.3 |
| 66 | Cl | 6-CH3 | 7.75 ± 0.13 | 18 | 1.1 ± 0.3 |
| 67 | Cl | 6-CH2F | 7.74 ± 0.04 | 18 | 1.4 ± 0.4 |
| 68 | Cl | 6-CHF2 | 7.44 ± 0.15 | 36 | 1.1 ± 0.4 |
| 69 | Cl | 6-CF3 | 6.51 ± 0.14 | 309 | 1.5 ± 0.2 |
| 70 | Cl | 6-OCH3 | 7.27 ± 0.04 | 53 | 1.3 ± 0.2 |
| 71 | Cl | 4-CH3, 5-F | 5.49 ± 0.04 | 3250 | 10.2 ± 7.7 |
| 72 | Cl | 5-F, 6-CH3 | 7.30 ± 0.08 | 50 | 1.1 ± 0.3 |
| 73 | Cl | 5-F, 6-CH2F | 6.84 ± 0.10 | 145 | 1.2 ± 0.3 |
| 74 | Cl | 5-F, 6-CHF2 | 6.30 ± 0.11 | 502 | 1.6 ± 0.3 |
Calcium mobilization mGlu5 assay; values are average of n ≥ 3
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3
CRC does not plateau
average of n=2
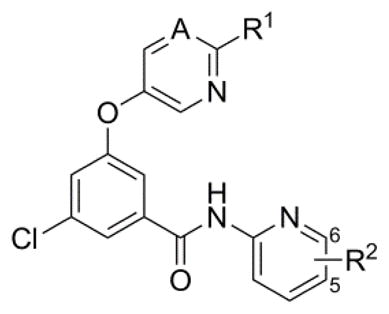
With a number of interesting analogs in hand, attention returned to preparing a limited number of new analogs in the northern heteroaryl ether portion of the chemotype. Representative SAR is presented here in the context of the 5-fluoropyridine and 6-methylpyridine amide groups exemplified in previously discussed compounds 28 and 66 (Table 6). Substitution of the pyrimidine ether at the 2-position (R1 = CH3) resulted in a dramatic loss of potency (>100-fold) in the context of the 5-fluoropyridine amide (75 vs. 28); however, the decrease in potency was much less severe (5-fold) in the context of the 6-methylpyrimidine (76 vs. 66). Installation of substituted pyridine ethers (77–80) proved more advantageous for maintaining or enhancing potency as evidenced by fluoropyridine ethers 77–78 and cyanopyridine ethers 79–80.
Table 6.
Head Group SAR
| ||||||
|---|---|---|---|---|---|---|
| cpd | A | R1 | R2 | mGlu5 pIC50 (± SEM)a | mGlu5 IC50 (nM) | % Glu Max (± SEM)a,b |
| 28 | N | H | 5-F | 7.49 ± 0.02 | 33 | 1.2 ± 0.2 |
| 66 | N | H | 6-CH3 | 7.75 ± 0.13 | 18 | 1.1 ± 0.3 |
| 75 | N | CH3 | 5-F | 5.35 ± 0.04 | 4470 | 2.6 ± 0.1 |
| 76 | N | CH3 | 6-CH3 | 7.02 ± 0.06 | 96 | 1.6 ± 0.2 |
| 77 | CF | H | 5-F | 7.61 ± 0.04 | 25 | 1.1 ± 0.1 |
| 78 | CF | H | 6-CH3 | 8.13 ± 0.08 | 7.5 | 1.8 ± 0.4 |
| 79 | CCN | H | 5-F | 7.57 ± 0.08 | 27 | 1.7 ± 0.2 |
| 80 | CCN | H | 6-CH3 | 7.99 ± 0.15 | 10 | 1.6 ± 0.1 |
Calcium mobilization mGlu5 assay; values are average of n ≥ 3
Amplitude of response in the presence of 30 μM test compound as a percentage of maximal response (100 μM glutamate); average of n ≥ 3
Throughout the course of a typical discovery project, we prefer to triage our compounds for advanced assays and to inform design of subsequent analogs using a number of important in vitro assays. Such assays can help predict future liabilities or inform us as to the drug-likeness of our analogs. Typically, measuring intrinsic clearance in rodent and human liver microsomes is a useful means of assessing metabolism that is mediated by cytochrome P450; however, internal research with compounds in this chemical series has demonstrated a major role for non-P450 mediated metabolism with compounds containing the pyrimidine ether in the northern region of the chemotype. A detailed description of this metabolic pathway has been elucidated and characterized with analog 24 (VU0409106) and has recently been published in the literature.31 Still, other in vitro assays did prove useful, including assessment of cytochrome P450 inhibition32 and non-specific binding to rodent brain homogenates (BHB).33 CYP3A4 is the major P450 present in the human liver and responsible for the metabolism of approximately half of the drugs in clinical use.34 Inhibition of CYP3A4 by representative compounds from this series of mGlu5 NAMs is presented here (Table 7). Also shown for each compound is the unbound fraction in mouse brain homogenates, which provides an indication of free drug available to interact with the receptor. In the context of the 4-methylthiazole amide, the halogen on the phenyl core proved important. The more lipophilic chlorophenyl analog 25 was both more highly protein bound and a more potent inhibitor of CYP3A4 than fluorophenyl analog 24 (VU0409106). Unbound fraction in brain homogenate was increased, and CYP3A4 inhibition was reduced by moving from the 4-methylthiazole 25 to the 5-fluoropyridine amide 28 (VU0415303). The 6-substituted pyridine analogs (64–66) demonstrated some interesting SAR. 6-Fluoropyridine 64 was an extremely potent inhibitor of CYP3A4; however, this liability could be mitigated by installation of larger chloro (65) and methyl (66) groups at the 6-position. Not surprisingly the more lipophilic analog 65 was the more highly protein bound among this group of 6-substituted analogs. The pyridyl ether compounds (77–79) were generally more highly protein bound and more potent inhibitors of CYP3A4 than their corresponding pyrimidine ether counterparts.
Table 7.
CYP3A4 Inhibition and Brain Homogenate Binding
| compound | cLogPa | mGlu5 IC50 (nM) | CYP3A4 IC50 (μM)b | Mouse BHB (Fu)c |
|---|---|---|---|---|
| 24 | 1.43 | 24 | 22.3 | 0.082 |
| 25 | 1.89 | 11 | 2.2 | 0.025 |
| 28 | 2.44 | 33 | 6.2 | 0.080 |
| 64 | 2.53 | 10 | < 0.1 | 0.057 |
| 65 | 2.99 | 14 | 1.9 | 0.019 |
| 66 | 2.59 | 18 | 4.3 | 0.052 |
| 77 | 3.65 | 25 | 1.3 | 0.007 |
| 78 | 3.80 | 7.5 | 0.7 | 0.003 |
| 79 | 3.22 | 27 | 1.7 | 0.016 |
Calculated using ADRIANA. Code (www.molecular-networks.com)
Inhibition of CYP3A4 assayed in pooled HLM+NADPH
BHB = brain homogenate binding; Fu = fraction unbound
Consideration of the potency and in vitro data presented herein identified compounds 24 (VU0409106) and 28 (VU0415303) as promising analogs for evaluation in mouse pharmacokinetic (PK) studies (Table 8). Prior PK studies in rats had demonstrated that VU0409106 was a moderate to high clearance compound;31 thus, intraperitoneal (IP) dosing was chosen as a convenient route for assessing exposure in mice.35 Both compounds demonstrated good brain to plasma ratios near unity; however, 24 (VU0409106) had nearly 2-fold better overall exposure than 28 (VU0415303). Using the brain homogenate binding data for 24 (VU0409106), the unbound drug levels in the brain are calculated to be 335 nM at 15 minutes post dose and 38 nM at one hour post dose. Given that these unbound brain levels are in excess of the functional potency of 24 (VU0409106), the compound was deemed an excellent candidate for evaluation in an acute behavioral study in mice. Prior to undertaking such studies, it was considered prudent to better understand the molecular pharmacology of 24 (VU0409106). The ability of the compound to compete with the equilibrium of [3H]3-methoxy-5-(pyridin-2-ylethynyl)pyridine,36 a close structural analog of MPEP, confirmed the interaction of the compound with the known mGlu5 allosteric binding site (mGlu5 Ki = 6.8 nM). 24 (VU0409106) was also examined in cell based functional assays at 10μM for its selectivity versus the other seven mGlus and was determined to be inactive against each.37 The functional activity of 24 (VU0409106) at human mGlu5 was also evaluated, and little difference was found across species (human mGlu5 IC50 = 49 nM). Finally, 24 (VU0409106) was submitted to a commercially available radioligand binding assay panel of 68 clinically relevant GPCRs, ion channels, kinases, and transporters,38 and no significant responses were found at 10 μM compound.39
Table 8.
Mouse PK Resultsa
| 24 VU0409106b | 28 VU0415303c | |
|---|---|---|
| Plasma Tmax (h) | 0.25 | 0.5 |
| Plasma Cmax (ng/mL) | 1450 | 303 |
| Brain Tmax (h) | 0.25 | 0.25 |
| Brain Cmax (ng/g) | 1350 | 311 |
| AUCplasma (ng·h/mL) | 702 | 391 |
| AUCbrain (ng·h/g) | 696 | 365 |
| B/P ratio | 0.99 | 0.93 |
10 mg/kg IP dose; 10% Tween 80 formulation
CD-1 mice (n=3 per time point)
CD-1 mice (n=2 per time point)
It has been established that mice will bury foreign objects such as glass marbles in deep bedding. Pretreatment of mice with low doses of anxiolytic benzodiazepines have been shown to inhibit this behavior.40 Furthermore, the known mGlu5 NAMs MPEP and fenobam are also effective in this model.7a,d Additionally, novel tool compounds developed in our laboratory have also demonstrated efficacy in this model.24b, 25 As this is also a convenient and rapid assay that is performed with na ve mice, we have found it to be a useful in vivo screening paradigm.41 Given these facts, a dose response marble burying study using IP dosing of 24 (VU0409106) was conducted using a 15 minute pretreatment (Fig. 3). To relate the observed results to compound exposure, brain samples were collected and analyzed immediately following each experiment. Gratifyingly, dose-dependent inhibition of marble burying that correlated with increased brain concentration of drug was observed with 24 (VU0409106). Statistically significant inhibition was noted at all doses greater than or equal to 3 mg/kg.
Figure 3.
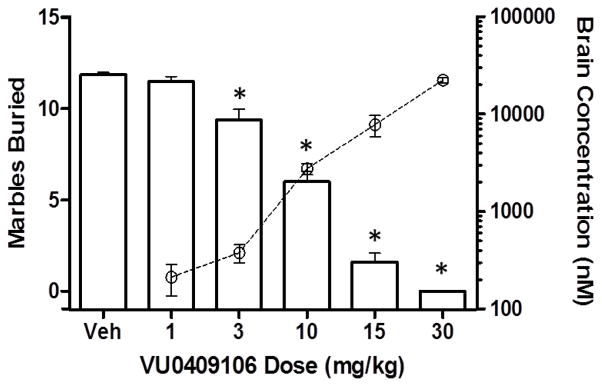
Dose dependent inhibition of marble burying with 24 (VU0409106) correlates with increased total brain exposure. n = 8 per dose; *, P < 0.001 vs. vehicle control group, Dunnett’s test. Bars denote marbles buried. Open circles denote brain exposure.
In conclusion, substantial SAR has been developed in an aryl ether series of mGlu5 NAMs leading to the identification of tool compound 24 (VU04019106). The compound is a potent and selective non-competitive antagonist of mGlu5 that binds at the known allosteric binding site and demonstrates good CNS exposure following intraperitoneal dosing in mice. Compound 24 (VU0409106) also proved efficacious in a mouse marble burying model of anxiety, an assay known to be sensitive to mGlu5 NAMs, and that efficacy appears to correlate with drug exposure in the brain. Many additional in vivo experiments with this new tool compound are ongoing, including behavioral models of other mGlu5 related diseases, and will be the subject of forthcoming communications.
Acknowledgments
We thank NIDA (RO1 DA023947) and Seaside Therapeutics (VUMC33842) for their support of our programs in the development of non-competitive antagonist of mGlu5. We also thank Tammy S. Santomango for technical contributions with the protein binding assays.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Schoepp DD, Jane DE, Monn JA. Neuropharmacology. 1999;38:1431. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]; (b) Conn PJ, Pin JP. Annu Rev Pharmacol Toxicol. 1997;37:205. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 2.(a) Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. J Med Chem. 2012;55:1445. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Conn PJ, Christopolous A, Lindsley CW. Nat Rev Drug Discovery. 2009;8:41. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bridges TM, Lindsley CW. ACS Chem Biol. 2008;3:530. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]; (d) Ritzén A, Mathiesen JM, Thomsen C. Basic Clin Pharmacol Toxicol. 2005;97:202. doi: 10.1111/j.1742-7843.2005.pto_156.x. [DOI] [PubMed] [Google Scholar]; (e) Kew JNC. Pharmacol Ther. 2004;104:233. doi: 10.1016/j.pharmthera.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 3.(a) Emmitte KA. Expert Opin Ther Pat. 2013;23:393. doi: 10.1517/13543776.2013.760544. [DOI] [PubMed] [Google Scholar]; (b) Emmitte KA. ACS Chem Neurosci. 2011;2:411. doi: 10.1021/cn2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rocher JP, Bonnet B, Boléa C, Lütjens R, Le Poul E, Poli S, Epping-Jordan M, Bessis AS, Ludwig B, Mutel V. Curr Top Med Chem. 2011;11:680. doi: 10.2174/1568026611109060680. [DOI] [PubMed] [Google Scholar]; (d) Lindsley CW, Emmitte KA. Curr Opin Drug Discov Dev. 2009;12:446. [PubMed] [Google Scholar]; (e) Gasparini F, Bilbe G, Gomez-Mancilla B, Spooren W. Curr Opin Drug Discov Dev. 2008;11:655. [PubMed] [Google Scholar]
- 4.Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Veliocelebi G, Kuhn R. Neuropharmacology. 1999;38:1493. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 5.Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, Rao S, Washburn M, Varney MA. J Med Chem. 2003;46:204. doi: 10.1021/jm025570j. [DOI] [PubMed] [Google Scholar]
- 6.Zhu CZ, Wilson SG, Mikusa JP, Wismer CT, Gauvin DM, Lynch JJ, Wade CL, Decker MW, Honore P. Eur J Pharmacol. 2004;506:107. doi: 10.1016/j.ejphar.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 7.(a) Nicolas LB, Kolb Y, Prinssen EPM. Eur J Pharmacol. 2006;547:106. doi: 10.1016/j.ejphar.2006.07.015. [DOI] [PubMed] [Google Scholar]; (b) Pietraszek M, Sukhanov I, Maciejak P, Szyndler J, Gravius A, Wislowska A, Plaznik A, Bespalov AY, Danysz W. Eur J Pharmacol. 2005;514:25. doi: 10.1016/j.ejphar.2005.03.028. [DOI] [PubMed] [Google Scholar]; (c) Busse CS, Brodkin J, Tattersall D, Anderson JJ, Warren N, Tehrani L, Bristow LJ, Varney MA, Cosford NDP. Neuropsychopharmacology. 2004;29:1971. doi: 10.1038/sj.npp.1300540. [DOI] [PubMed] [Google Scholar]; (d) Klodzinska A, Tatarczynska E, Chojnacka-Wójcik E, Nowak G, Cosford NDP, Pilc A. Neuropharmacology. 2004;47:342. doi: 10.1016/j.neuropharm.2004.04.013. [DOI] [PubMed] [Google Scholar]; (e) Spooren WPJM, Vassout A, Neijt HC, Kuhn R, Gasparini F, Roux S, Porsolt RD, Gentsch C. J Pharmacol Exp Ther. 2000;295:1267. [PubMed] [Google Scholar]
- 8.(a) Jensen J, Lehmann A, Uvebrant A, Carlsson A, Jerndal G, Nilsson K, Frisby C, Blackshaw LA, Mattsson JP. Eur J Pharmacol. 2005;519:154. doi: 10.1016/j.ejphar.2005.07.007. [DOI] [PubMed] [Google Scholar]; (b) Frisby CL, Mattsson JP, Jensen JM, Lehmann A, Dent J, Blackshaw LA. Gastroenterology. 2005;129:995. doi: 10.1053/j.gastro.2005.06.069. [DOI] [PubMed] [Google Scholar]
- 9.Morin N, Grégoire L, Gomez-Mancilla B, Gasparini F, Di Paolo T. Neuropharmacology. 2010;58:981. doi: 10.1016/j.neuropharm.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 10.(a) de Vrij FMS, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Neurobiol Dis. 2008;31:127. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Neuropharmacology. 2005;49:1053. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Lindemann L, Jaeschke G, Michalon A, Vieira E, Honer M, Spooren W, Porter R, Hartung T, Kolczewski S, Büttelmann B, Flament C, Diener C, Fischer C, Gatti S, Prinssen EP, Parrott N, Hoffman G, Wettstein JG. J Pharmacol Exp Ther. 2011;339:474. doi: 10.1124/jpet.111.185660. [DOI] [PubMed] [Google Scholar]
- 12.Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L. Neuron. 2012;74:49. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Silverman JL, Smith DG, Sukoff Rizzo SJ, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, Crawley JN. Sci Transl Med. 2012;4:131ra51. doi: 10.1126/scitranslmed.3003501. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Silverman JL, Tolu SS, Barkan CL, Crawley JN. Neuropsychopharmacology. 2010;35:976. doi: 10.1038/npp.2009.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes ZA, Neal SJ, Smith DL, Sukoff Rizzo SJ, Pulicicchio CM, Lotarski S, Lu S, Dwyer JM, Brennan J, Olsen M, Bender CN, Kouranova E, Andree TH, Harrison JE, Whiteside GT, Springer D, O’Neil SV, Leonard SK, Schechter LE, Dunlop J, Rosenzweig-Lipson S, Ring RH. Neuropharmacology. 2013;66:202. doi: 10.1016/j.neuropharm.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 15.(a) Martin-Fardon R, Baptista MAS, Dayas CV, Weiss F. J Pharmacol Exp Ther. 2009;329:1084. doi: 10.1124/jpet.109.151357. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumaresan V, Yuan M, Yee J, Famous KR, Anderson SM, Schmidt HD, Pierce RC. Behav Brain Res. 2009;202:238. doi: 10.1016/j.bbr.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Platt DM, Rowlett JK, Spealman RD. Psychopharmacology. 2008;200:167. doi: 10.1007/s00213-008-1191-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bäckstrom P, Hyytiä P. Neuropsychopharmacology. 2006;31:778. doi: 10.1038/sj.npp.1300845. [DOI] [PubMed] [Google Scholar]; (e) Lee B, Platt DM, Rowlett JK, Adewale AS, Spealman RD. J Pharmacol Exp Ther. 2005;312:1232. doi: 10.1124/jpet.104.078733. [DOI] [PubMed] [Google Scholar]; (f) Kenny PJ, Boutrel B, Gasparini F, Koob GF, Markou A. Psychopharmacology. 2005;179:247. doi: 10.1007/s00213-004-2069-2. [DOI] [PubMed] [Google Scholar]; (g) Tessari M, Pilla M, Andreoli M, Hutcheson DM, Heidbreder CA. Eur J Pharmacol. 2004;499:121. doi: 10.1016/j.ejphar.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 16.Tronci V, Vronskaya S, Montgomery N, Mura D, Balfour DJK. Psychopharmacology. 2010;211:33. doi: 10.1007/s00213-010-1868-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gass JT, Osborne MPH, Watson NL, Brown JL, Olive MF. Neuropsychopharmacology. 2009;34:820. doi: 10.1038/npp.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kotlinska J, Bochenski M. Eur J Pharmacol. 2007;558:113. doi: 10.1016/j.ejphar.2006.11.067. [DOI] [PubMed] [Google Scholar]
- 19.(a) Adams CL, Short JL, Lawrence AJ. Br J Pharmacol. 2010;159:534. doi: 10.1111/j.1476-5381.2009.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Besheer J, Grondin JJM, Salling MC, Spanos M, Stevenson RA, Hodge CW. J Neurosci. 2009;29:9582. doi: 10.1523/JNEUROSCI.2366-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gass JT, Olive MF. Psychopharmacology. 2009;204:587. doi: 10.1007/s00213-009-1490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schroeder JP, Spanos M, Stevenson JR, Besheer J, Salling M, Hodge CW. Neuropharmacology. 2008;55:546. doi: 10.1016/j.neuropharm.2008.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lominac KD, Kapasova Z, Hannun RA, Patterson C, Middaugh LD, Szumlinski KK. Drug Alcohol Depend. 2006;85:142. doi: 10.1016/j.drugalcdep.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 20.(a) Rohof WO, Lei A, Hirsch DP, Ny L, Astrand M, Hansen MB, Boeckxstaens GE. Aliment Pharmacol Ther. 2012;35:1231. doi: 10.1111/j.1365-2036.2012.05081.x. [DOI] [PubMed] [Google Scholar]; (b) Keywood C, Wakefield M, Tack J. Gut. 2009;58:1192. doi: 10.1136/gut.2008.162040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Jacquemont S, Curie A, des Portes V, Torrioli MG, Berry-Kravis E, Hagerman RJ, Ramos FJ, Cornish K, He Y, Paulding C, Neri G, Chen F, Hadjikhani N, Martinet D, Meyer J, Beckmann JS, Delange K, Brun A, Bussy G, Gasparini F, Hilse T, Floesser A, Branson J, Bilbe G, Johns D, Gomez-Mancilla B. Sci Transl Med. 2011;3:64ra1. doi: 10.1126/scitranslmed.3001708. [DOI] [PubMed] [Google Scholar]; (b) Berry-Kravis EM, Hessl D, Coffey S, Hervey C, Schneider A, Yuhas J, Hutchinson J, Snape M, Tranfaglia M, Nguyen DV, Hagerman R. J Med Genet. 2009;46:266. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Tison F, Durif F, Corvol JC, Eggert K, Trenkwalder C, Lew M, Isaacson S, Keywood C, Rascol O. Neurology. 2013;80(Meeting Abstracts 1):S23.004. [Google Scholar]; (b) Berg D, Godau J, Trenkwalder C, Eggert K, Csoti I, Storch A, Huber H, Morelli-Canelo M, Stamelou M, Ries V, Wolz M, Schneider C, Di Paolo T, Gasparini F, Hariry S, Vandemeulebroecke M, Abi-Saab W, Cooke K, Johns D, Gomez-Mancilla B. Movement Disorders. 2011;26:1243. doi: 10.1002/mds.23616. [DOI] [PubMed] [Google Scholar]
- 23.(a) Amato RJ, Felts AS, Rodriguez AL, Venable DF, Morrison RD, Byers FW, Daniels JS, Niswender CM, Conn PJ, Lindsley CW, Jones CK, Emmitte KA. ACS Chem Neurosci. 2013;4:1217. doi: 10.1021/cn400070k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Felts AS, Saleh SA, Le U, Rodriguez AL, Weaver CD, Conn PJ, Lindsley CW, Emmitte KA. Bioorg Med Chem Lett. 2009;19:6623. doi: 10.1016/j.bmcl.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou Y, Rodriguez AL, Williams R, Weaver CD, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2009;19:6502. doi: 10.1016/j.bmcl.2009.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rodriguez AL, Williams R, Zhou Y, Lindsley SR, Le U, Grier MD, Weaver CD, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2009;19:3209. doi: 10.1016/j.bmcl.2009.04.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Lamb JP, Engers DW, Niswender CM, Rodriguez AL, Venable DF, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2011;21:2711. doi: 10.1016/j.bmcl.2010.11.119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lindsley CW, Bates BS, Menon UN, Jadhav SB, Kane AS, Jones CK, Rodriguez AL, Conn PJ, Olsen CM, Winder DG, Emmitte KA. ACS Chem Neurosci. 2011;2:471. doi: 10.1021/cn100099n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Felts AS, Lindsley SR, Lamb JP, Rodriguez AL, Menon UN, Jadhav S, Jones CK, Conn PJ, Lindsley CW, Emmitte KA. Bioorg Med Chem Lett. 2010;20:4390. doi: 10.1016/j.bmcl.2010.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller R, Dawson ES, Meiler J, Rodriguez AL, Chauder BA, Bates BS, Felts AS, Lamb JP, Menon UN, Jadhav SB, Kane AS, Jones CK, Gregory KJ, Niswender CM, Conn PJ, Olsen CM, Winder DG, Emmitte KA, Lindsley CW. Chem Med Chem. 2012;7:406. doi: 10.1002/cmdc.201100510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.HEK293A cells expressing rat mGlu5 were cultured and plated. The cells were loaded with a Ca2+ sensitive fluorescent dye and the plates were washed and placed in the Functional Drug Screening System (Hamamatsu). Test compound was applied to cells 3 seconds after baseline readings were taken. Cells were incubated with the test compounds for 140 seconds and then stimulated with an EC20 concentration of glutamate; 60 seconds later an EC80 concentration of agonist was added and readings taken for an additional 40 seconds. Allosteric modulation by the compounds was measured by comparing the amplitude of the responses at the time of glutamate addition plus and minus test compound. For a more detailed description of the assay, see Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. Bioorg Med Chem Lett. 2008;18:4098. doi: 10.1016/j.bmcl.2008.05.091.
- 27.Detailed synthetic procedures for the preparation of analogs and characterization are described in Conn PJ, Lindsley CW, Emmitte KA, Weaver CD, Rodriguez AL, Felts AS, Jones CK. 2011/0152299 US Patent Appl.
- 28.Scalable synthesis of VU0409106: (a) 3,5-Difluorobenzonitrile (1.0 g, 7.2 mmol, 1.0 eq), 5-hydroxypyrimidine (691 mg, 7.19 mmol, 1.0 eq), K2CO3 (1.2 g, 8.7 mmol, 1.2 eq) and DMF (17 mL) were added to a microwave vial and heated at 150 °C for 15 min. The reaction was filtered and concentrated on silica gel. The silica gel with absorbed compound was loaded on top a fresh pad of silica gel and washed with 50% ethyl acetate/hexane. The solvents were removed in vacuo and the crude mixture was purified by flash chromatography on silica gel to afford 750 mg (48%) of the desired compound. 1H NMR (400 MHz, CDCl3) δ 9.08 (s, 1H), 8.75 (s, 2H), 7.71 (ddd, J = 8.4, 2.2, 1.2 Hz, 1H), 7.62–7.61 (m, 1H), 7.56 (dt, J = 10.0, 2.3 Hz, 1H); [M+H]+: 216.1. (b) 3-Fluoro-5-(pyrimidin-5-yloxy)benzonitrile (1.36 g, 6.32 mmol, 1.0 eq) was dissolved in dioxane (32 mL) and 2N NaOH (17 mL) in a sealed tube. The mixture was heated for 18 h at 100 °C. After cooling the reaction was neutralized with 2N HCl (11 mL), and the mixture was concentrated in vacuo. The crude mixture was dissolved in 10% MeOH/CH2Cl2 and the undissolved salt was filtered off, and the solvents were removed in vacuo to afford 1.47 g (99%) of the desired compound. 1H NMR (400 MHz, CDCl3) δ 9.06 (s, 1H), 8.72 (s, 2H), 7.44 (ddd, J = 9.2, 2.4, 1.1 Hz, 1H), 7.34–7.32 (m, 1H), 7.22 (dt, J = 9.5, 2.4 Hz, 1H); [M+H]+: 235.1. (c) 3-Fluoro-5-(pyrimidin-5-yloxy)benzoic acid (5.18 g, 22.1 mmol, 1.0 eq) and 2-amino-4-methylpyridine (2.78 g, 24.3 mmol, 1.1 eq) were dissolved in pyridine (147 mL) and cooled to −15 °C. POCl3 (2.27 mL, 24.3 mmol, 1.1 eq) was added dropwise keeping the temperature below −15 °C. The reaction was stirred an additional 30 minutes at −15 °C and quenched with water and 10% aqueous K2CO3 (37 mL). The reaction was extracted with EtOAc (3x), dried with MgSO4 and concentrated in vacuo. Purification using reverse phase chromatography afforded 4.76 g (65%) of the desired compound as a white solid. 1H NMR (400 MHz, CDCl3) δ 9.09 (s, 1H), 8.77 (s, 2H), 7.74–7.70 (m, 1H), 7.62 (t, J = 1.6 Hz, 1H), 7.43 (dt, J = 9.6, 2.3 Hz, 1H), 6.83 (d, J = 1.0 Hz, 1H), 2.28 (s, 3H). [M+H]+: 331.0.
- 29.Calculated LogP values were determined using ADRIANA. Code. ( http://www.molecular-networks.com)
- 30.Pajouhesh H, Lenz GR. NeuroRx®. 2005;2:541. doi: 10.1602/neurorx.2.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison RD, Blobaum AL, Byers FW, Santomango TS, Bridges TM, Stec D, Brewer KA, Sanchez-Ponce R, Corlew MM, Rush R, Felts AS, Manka J, Bates BS, Venable DF, Rodriguez AL, Jones CK, Niswender CM, Conn PJ, Lindsley CW, Emmitte KA, Daniels JS. Drug Metab Dispos. 2012;40:1834. doi: 10.1124/dmd.112.046136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.CYP3A4 inhibition assay was carried out according to methods described in Zientek M, Miller H, Smith D, Dunklee MB, Heinle L, Thurston A, Lee C, Hyland R, Fahmi O, Burdette D. J Pharmacol Toxicol Methods. 2008;58:206. doi: 10.1016/j.vascn.2008.05.131.Kuresh AY, Lyons R, Payne L, Jones BC, Saunders K. J Pharm Biomed Anal. 2008;48:92. doi: 10.1016/j.jpba.2008.05.011.
- 33.Binding to mouse brain homogenates were measured using equilibrium dialysis according to methods similar to those described in Kalvass JC, Maurer TS. Biopharm Drug Dispos. 2002;23:327. doi: 10.1002/bdd.325.
- 34.Flück CE, Mullis PE, Pandey AM. Biochem Biophys Res Commun. 2010;401:149. doi: 10.1016/j.bbrc.2010.09.035. [DOI] [PubMed] [Google Scholar]
- 35.Compounds were formulated as 10% Tween 80 microsuspension in sterile water and administered intraperitoneally to male CD-1 mice weighing around 30 g at the dose of 10 mg/kg. The mice blood and brain samples were collected at 15, 30, 60, 180 and 360 min after dose administration. Mice were euthanized and decapitated, and both blood and brain samples were collected. Following protein precipitation, the supernatants of all plasma and brain homogenate samples were analyzed by means of LC-MS/MS. PK studies were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee.
- 36.Cosford NDP, Roppe J, Tehrani L, Schweiger EJ, Seiders TJ, Chaudary A, Rao S, Varney MA. Bioorg Med Chem Lett. 2003;13:351. doi: 10.1016/s0960-894x(02)00997-6. [DOI] [PubMed] [Google Scholar]
- 37.mGlu selectivity assays are described in Noetzel MJ, Rook JM, Vinson PN, Cho H, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels JS, Lindsley CW, Weaver CD, Conn PJ. Mol Pharmacol. 2012;81:120. doi: 10.1124/mol.111.075184.
- 38.LeadProfilingScreen®. Eurofins Panlabs, Inc; ( http://www.eurofinspanlabs.com) [Google Scholar]
- 39.Significant responses are defined as those that inhibited more than 50% of radioligand binding. In the case of VU0409106, no inhibition greater than 35% was observed.
- 40.(a) Njung’e K, Handley SL. Brit J Pharmacol. 1991;104:105. doi: 10.1111/j.1476-5381.1991.tb12392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Broekkamp CL, Rijk HW, Joly-Gelouin D, Lloyd KL. Eur J Pharmacol. 1986;126:223. doi: 10.1016/0014-2999(86)90051-8. [DOI] [PubMed] [Google Scholar]
- 41.Marble burying experiments were conducted in accordance with the National Institute of Health regulations of animal care covered in Principles of Laboratory Animal Care and were approved by the Vanderbilt University Medical Center Animal Care and Use Committee. For a detailed experimental procedure for the marble burying assay see reference 24b.