

Abstract
Recently, ultra high-throughput sequencing of RNA (RNA-Seq) has been developed as an approach for analysis of gene expression. By obtaining tens or even hundreds of millions of reads of transcribed sequences, an RNA-Seq experiment can offer a comprehensive survey of the population of genes (transcripts) in any sample of interest. This paper introduces a statistical model for estimating isoform abundance from RNA-Seq data and is flexible enough to accommodate both single end and paired end RNA-Seq data and sampling bias along the length of the transcript. Based on the derivation of minimal sufficient statistics for the model, a computationally feasible implementation of the maximum likelihood estimator of the model is provided. Further, it is shown that using paired end RNA-Seq provides more accurate isoform abundance estimates than single end sequencing at fixed sequencing depth. Simulation studies are also given.
Key words and phrases: Paired end RNA-Seq data analysis, Minimal sufficiency, Isoform abundance estimation, Fisher information
1. INTRODUCTION
1.1 Biological Background
All cells in an individual mammal have almost identical DNA. Yet, cell function within an organism has huge variation. One mechanism that differentiates cell function is its gene expression pattern. Recent research has shown that this differentiation may be on a fine scale: that subtle sequence variants of expressed genes (also referred to as transcripts), called isoforms, have significant impact on the function of the proteins encoded by the RNA and hence their function in the cell (see eg. Wang et al. (2008)). The purpose of this paper is to develop and analyze statistical methodology for measuring differential expression of isoforms using an emerging powerful technology called Ultra High Throughput Sequencing (UHTS). Such study has the potential to help reveal new insights into cellular isoform level gene expression patterns and mechanisms, including characteristics of cell specific specialization.
The central dogma in biology describes the information transfer that allows cells to generate proteins, the building blocks of biological function. This dogma states that DNA is transcribed to messenger RNA (mRNA) which is in turn translated into proteins. Recent discoveries have highlighted the importance of regulation at the level of mRNA, showing that protein levels and function can be regulated by subtle differences in the sequence of mRNA molecules in a cell.
In bacteria, short DNA sequences are transcribed in a one to one fashion to mRNA. This mRNA is referred to as a gene or a transcript. Like DNA, each mRNA is a string of nucleotides, each position taking four possible values. Mammalian cells commonly generate a large class of mRNA molecules from a single relatively short DNA sequence. The set of such mRNA molecules are called isoforms of a gene. This paper concentrates on one common mechanism generating isoforms called alternative splicing. An example of alternative splicing is depicted in Figure 1: two isoforms can arise from the same gene when the DNA, which is comprised of three sequence blocks (called exons), can be transcribed into two different mRNA molecules: one of which contains all three exons and one of which only contains the first and third exon. As this example shows, isoforms typically have highly similar sequence. Despite this sequence similarity, isoforms can encode proteins which may have different functional roles. Further, most genes have more than three exons, and alternative use of exons can give rise to large numbers of isoforms. Thus, it has been historically difficult for technology and statistical methods to allow researchers to distinguish between different isoforms of the same gene.
Fig 1.
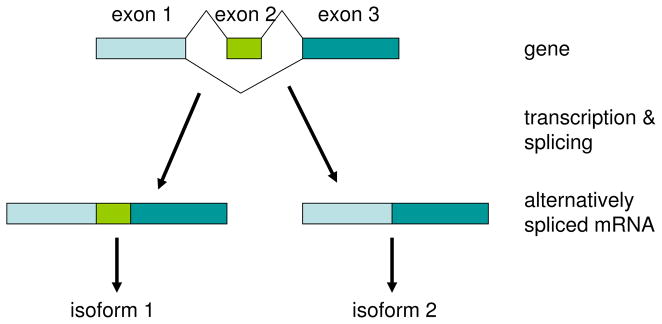
A gene (DNA sequence) with three exons. During transcription, two isoforms are generated. The first isoform contains all of the gene’s three exons. The second isoform contains the first and third exon, skipping the middle exon. This process is called alternative splicing and the middle exon is called an alternatively spliced exon.
1.2 Ultra High Throughput Sequencing
Ultra High Throughput Sequencing (UHTS or simply ‘sequencing’) is an emerging technology which promises to become as (or more) powerful, popular and cost-effective than current microarray technology for several applications, including isoform estimation. When used to study mRNA levels, UHTS is referred to as RNA-Seq. In the past year, studies using UHTS to study genome organization, including isoform expression, have been prominent (see Pan et al. (2008), Zhang et al. (2009), Wahlstedt et al. (2009), Hansen et al. (2009), Maher et al. (2009)) and featured in the journals Science and Nature (see Sultan et al. (2008), Wang et al. (2008)), which dubbed 2007 as the “year of sequencing” (see Chi (2008)).
Briefly, UHTS is a method that relies on directly sequencing the nucleotides in a sample rather than inferring abundance of mRNA by measuring intensities using pre-determined homologous probes as microarrays do. Thus, the data generated from an UHTS experiment are large numbers of discrete strings of nucleotides, called base pairs (bp), which can take values of A,C,G or T. In 2010, each experiment produced tens of millions of up to 100bp reads. The throughput of this technology is expected to continue its rapid growth.
Two experimental protocols for RNA-Seq are in common use: a) single end and b) paired end sequencing experiments. For single end experiments, one end (typically about 50–100 bp) of a long (typically 200–400 nucleotide) molecule is sequenced. For paired end experiments, typically 50–100 bp of both ends of a typically 200–400 nucleotide molecule are sequenced. Using current Illumina technology, each time the sequencing machine is operated, eight samples (e.g. potentially eight different catalogues of gene expression) can be interrogated (essentially) independently and tens of millions of reads are produced in each sample.
1.3 Related Works
An important application and use of UHTS technology is to quantify the abundance of mRNA in a cell (RNA-Seq). This is done by matching the sequences generated in an UHTS experiment to a database of known mRNA sequences (called alignment) and inferring the abundance of each mRNA from the number of experimental reads (fragments of the original mRNA molecules) aligning to it. Sometimes, a statistical model is used for this estimate. Importantly, experimental steps involved in an UHTS experiment can affect the probability of each fragment being observed, although modeling of these processes is not the focus of this paper.
The rapid technological advances in sequencing have spawned a large number of algorithms for analyzing sequence data (see Langmead et al. (2009), Trapnell et al. (2009), Trapnell et al. (2010), Mortazavi et al. (2008)), some of which aim to estimate mRNA abundance. To date, inference on the abundance of mRNA has been made by aligning reads to known genes and estimating a gene’s expression by averaging the number of reads which map uniquely to it using the simplifying assumption that the transcript is sampled uniformly (see Jiang and Wong (2009); Mortazavi et al. (2008)), and sometimes using heuristic approaches to accommodate reads which map to multiple locations (see Mortazavi et al. (2008)). These models do not provide optimal estimators of isoform-specific expression levels and do not accommodate modeling of important steps in the experimental procedure. The work in this paper significantly extends a basic Poisson model developed in Jiang and Wong (2009) to allow for more flexible and efficient inference and establish rigorous statistical theory. In particular, the model in Jiang and Wong (2009) does not work with paired end sequencing data, or read-specific sample rate in a sequencing protocol.
This paper introduces a statistical model for estimating isoform abundance from RNA-Seq data. By grouping the reads into categories and model the read counts within each categories as Poisson variables, the model is flexible enough to accommodate both single end and paired end RNA-Seq data. Based on the derivation of minimal sufficient statistics, a computationally feasible implementation of the maximum likelihood estimator of the model is provided. Using a study of the Fisher information and also numerical simulation, it is shown that using paired end RNA-Seq one can get more accurate isoform abundance estimates. To the best of our knowledge, this is the first such statistically rigorous methodology and analysis to be developed.
2. RNA-SEQ
Isoforms of a gene are subtle differences in a gene sequence, sometimes resulting from inclusion or exclusion of a single exon, a discrete piece of sequence depicted in Figure 1. In principle, compared to microarrays, UHTS has the potential to provide high resolution estimates of isoform use. However, signal deconvolution must take place for these estimates to be accurate.
In order to estimate the expression of different isoforms of the same gene, several measurements of that gene’s expression, whether from a microarray or sequencing, must be deconvolved. Several studies have investigated this deconvolution problem when measurements are made from a microarray (see Hiller et al. (2009) or She et al. (2009)). This paper presents an estimator for deconvolution for ultra high throughput sequencing experiments.
As mentioned, two experimental approaches for RNA-Seq are in wide use. In single end read experiments, reads are generated from one end of a molecule (depicted schematically in Figure 2); in paired end reads, reads are generated from both ends of a molecule, but typically a large number of nucleotides interior to the molecule are left unsequenced (depicted schematically in Figure 3). The length of the whole molecule being sequencing is called the insert size or insert length.
Fig 2.
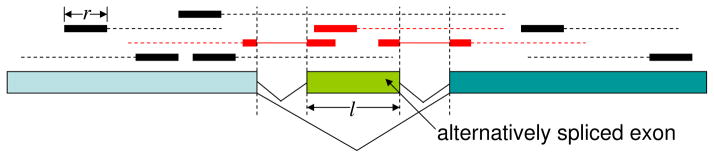
Single end sequencing. A genes of three exons is shown with the middle exon of length l being alternatively spliced. Reads that come from this gene are shown above the gene in solid bars and the parts that are not sequenced are shown in broken lines. Reads that span an exon-exon junction are shown in solid bars connected by thin lines. Reads that are related to the AS exon are shown in red color. In this case only the reads in red are isoform informative.
Fig 3.
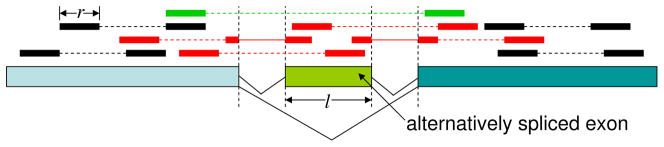
Paired end sequencing. A genes of three exons is shown with the middle exon of length l being alternatively spliced. Paired end reads that come from this gene are shown above the gene in solid bars and the parts that are not sequenced are shown in broken lines. Reads that span an exon-exon junction are shown in solid bars connected by thin line. Reads that are directly related to the AS exon are in red as before. Reads that provide indirect information for separating isoform expressions are in green.
To appreciate the additional information provided by the paired end reads, consider Figure 2 which depicts single end reads randomly sampled from a transcript of a gene. Suppose there are two possible isoforms for the transcript of this gene depending on whether an exon of length l is retained or skipped. In this case, only the reads that come from the alternatively spliced exon (AS exon), or come from junctions involving either the AS exon or the two neighboring exons, can provide information to distinguish the two isoforms from each other, i.e. only these reads are isoform informative. If the AS exon is short compared to the transcript, then the majority of the single end reads contain information only on gene level expression but not isoform level expression. Assuming uniform distribution on the reads’ positions in the gene, it is evident that a read is related to the AS exon with probability if the read comes from the AS exon inclusive isoform, where L is the length of the whole gene (without the intronic regions), r is the length of the reads. Thus, P is a strictly increasing function with respect to the read length r as well as the AS exon length l. As an example, for a gene of length 2000bp with a short AS exon of length 50bp; P = 0.0406 for reads of length 30bp; P = 0.0513 for reads of length 50bp, and P = 0.0789 for reads of length 100bp.
Currently, technical limitations limit the length of sequenced reads. These limitations vary by particular platform used for UHTS. The two platforms in widest use are the Illumina platform and the ABI SOLiD platform. To date, The longest read that can be sequenced on the Illumina platform is roughly 100bp, and the most reliable read length is still roughly 70 bp1.
Paired end reads are an attractive way to decouple the isoform specific gene expression. By performing paired end sequencing, reads are produced from both ends of the fragments, but the interior of the fragment remains unsequenced. This method of sequencing both sides of the fragment increases the number of isoform-informative reads as illustrated in Figure 3. Paired end reads that are mapped to the genes are shown in solid bars above the gene, with read pairs connected by broken lines.
As shown in Figure 3, some read pairs (colored red) are directly informative on the retention or skipping of the AS exon. In addition, some read pairs span both sides of the AS exon (colored green). For these read pairs, the length of the fragment that they span (a.k.a. the insert size or insert length) depends on whether the AS exon is used or skipped in the transcript. If the distribution of the insert size is given, then these read pairs can also provide discriminatory information on the isoforms as shown in Figure 3 and developed rigorously through the insert length model in Section 3.4.2. For illustration, suppose the experimental protocol selects fragments of sizes around 200bp for pair-end sequencing 2. In such an experiment, if the insert size of a read pair is either 200bp or 350bp depending on whether the read pair came from a transcript that included or excluded an exon of length 150bp, then this read pair is unlikely to have come from a transcript that retained the AS exon.
It is easy to see from Figure 3 that the fraction of reads that contain information to distinguish the two isoforms from each other increases not only with the read length and the length of the AS exon, but also with the insert size (when the insert size distribution is a point mass). Since it is possible to have a much longer insert size than read length3, a considerable amount of information can be extracted from the paired end reads for decoupling the isoform-specific gene expression. This concept is developed precisely in the following sections.
3. THE MODEL
3.1 Notation
The notation in Table 1 is used to present the statistical model.
Table 1.
Notation
| Symbol | Meaning |
|---|---|
| I | Total number of unique transcripts (nucleotide sequences) in the sample. |
| J | Total number of unique reads. |
| θi | The abundance of transcript type i, i = 1, …, I. |
| θ | The isoform abundance vector [θ1, θ2, …, θI]. |
| sj | Read type j, j = 1, …, J. |
| ni;j | The number of reads sj that are generated from transcripts i. |
| nj | The number of read sj that are generated from all the transcripts, i.e. . |
| ai;j | Up to proportionality, the sampling rate of ni;j, i.e., the rate that read sj is generated from each individual transcript i. |
| aj | The sampling rate vector [a1,j, a2,j, …, aI,j] for read sj. |
| θ·aj | The sampling rate of nj, i.e., the rate that read sj is generated from all the transcripts. |
| A | The I × J matrix of the sampling rates . |
| ci | The number of copies of the ith transcript in the sample. |
| li | The length of the ith transcript in the sample. |
| n | The total number of reads. |
3.2 Assumptions
The following assumptions on the process of UHTS are used in this paper.
The sample contains I unique transcripts. In this paper we deal with one gene at a time and consider all the isoforms of the genes of interest as the set of I transcripts. The abundances for the transcripts are the parameters of interest and denoted .
After sequencing the sample, there are J distinct reads denoted as . A type of read refers to single end read that is mapped to a specific position (which can be denoted as the 5′ end of the read) in a transcript in single end sequencing, or a pair of reads that are mapped to two specific positions (which can be denoted as the 5′ end of the first read and the 3′ end of the second read) in paired end sequencing. The θi is a rate parameter representing the expression of the ith isoform.
Each transcript is independently processed and then sequenced.
ni,j, the number of reads of type sj that are generated from transcript i, are approximated as Poisson with parameter θiai,j, where ai,j is the relative rate that each individual transcript i generates read sj, called the sampling rate defined below.
Given {θi}1≤i≤I, {ni,j}1≤i≤I, 1≤j≤J are independent random variables.
If transcript i can not generate read sj, ai,j is set to zero: ai,j = 0. More specifically, for 1 ≤ i1, i2 ≤ I, 1 ≤ j1, j2 ≤ J, assuming none of the aikjk for k = 1, 2 are zero, aikjk are defined so that
| (1) |
Therefore, up to a multiplicative constant, ai,j is the sampling rate of the jth read from the ith transcript. This constant is chosen so that the estimates of θi are normalized in order to be comparable across experiments. Two such choices are described in Section 3.4. With appropriate choice of ai,j, the probabilistic interpretation of ai,j can be maintained across different experiments4.
Example 1
Suppose a gene has three exons and two isoforms, as shown in Figure 2 and Figure 3. Suppose the three exons have lengths 200 bp, 100 bp and 200bp. Suppose the read length is 50 bp and single end reads are generated from a transcript uniformly. There are totally 500 different reads. 302 of them are from regions shared by the two isoforms, 149 of them are from isoform 1 only and 49 of them are from isoform 2 only. In this case, I = 2, J = 450 and the matrix A, up to a multiplicative constant, is
where A has 302 columns of , 149 columns of and 49 columns of .
3.3 Likelihood Function
The challenge of estimating isoform abundance arises from the fact that different isoforms of a gene can have common sequence characteristics, and therefore different isoforms may generate common read types. Thus, the ni,j’s can not be directly observed. Rather, the observed quantities are sequences that are necessarily collapsed over the potentially multiple transcripts generating them. The observed quantities in an RNA-Seq experiment are therefore nj where
denoted as nj for simplicity.
Since {ni,j}1≤i≤I, 1≤j≤J are assumed to be independent, and it is assumed that the number of reads of type sj that are generated from transcript i follows a Poisson distribution with parameter θiai,j, nj follows a Poisson distribution with parameter , where θ is the vector of isoform abundance [θ1, θ2, …, θI] and aj is the vector of sampling rates [a1,j, a2,j, …, aI,j] for read sj, in which there is a component for each isoform.
Under the assumption that each read is independently generated, given are independent Poisson random variables, and therefore have the joint probability density function
| (2) |
Note that since , for all i, j, θ, the density (2) is a curved exponential family: the natural parameter of the model is in ℝJ while the underlying parameter is in ℝI with J > I.
3.4 Statistical Models for the Sampling Rate: ai,j
This paper focuses on two choices of ai,j and illustrates the assumptions and interpretation of the resulting parameters. The two choices give rise to two different models: the first is the uniform sampling model, and the second is the insert length model.
While these models differ by whether insert length is taken into consideration, both are motivated by the same model of sample preparation below. To facilitate such modeling, the biochemical steps preparing a sample for sequencing are represented schematically as the composition of the following:
Transcript fragmentation: each full length mRNA is fragmented at positions according to a Poisson process with rate parameter λ.5
Size selection: each fragment is selected with some probability depending on only its length.
Sequence specific amplification or selection: each sequence is amplified or further selected based on sequence characteristics.
The sampling rate matrices A for the uniform sampling model and the insert length model presented below are approximated from the same statistical model for steps (a) and (b) above. Namely, transcript fragmentation (positions where the transcript is cut) is modeled as a Poisson point process. Let p(·) denote the probability mass function of fragment lengths obtained from this process. Note that p(·) is an unobserved quantity because the sample is subject to a size selection step after fragmentation and before sequencing. The size selection step is modeled as follows: a length l fragment of transcript is obtained with probability r(l) independently of the identity of the molecule. r(·) is called the filtering function.
While the model in steps (a) and (b) are realistic across experiments, modeling step (c) is more involved and variable across experiments. Modeling how the specific nucleotide sequences affect the probability of being amplified and selected for sequencing varies significantly by experiment and is beyond the scope of this work. However, it is important to emphasize that the model presented in this section is flexible enough to account for estimation of the effect of step (c). Moreover, the model can be adapted to accommodate different model choices in any of steps (a), (b), or (c). In the two models presented below, it is assumed that sequence selection and amplification is uniform.
Modeling the random processes (a) and (b) above as independent and only dependent on a fragment’s length and assuming that sequence selection and amplification is uniform produces a model for the distribution of fragment lengths in the sample. This distribution is represented by q(·) and can be estimated empirically from a paired end sequencing run: namely, mapping both pairs from each read to a database and inferring the insert length.6 Such an empirical function q̂(·) is depicted in Figure 5 and represents a reasonable approximation to the overall distribution of molecule sizes sequenced in an experiment. Further, note that a consequence of the modeling in steps (a), (b) and (c) above produces the expression q(l) = r(l)p(l).
Fig 5.

A typical empirical mass function of the insert length.
Some mapping programs (such as introduced in Langmead et al. (2009)) have options that take advantage of a user specified expected insert size to help improve mapping performance, which may lead to biases in the mapping. The mapping procedure described in this manuscript performs each paired end alignment by aligning the first and second read separately, which does not bias the insert length model and allows for the calculation of minimal sufficient statistics for the model and to perform statistical inference on isoform abundance without such bias.
3.4.1 Uniform sampling model
The uniform sampling model is appropriate for single read data. It assumes that during the sequencing process, each read (regarded as a point) is sampled independently and uniformly from every possible nucleotide in the biological sample. Uniform sampling is a good approximation to sampling from a Poisson fragmentation process and subsequent filtering step when the filtering function r(·) has support on a set that is small compared to the transcript lengths; under these conditions, the process is approximately stationary.
To investigate if the uniform sampling model satisfactorily approximates the Poisson fragmentation and filtering above for numerical regimes of transcript length and fragmentation rate encountered in sequencing, the following three simulations were performed: reads were generated from 10, 100 or 1000 copies of a transcript of length 2,000bp with λ = 0.005. All the fragments of length 200 ± 20bp were retained and the fragment ends were then compared to the sampled read positions as modeled by the uniform sampling model (see Figure 4). It can be seen that as the sample size increases, the two models are very similar except at the two ends of the transcript. At the two ends the Poisson process has some boundary effects, and the sequencing protocol cannot be explained by a simple model. For most situations, these effects will be small, and hence are ignored in the uniform sampling model.
Fig 4.

Uniform Q-Q plot with sampled read positions. (a), (b) and (c) are generated by simulations with 10, 100 and 1000 copies of transcripts, respectively.
Thus, the uniform sampling model is appropriate for sequencing single short reads where the sequencing process can be regarded as a simple random sampling process, during which each read (regarded as a point) is sampled independently and uniformly from every possible nucleotide in the sample. The assumption of uniformity implies that a constant sampling rate for all ai,j > 0 is used. Specifically, let ai,j = 0 if transcript i can not generate read sj, and otherwise, ai,j = n, where n is the total number of reads. As seen below, n serves as a normalization factor.
To motivate this choice of ai,j, consider the interpretation of induced by A. Under the uniform model, the (unobserved) counts from the jth nucleotide which is generated from the ith transcript are modeled as a Poisson random variable with paramete ai,jθi, i.e.:
Computing E (ni,j) using the uniform sampling model with n total reads,
where li is the length of the ith transcript and ci is the number of copies of the ith transcript in the sample. Thus, setting ai,j = n iff transcript i can generate read j produces the identity
so the uniform sampling model has parameter
This choice of A has the property that it normalizes so that
that is, it normalizes θi as a fraction of the total nucleotides sequenced, as shown in Jiang and Wong (2009), making it conceptually compatible with the RPKM (Reads Per Kilobase of exon model per Million mapped reads) normalization scheme in Mortazavi et al. (2008), which is widely used by the RNA-Seq community. This normalization convention assumes the number of nucleotides in the sequenced RNA of each cell do not vary between samples. Modifying these assumptions to be more realistic yield better choices for normalizing constants (see eg. Bullard et al. (2010)) and can easily be incorporated into the normalization of the sampling rate vector.
3.4.2 Insert length model
This model is applicable to paired end sequencing data. In paired end sequencing, the insert length is usually controlled to have a small range. Therefore, as suggested in Figure 3, besides read positions, information can also be extracted from insert lengths inferred from reads. By modeling insert lengths properly, this piece of information can be utilized and statistical inference can be improved. Example 2 below illustrates this concept and Section 6 quantifies the gain in statistical efficiency using the pairing information.
The insert length model models the sampling of transcripts, conditional on insert length, as uniform. The insert length model sets each ai,j using the empirical distribution of the insert lengths of the sample (see Figure 5) such that conditional on the insert length, reads are sampled from transcripts uniformly. This is specified mathematically as
| (3) |
where li,j is the length of corresponding fragment of sj on the ith transcript, n is the total number of read counts and q(l) is the probability of a fragment of length l in the sample after the filtering. In application, for the insert length model, q(·) is taken as q̂(·), the empirical probability mass function computed from all the mapped read pairs. A typical mass function is illustrated in Figure 5. Although usually this function is uni-modal (as in this case) which favors our isoform estimation approach, our approach is flexible enough to allow other type of functions, such as bi-modal functions and etc.
To see the relationship between this choice of sampling rate matrix and a model where reads are subject to Poisson fragmentation and length dependent filtering, suppose that paired end read sj is mapped to transcript 1 at coordinates (x1, y1) and transcript 2 at coordinates (x2, y2) and both reads are in the forward direction. Then, assuming none of x1, x2, y1, y2 is at the boundary of a transcript, under the Poisson fragmentation model (a) and length dependent size selection (b),
Thus, the ratio
is approximately the same as defined by the sampling rate matrix A for the insert length model, with the assumption that none of x1, x2, y1 or y2 is on the boundary of the transcript. As long as the insert length distribution has support which is small compared to transcript length, relatively few transcripts map exactly to the boundary, and little data is lost by ignoring them; doing so allows the above conditions to be satisfied. Further, the argument above shows that the insert length model is consistent with assumptions (a), (b) and (c) of the sample preparation.
The insert length model yields a similar interpretation for the normalization of as in the uniform sampling model, illustrated in the following computation: The paired end read model specifies that the reads of type j from transcript i are Poisson with parameter
The insert length model assumes that reads are filtered based on length independent of their sequence. This produces a method of estimating the expectation of ni,j. The following approximates E(ni,j) under the insert length model:
where A,B and C are defined as follows. Let Y be a random variable representing a read in the sample after fragmentation. Let A be the event that Y is a fragment of transcript i, B the event that Y is read j of transcript i and C the event that Y is a fragment of length li,j and is observed after filtering. Using the product rule,
Each term is analyzed separately. Assuming uniform fragmentation across the transcript and length dependent filtering,
The basic assumption of the insert length model is that the probability of observing a transcript of length li,j does not depend on the transcript and is equal to the empirical insert length, q(li,j), hence
To estimate Pr(A), consider the random variables Xi, the number of fragments in the sample from transcript i and X, the total number of transcript fragments in the sample. Then, assuming transcript i is sufficiently and not overly abundant in the sample,
Assuming a Poisson fragmentation model, up to a boundary effect which has small impact on the approximation,
Combining these approximations yields
Thus, if is close to 1, θi is identified in this model as
Thus, in both models, the choice of ai,j is consistent with its definition in Equation (1). To illustrate the difference between the insert length and uniform sampling models, consider the following example:
Example 2
Consider a case of two isoforms labeled 1 and 2 with an alternative included exon as in Figure 1. Suppose the middle exon 2 has length 50 for concreteness. Suppose pair end read sj has an imputed length of 50 when mapped to 2 and of 100 when mapped to 1 as will be the case if one of the ends is in exon 1 and one in exon 3. Suppose the empirical insert length function is modelled as uniform [60, 140). Then, in the uniform model, because n total reads have been sequenced and mapped,
whereas in the insert length model,
Note that although the denominator 80 in a1j in the insert length model seems arbitrary, because there are 80 different paired end reads that start at the same position as sj, having all of them in the model gives consistent gene expression estimates as in the uniform model.
3.5 Maximum Likelihood Estimation
In this paper, θ is estimated using the MLE. Standard theory shows that the MLE of model (2) will be asymptotically unbiased and consistent provided the parameters in the model is in the interior of the parameter space (see Theorem 6.3.10 of Lehmann (1998)). Computationally efficient procedures are needed to solve for these estimates in practice.
The fact that the distribution of minimum sufficient statistics are Poisson allows for a simplification of the calculation of the MLE by regarding the distribution of the minimum sufficient statistics as a generalized linear model (GLM) problem with Poisson density and identity link function (see McCullagh and Nelder (1989)) with extra linear constraints that require all the parameters to be non-negative. The optimization problem in matrix form is
| (4) |
where n is a J × 1 column vector for the observed read counts [n1, n2, …, nJ], A is a I × J matrix for the sampling rates and θ is the I × 1 isoform abundance vector [θ1, θ2, …, θI]. log(·) takes logarithm over each element of a vector and sum(·) takes summation over all the elements of a vector.
As shown in Jiang and Wong (2009), the log-likelihood function
is always concave and therefore any linear constraint convex optimization method can be used to solve this non-negative GLM problem7.
4. SUFFICIENCY AND MINIMAL SUFFICIENCY
Because J is usually very large, it is extremely inefficient to work with the statistics in (2) directly: in single end sequencing of a human or mouse cell, J can exceed 2, 000 for a typical gene, and in paired end sequencing with variable insert length it can easily reach 100, 000. For computational purposes, it is therefore necessary to use sufficient statistics for the likelihood function (2). Because these statistics have an intuitive interpretation, they are referred to as a collapsing. This section analyzes sufficiency and minimal sufficiency in model (2) and its relation to collapsing.
4.1 Sufficient Statistics and Collapsing
As will be shown below, sufficient statistics have a natural interpretation as collapsing read counts. Proposition 2 shows that to group reads j and k into the same category, it is sufficient that reads have the same normalized sampling rate vector (i.e.,
where || · || is the vector 2-norm).
Such grouping of reads will be called (maximal) collapsings: reads with the same normalized sampling rate vector are grouped together. Intuitively, a maximal collapsing reduces the number of such groups to be as small as possible.
Definition 1
Let Ck be a collection of mk reads so
A set
is called a collapsing, if for any Ck ∈
 and any sj1, sj2 ∈ Ck,
and any sj1, sj2 ∈ Ck,
for some positive number c.
Furthermore, if for any k1 ≠ k2 and any sj1 ∈ Ck1, sj2 ∈ Ck2,
for any positive number c, then is called a maximal collapsing. In a collapsing, each Ck is called a category.
As will be seen in Theorem 3, the maximal collapsing gives rise to a set of minimal sufficient statistics making it useful from a computational perspective. A real data example of such a collapsing is provided in Section 5. The collapsed read counts also has a standard statistical interpretation as the sum of independent Poisson random variables. Suppose categories
are non-overlapping, i.e., Ck1 ∩ Ck2 = ∅ when k1 ≠ k2. Then, assuming each nj follows a Poisson distribution with parameter θ · aj, nCk, the number of observed reads that belong to category Ck (i.e., nCk = Σsj∈Cknj) follows a Poisson distribution with parameter
where and for 1 ≤ j ≤ mk, is the sampling rate vector of the jth read in category k.
Proposition 1
The maximal collapsing is unique.
Proof
The relation satisfied by two types of reads in a category in the maximal collapsing is an equivalence relation. This makes the maximal collapsing a grouping of reads into equivalence classes which is always uniquely determined. To show a relation is a equivalence relation, it suffices to show that the reflexivity, symmetry and transitivity hold.
Reflexivity
For any sj,
i.e., sj ~ sj.
Symmetry
For any sj and sk,
i.e., sj ~ sk ⇒ sk ~ sj.
Transitivity
For any sj, sk and sl,
and
i.e., sj ~ sk and sk ~ sl ⇒ sj ~ sl.
To illustrate how maximal collapsing can be derived from the choice of ai,j in the uniform model to produce the maximal collapsing, reads with the same normalized sampling rate vector are grouped into one category. Because ai,j is either 0 or n, two reads sj1 and sj2 will have the same normalized sampling rate vector, i.e., aj1/||aj1|| = aj2/||aj2||, if and only if they can be generated by the same set of transcripts.
Example 3
Consider a continuation of the setup in Example 2. Suppose a uniform sampling model and suppose reads s1 and s2 can be generated by both transcripts 1 and 2, whereas read s3 can only be generated by transcript 1. Then
and
Grouping s1 and s2 together produces the maximal collapsing
= {{s1, s2}, {s3}}; the first category containing reads that can be produced by both transcripts and the second category containing reads only generated by transcript 1.
4.1.1 Collapsing and Sufficiency
Analysis of the likelihood function (2) shows that collapsing the reads produces sufficient statistics and maximal collapsings are equivalent to minimal sufficient statistics.
Recall that a statistic T(X) is sufficient for the parameter θ in a model with likelihood function fθ(x) if
It is clear that the observed count vector n = [n1, n2, …, nJ ] is sufficient for θ. The collapsed read count vector is also sufficient for θ, as detailed in the next proposition:
Proposition 2
For any collapsing C = [C1, C2, …, CK], the observed read count vector nC = [nC1, nC2, …, nCK] is a sufficient statistic for θ.
Proof
From the definition of collapsing, consider the kth category Ck with the re-enumerated reads , the reads in category k are enumerated
Define to be the sampling rate vector for , 1 ≤ j ≤ mk. By definition, for all 1 ≤ j ≤ mk, for some scalar ,
Therefore,
Rearranging the product in the RHS of equation (2) as a product over each read by the category into which it falls, and denoting the ith read ni and parameter θ · ai as with parameter when it falls as the jth enumerated read in the kth category,
| (5) |
where, since ,
and
establishing the sufficiency of nC = [nC1, nC2, …, nCK].
In addition to the sufficiency proved in Proposition 2, nC is minimal sufficient if the corresponding collapsing C is a maximal collapsing. This is detailed in the next section.
4.2 Minimal Sufficiency
To prove that the read counts derived from a maximal collapsing are minimal sufficient statistics, recall
Definition 2 (Definition 6.2.13 of Casella and Berger (2002)). for the family of densities fθ(·), the statistic T (X) is minimal sufficient if and only if
Theorem 3
In the likelihood specified by Equation (2), counts on maximally collapsed categories are minimal sufficient statistics.
Proof of Theorem 3
Let T(X) be the collapsed vector of counts xC1, xC2, …, xCK and let T(Y) be the vector of counts yC1, yC2, …, yCK each of which are maximal collapsings. If T(X) = T(Y), Equation (5) shows that the ratio of densities
does not depend on θ. To show the reverse implication, suppose T(X) ≠ T(Y). To show that
it suffices to show that
It is possible to simplify this ratio as
| (6) |
where {ng}g∈G and {nh}h∈H are positive numbers and G and H are subsets of the categories and are disjoint since if G and H share a common j, the ratio in Equation (6) can be reduced. Further, since the collapsings are maximal, for any ai, aj appearing in any product in the numerator or denominator, there is no c so that ai = caj. Using these properties, it will be shown that the ratio of densities must depend on θ by contradiction.
Suppose for some (now fixed) T(X) ≠ T(Y), Equation (6) does not depend on θ and is equal to a constant c. Note that since θ can be the vector of all 1s, if Equation (6) does not depend on θ, c > 0 as when θ is the vector of all 1’s both the numerator and denominator of Equation (6) are positive. Then Equation (6) is equivalent to a polynomial equation
| (7) |
∀θ ∈ (ℝ+)I. By basic algebraic geometry, any polynomial in θ which is identically zero in the space (ℝ+)I is identically zero in all of ℝI. Therefore, the last step is to show that the right hand side of Equation (7) is not actually zero for some θ ∈ ℝI. To proceed, fix h ∈ H. The claim is that there exists v ∈ ℝI with 〈v, ah〉 = 0 but ∀g ∈ G,
This v will be be the choice of θ producing the contradiction. For a vector z ∈ ℝI, let z⊥ denote the I − 1 dimensional subspace of vectors orthogonal to it. Then, to finish the proof, it suffices to showing that there is some vector in which is not in . It is equivalent to show there is a strict containment
Strict containment follows since for any h ∈ H,
is a subspace of dimension at most I − 2, thus a countable union of such spaces cannot equal a subspace of dimension I − 1.
Using Theorem 3, the optimization problem (Equation (4)) is reduced to
| (8) |
where n is a K × 1 column vector for the collapsed read counts for categories C1, C2, …, CK, A is a I × K matrix for the collapsed sampling rates and θ is the isoform abundance vector.
The next section illustrates the relationship of minimal sufficient statistics to raw data observed in sequencing experiments.
5. APPLICATION
This section illustrates how minimal sufficient statistics are calculated in an example with real RNA-Seq data from an experiment on cultured mouse B cells. After the sequencing reads were generated, they were mapped to a database of known mouse mRNA transcripts using the RefSeq annotation database (see Pruitt et al. (2005)) and the mouse reference genome (mm9, NCBI Build 37). The reads were mapped using SeqMap, a short sequence mapping tool developed in Jiang and Wong (2008). The two ends of the paired end reads were mapped separately and then a filtering step was applied during which only the pair of reads which were mapped to the same transcript and on the right direction were retained. Further, in the analysis of this section, reads that map to multiple genes were also discarded for computational ease. Because we are mapping the reads to transcript sequences rather than the whole genome, the positions that can not be uniquely mapped are less than 1% which is not likely to change our results significantly. Of course, the model presented in Section 3 can accommodate reads which map to multiple genes because of the statistical equivalence of this problem to that of deconvolving the expression levels of multiple isoforms. We have chosen not to implement this approach because only a small number of genes are impacted and because as rapid growth of the technology continues to produce longer reads, the problem will become negligible. A total of 2,789,546 read pairs (32bp for each end) passed the filtering. The empirical distribution of the insert length was inferred. This distribution has a mean of 251bp and a single mode of 234bp (See Figure 5).
Because more than 99% of the data has an inferred length between 73 bp and 324 bp, reads outside of this range are not considered in subsequent analysis for this example as it is likely these reads come from unannotated isoforms. This resulted in 27,118 (about 1%) read pairs being excluded and the rest 2,762,428 (denoted as n below) read pairs were used in the computation.
The mouse gene Rnpep is used to demonstrate the computation of minimal sufficient statistics. Rnpep has an alternatively spliced exon which gives rise to two different isoforms (see Figure 6). The gene itself is an amino peptidase, meaning that it is used to degrade proteins in the cell. After mapping, 116 read pairs were assigned to this gene, out of which 113 read pairs were used in the computation after outlier removal. Figure 6 presents the positions where the reads are mapped. The gene was picked because it has two alternatively spliced isoforms with a structure that makes distinguishing reads from each isoform challenging, and because the number of reads was small enough to visualize all of them in a simple figure.
Fig 6.

Visualization of RNA-Seq read pairs mapped to the mouse gene Rnpep in CisGenome Browser (see Jiang et al. (2010)). From top to bottom: genomic coordinates, gene structure where exons are magnified for better visualization, read pairs mapped to the gene. Reads are 32bp at each ends. A read that spans a junction between two exons is represented by a wider box.
5.1 Uniform Sampling Model
Any paired end read experiment can be treated as a single end read experiment by taking each paired end read and treating it as two distinct single end reads, one from each side of the pair. In this, the 113 paired end reads become 226 single end reads (without pairing information).
In the uniform sampling model, for either isoform, the sampling rate vector for each read sj can take at most two values: 2n when the isoform can generate read j and 0 when it cannot. Because there are only two isoforms, one of which (isoform 2) excludes one of the exons of the other (isoform 1), it is evident that in the uniform sampling model, there are only three categories for the two isoforms.
The total length of isoform 1 is 2,300. The total length of isoform 2 is 2,183. Hence, computing ai,j by summing over the sampling rate vectors of the reads in the same category, the three categories can be represented by their sampling vectors: [4242n, 4242n], [296n, 0], [0, 62n]. Using minimal sufficient statistics reduces the the data from a vector representing counts on the 2, 300 possible reads sj from the two isoforms to the 3 minimal sufficient statistics which are counts on these categories.
The three categories representing minimal sufficient statistics are tabulated in Table 2. Each category refers to a group of reads that are generated by a particular set of isoforms. For example, category 1 consists of reads generated by both isoforms and category 3 consists of reads generated by isoform 2 only. Using these statistics to solve the optimization problem (4), the MLE for the two isoforms is [θ̂1, θ̂2] = [15.47, 2.70]8. Bayesian credible intervals for these estimates can be obtained by sampling from the posterior space of the parameters (as outlined in Jiang and Wong (2009)), the marginal 95% credible intervals for θ1 and θ2, are (7.89, 18.81) and (0.25, 10.83) respectively.
Table 2.
Single end read categories for Rnpep
| Category ID | Sampling rate vector | Read count |
|---|---|---|
| 1 | [4242n, 4242n] | 216 |
| 2 | [296n, 0] | 10 |
| 3 | [0, 62n] | 0 |
5.2 Insert Length Model
To visualize how the insert length model can be used to produce potentially stronger statistical inference as compared to the uniform sampling model, consider Figure 6. Each paired end read is depicted by two boxes with arrows joining pairs of reads. The direction of the arrows represent which side of the read was sequenced first. For those interested, the direction of the arrows in the Rnpep gene itself indicates the transcriptional direction of the gene in genomic coordinates, although this concept can be ignored for the purposes here. Note that there is no direct evidence that isoform 2 is present in the sample, as no read crosses the junction between the two exons which are adjacent in isoform 2 but not in isoform 1. There is direct evidence of the presence of isoform 1, for example, as depicted in the fifth read from the left in the first row which directly crosses a junction between two exons only adjacent in isoform 1.
Because of the small gap between exons in the figure, reads spanning exons will be slightly longer than reads not spanning exons. Also, some inserts are very short, and absence of the arrow connecting two reads indicates that the entire insert has been fully sequenced. Note that several of the reads spanning the alternatively spliced exon are exceedingly long. This suggests that such reads are actually generated from isoform 2 rather than isoform 1. If such reads are generated from isoform 2, they would likely have a smaller insert length than the inferred insert length when generated by isoform 1, which are the lengths depicted in the figure. Because the empirical insert length distribution has its only mode near 250 bp, conditional on observing the 6th and 7th reads from the top of the figure spanning the alternatively spliced exon, the read is more likely to come from isoform 2. Thus, there is indirect evidence of the presence of isoform 2 in the sample.
Such indirect evidence is utilized by the insert length model; the model produces quantitative estimates of the relative abundance of the two isoforms. As will be seen in the next section, the abundance estimates from the insert length model have larger Fisher information than the estimates from the uniform sampling model.
In the insert length model, each of the possible insert lengths where q(·) has support produce a unique read sj yielding a total of 569, 205 possible reads from the two isoforms. The maximal collapsing produces a total of 138 categories some of which are represented in Table 3. For intuition, all of the reads with a fixed insert length where both ends fall in the left most 7 or right most 3 exons of Rnpep will be in the same category as they have the same probability of being sampled.
Table 3.
Paired read categories for Rnpep
| Category ID | Sampling rate vector | Read count |
|---|---|---|
| 1 | [1681.82n, 1681.82n] | 95 |
| 2 | [294.60n, 0] | 10 |
| 3 | [0, 245.80n] | 2 |
| ⋮ | ⋮ | ⋮ |
| 138 | [0.0057n, 0.0018n] | 0 |
Using the minimal sufficient statistics, the MLE is computed to be [θ̂1, θ̂ 2] = [16.73, 3.43]. The marginal 95% credible intervals for θ1 and θ2 are (11.22, 21.02) and (1.03, 9.29) respectively. The computed marginal 95% credible intervals for θ1 and θ2 are non-overlapping, whereas in the single end read model, one cannot conclude that the expression of isoform 1 and 2 differ. Further, the insert length model has slightly smaller marginal credible intervals for each parameter.
This example suggests that although the uniform sampling model for single end reads has twice the sample size compared with the insert length model for paired end reads, the insert length model actually provides estimates with smaller standard errors than those generated by the uniform sampling model, because the insert length model can utilize the extra information from the insert sizes of the reads. This difference can be quantified by analyzing the Fisher information of each model, the subject of Section 6.
5.3 Practical Implementation Issues
In general, to apply Theorem 3 one needs to enumerate all the read types before collapsing, as shown in the example of mouse gene Rnpep. This might be a time consuming step, especially when the number of read types is large. In practice, however, under some suitable sampling rate models (which include both our uniform model and insert length model), it is sufficient to enumerate only the read types that have at least one read being mapped. This can reduce the computation when the number of mapped read for the gene is small, or in other words, when the gene is lowly expressed.
To see how this works, rearrange the RHS of equation (2) as follows,
| (9) |
where only the term depends on the sampling rates of read types with read counts nj = 0. Therefore, if we can compute this term without knowing each particular sampling rate ai,j, the enumeration of all the read types is no longer necessary. Fortunately, it is possible under some suitable sampling rate models, including both our uniform model and insert length model. For example, in the uniform model, , where n is total number of mapped reads, li is the length of transcript i and r is the read length. Similarly, in the insert length model, .
Using this trick, we can take only the read types with at least one read being mapped and collapse them to categories C1, C2, …, CK. Accordingly, the optimization problem (Equation (4)) is reduced to
| (10) |
where n is a K × 1 column vector for the collapsed read counts for categories C1, C2, …, CK, A is a I × K matrix for the collapsed sampling rates and θ is the isoform abundance vector. W is a I×1 vector with the i-th element computed based on the corresponding sampling rate model.
In a more complex sampling rate model, e.g., when ai,j depends on the particular nucleotide sequence of read sj, the optimization problem (Equation (10) can still be solved. However, all the read types (including the read types with nj = 0) will have to be enumerated and each sampling rate ai,j will have to be computed.
6. INFORMATION THEORETIC ANALYSIS
Many considerations impact the choice of sequencing protocol in an experimental design. One such choice is relative cost of sequencing. In this case, the experimentalist may be interested in choosing the sequencing protocol (paired end or single end) that provides the best estimate of isoform abundance at least relative cost. This section outlines the statistical argument for why, in typical situations, paired end sequencing can produce better estimates of transcript abundance compared to single end sequencing at a fixed number of sequenced nucleotides (cost). The theoretical analysis aims to show that for the same number of sequenced nucleotides, the Fisher information in the insert length model is more than double the Fisher information in the single end read model. Since estimates in RNA-Seq are maximum likelihood estimators, their variance of the estimator converges to the reciprocal of the Fisher information. Thus, larger Fisher information produces estimators with improved accuracy.
6.1 Theoretical Analysis
Consider the following quite simple example showing the increase in information as the fraction of reads unique to each isoform grows:
Example 4
Continuing Example 1, suppose that isoform 1 and isoform 2 have Poisson rate parameters θ1 and θ2 respectively where θ2 = 1 − θ1 and probability 0 < α, β < 1 respectively of producing a read read unique to the isoform. Let n1 be the reads unique to 1, n2 the reads unique to 2 and n3 the reads which cannot be distinguished between the isoforms. Assume there are n total reads in the sample, and assume there is uniform fragmentation which gives rise to three categories.
Fix α < β as known and compute the information in this distribution with θ1 as the unknown parameter as a function of α using the definition that the information is equal to the variance of the derivative of the log likelihood with respect to θ1:
where x̄ = 1 − x. Thus, ᾱ − β̄ = β − α, and δ := β − α and α are re-parameterizations of β, α. The last equation shows that the partial derivatives of the information with respect to α and with respect to δ are positive.
Note that in the example above, no generality is lost by assuming β > α since θ1 and θ2 can be interchanged with no effect on the model.
To see that for a fixed cost of sequencing (number of sequenced nucleotides), the statistical model produced by paired end sequencing has more information than single end sequencing, it is necessary to show that the information obtained by twice as many single end reads in a single end sequencing experiment is smaller than that obtained by a paired end sequencing experiment. Such a comparison necessarily depends on each gene, its isoforms and their relative abundance. The computation of the Fisher information for a typical such example is presented below, and the computation shows that the examples easily generalizes to other configurations of isoforms.
Example 5
Continuing the running example, consider reads of length r = 100 bp and paired end insert size x = 200 bp in the schematic of three exons in Figure 1 where the length of exons 1 and 3 is 500 bp and exon 2 is e = 50 bp.
For a single end read experiment, αs is the probability that a read includes any part of the included exon (i.e. uniquely identifies isoform 2) so for the read length of r,
and βs is the probability that a read includes any part of the spliced junction (i.e. uniquely identifies isoform 1) so
For a paired end read experiment, with x the insert length, αp, the probability that a read uniquely identifies the second isoform is
and βp, the probability that a read uniquely identifies the first isoform is
For a concrete example, suppose θ1 = 2θ2. Assume further that there are twice as many single end reads (a sample size of 2n) compared to the n reads in a paired end run:
and the information in a paired end run for a fixed insert size is:
Plugging in numbers x = 200, e = 50, r = 30, gives
In other words, in the insert length model, the maximum likelihood estimator of θ1 has asymptotic variance roughly 3 times larger in the single end read experiment than in the paired end experiment.
Of course, this ratio will change if the parameters change. For instance, Is/Ip = 0.63 if x = 200, e = 50 and r = 70; Is/Ip = 0.47 if x = 200, e = 100 and r = 50.
The next section gives simulation results for a related example.
6.2 Simulation study
Simulations were used to study the following questions: 1) the quality of the proposed model at estimating isoform-specific gene expression, especially when the insert length is variable and 2) whether abundance estimates from paired end reads are more reliable than abundance estimates from single end reads.
To address these questions, reads were simulated from a “hard case” where a gene has three exons of lengths 500bp, 50bp and 500bp, respectively (See Figure 7); the middle exon can be skipped, producing two different isoforms of the gene. Since the middle exon is short, this case has been shown to be difficult for isoform-specific gene expression estimation in Jiang and Wong (2009).
Fig 7.
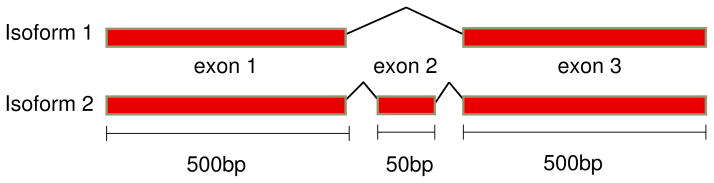
A model gene for study of Fisher information and accuracy of the single end and insert length models.
In the simulation, the two isoforms were assumed to have equal abundance. Reads were simulated using different models and parameters described in detail in below and estimate isoform abundances as described in Section 3.5. The relative error of estimation was computed based on the empirical relative L2 loss:
where is the true isoform abundance vector, and θ̂ = [θ̂1, θ̂2] is the estimated isoform abundance vector after normalization so that θ̂1 +θ̂2 = 1. Each simulation experiment was repeated 200 times to get the sample mean and standard error of the relative error.
6.2.1 Simulating single end reads with uniform sampling
To explore the quality of estimation in the uniform sampling approach, single end reads with length 30bp using the uniform sampling model were generated. Five separated experiments were performed to investigate the effect of sample size on the estimation procedure using sample sizes of 10, 50, 200, 1000 and 5000, respectively. The red curve in Figure 8(a) gives the sample mean and standard error of the relative error. It is clear that relative error decreases as the sample size increases.
Fig 8.

Relative error of different read generation models. X axis is the sample size, i.e., the number of reads that are generated in each simulation experiment. Y axis is the mean relative error based on 200 simulation experiments. The error bars give the standard errors of the sample means. In the figures, single 30bp reads generated with uniform sampling rate (red curves) is compared to (blue curves) (a) single 100bp reads, (b) single 30bp reads generated with lognormal sampling rate, (c) paired end 30bp reads generated with Gaussian insert size and (d) paired end 30bp reads generated with uniform insert size. When compared with n (e.g., 5000) single end reads, n/2 (i.e., 2500) pairs of paired end reads were used.
To examine whether longer reads can provide better estimates, all the simulation experiments were repeated with read length 100bp. Figure 8(a) shows the comparison between read lengths of 30bp and 100bp. As expected, 100bp reads produce smaller error than 30bp reads.
6.2.2 Simulating single end reads with non-uniform sampling
In real UHTS data, the read distribution is not uniform. To evaluate how well the RNA-Seq methodology performs in this regime, simulations were performed where the positions of reads were sampled from a log-normal distribution. Specifically, up to a scalar multiple, the true sampling rates ai,j are independently and identically distributed random variables which follows log-normal distribution with mean μ = 0 and standard deviation σ = 1.
Figure 8(b) gives the comparison between reads that were sampled from uniform distribution and reads that were sampled from log-normal distribution The figure shows that non-uniform reads produce estimates which appear consistent, albeit with larger error than with uniform reads.
6.2.3 Simulating paired end reads
This section investigates whether, in simulation, paired end reads can provide more information than single end reads. When insert lengths do not have a simple distribution, closed form expressions for the information are difficult to obtain. Simulation studies are thus important tools for analyzing such situations. For this purpose, paired end reads of length 30bp with insert size following a normal distribution with mean μ = 200bp and standard deviation σ = 20bp were generated. For a given insert size, read pairs were generated using a uniform sampling model. Figure 8(c) shows that the paired end reads produce smaller errors than single end reads with the same number of sequenced nucleotides: to make the comparison comparable on the level of total sequenced bases, n/2 pairs of paired end reads were used when compared with n single end reads.
When the insert size was generated using a uniform distribution, e.g., the effective insert size is uniform within 200 ± 20bp, similar results were produced (See Figure 8(d)). Comparing Figure 8(d) with Figure 8(a) shows that paired end 30bp reads produce similarly accurate estimates as 100bp single end reads, which means that on average paired end reads provide more information per nucleotide being sequenced.
6.2.4 Simulating with other parameters
We also performed simulations with other settings of parameters. For instance, with read length 70 bp, with true isoform expression vector (0.1, 0.9) or with exon lengths (500 bp, 200 bp, 500 bp). The results are shown in Figure 9. In all these simulations, the advantage of paired end sequencing over single end sequencing is obvious for moderate sampling (50 ≤ n ≤ 1000) as in typical cases for sequencing data.
Fig 9.

Relative error of single end reads (red curves) and paired end reads (blue curves) with different settings of parameters: (a) 70bp reads, true isoform expression vector (0.5, 0.5) (b) 30bp reads, true isoform expression vector (0.1, 0.9) (c) 30 bp reads, true isoform expression vector (0.5, 0.5), exon lengths (500 bp, 200 bp, 500 bp). When compared with n (e.g., 5000) single end reads, n/2 (i.e., 2500) pairs of paired end reads were used.
7. DISCUSSION
The insert length model presented in this paper is a flexible statistical tool. The model has the capacity to accommodate oriented reads from Illumina data and to model fragment specific biases in the probability of each fragment being sequencing. In Sections 3.2, the model has been derived when the experimental step of fragmentation is assumed to be approximated by a Poisson point process, and a transcript is assumed to be retained in the sample in proportion to the fraction of transcripts of its length estimated after sequencing. These assumptions are at once simplifying and realistic. As experimental protocols improve, it is likely they will better model RNA-Seq data.
At the current time, several improvements may be made to the model to increase its accuracy. Firstly, the read sampling rate is undoubtedly non-uniform, as it depends on biochemical properties of the sample and fragmentation process as experimental studies have highlighted (see Ingolia et al. (2009), Vega et al. (2009), and Quail et al. (2008)). This effect becomes more apparent for longer fragments such as those used in paired end library preparation. Explicit models for the sampling rates are difficult to obtain, but doing so is an area of future research. Recent research (see Hansen et al. (2010), Li et al. (2010)) has shown that the non-uniformity can be modelled and estimated quite well from the data. It may be possible to combine these models with our approach to improve the estimation performance.
Statistical tests of the reproducibility of the non-uniformity of reads shows a consistent sequence specific bias across biological and technical replicates of a gene. This effect could be due to bias in RNA fragmentation, bias in other biochemical sample preparation steps or boundary effects when a gene of fixed length is fragmented. The last cause of bias can be modeled using Monte Carlo simulations of a fixed length mRNA sequence subject to a Poisson fragmentation process and incorporated into the insert length model.
Similarly, the fragmentation and filtering steps have not been explicitly modeled in the insert length model presented here. Rather, the probability mass function of read lengths, what is necessary for defining the model, has been estimated empirically. Improvements to the model could be made by increasing the precision of the estimate of the probability mass function of read lengths, for example by simulating a fragmentation and filtering process by Monte Carlo and matching the output of the simulations to the empirical distribution function q(·). If such modeling were desired, as described in Section 3.2, the effects could be easily incorporated into the insert length model. On the other hand, as experimental protocols improve, they may reduce this bias and increase the accuracy of the insert length model as presented in this paper.
In reality sequencing mapping is another step that may affect the analysis. For instance, some reads can not be mapped because of sequencing errors and some can be mapped to multiple places. We have not focused on the issue of mapping fidelity because we restrict attention to the reads which did map uniquely. We are also not taking into account mapping errors which themselves require statistical modeling. We have chosen not to model these errors partly because some mapping errors are platform-dependent (i.e. different sequencing errors tend to be made by the Illumina vs. other platforms).
In some applications, the parameters of interest to biologists are not the RP-KMs for isoforms 1 and 2, but rather the relative expression ratio of both isoforms. One way to estimate the ratio is to re-parameterize the problem with θ1 as a first parameter and a second parameter μ = θ1/θ2. The re-parameterization will make the model no longer linear in the parameters therefore harder to solve. Also, the choice of μ is not straightforward when there are 3 or more isoforms. An easier way is to estimate μ indirectly after estimating θ1 and θ2.
We believe that technological improvements that produce longer read lengths will not diminish the relevance of the insert length model. Paired end models will be relevant at least until read lengths are comparable to the length of each transcript, and perhaps longer for reasons of cost. Since many transcripts are larger than 104 nucleotides, and longer in some important cases, such a time is unlikely to occur in the next few years. Further, longer insert lengths and reads combined with the insert length model in this paper will aid in discrimination of complex isoforms and estimation of isoform-specific poly-A tail lengths. Thus, we do not foresee any imminent obsolescence of this model.
While the model developed in this paper has the potential for great use and extends current methodology for isoform-specific estimation, the model assumes that the complete set of isoforms of a gene have been annotated. De novo discovery of isoforms from a sample is an important and difficult statistical problem that we have not addressed in this paper. Another shortcoming of the model is that in order statistical inference to be accurate, with the current short read technology, the number of isoforms should be relatively small (for example, 2–5). We expect these challenges to motivate methodological development in the field of RNA-Seq in the coming years.
In conclusion, this paper has presented a statistical model for RNA-Seq experiments which provides estimates for isoform specific expression. Finding such estimates is difficult using microarray technology, focusing interest in UHTS to address this question. In addition to modeling, the paper has presented an in depth statistical analysis. By using the classical statistical concept of minimal sufficiency, a computationally feasible solution to isoform estimation in RNA-Seq is provided. Further, statistical analysis quantifies the perceived gain in experimental efficiency from using paired end rather than single end read data to provide reliable isoform specific gene expression estimates. To the best of our knowledge, this is the first statistical model for answering this question.
Acknowledgments
The authors would like to acknowledge Jamie Geier Bates for providing the data used in Section 5, and Patrick O. Brown for useful discussions, as well as several anonymous referees whose comments improved the clarity of the manuscript. We thank Michael Saunders for his help in interfacing the PDCO package. Salzman’s research was supported in part by a grant from the National Science Foundation (DMS-0805157). Jiang’s research was supported in part by a grant from the National Institute of Health (2P01-HG000205). Wong is funded by a National Institute of Health grant (R01-HG004634). The computation in this project was performed on a system supported by NSF computing infrastructure grant DMS-0821823.
Footnotes
The read length is roughly the same for the ABI SOLiD platform. The 454 platform the read length can be several folds higher, but the throughput is much lower comparing to the other two platforms. Because sequencing technology is developing so rapidly, these numbers are likely to be out of date very soon. Our statistical models apply to all platforms and all read lengths.
The insert size can be controlled by tuning the parameters involved in the fragmentation, random priming and size selection steps in the sample preparation process.
Current technology allows a biochemical modification of sequenced molecules (via a circularization step) that can produce two short reads from two physical locations on a molecule that may be separated by up to several kilobases (using the ABI platform or a long-insert protocol from Illumina), which is also called the mate-pair sequencing. Although technologically it is different from the paired end sequencing, the analysis is the same from a statistical point of view.
The implementation of the model described in this paper ignores reads that align to multiple genes (while of course not ignoring reads that align to multiple isoforms). This detail does not impact the significant number of genes which contain no such reads that map to multiple genes, and a simple adaptation of the model can accomidate reads mapping to multiple genes.
Because genomic coordinates are discrete, the occurrence times in the Poisson process should be rounded to the nearest natural numbers.
In the traditional bioinformatics literature this is also called alignment, while the nomenclature “mapping” is more often used in the UHTS literature where the sequences being aligned are short reads.
In our experiments we used the PDCO (Primal-Dual interior method for Convex Objectives, http://www.stanford.edu/group/SOL/software/pdco.html) package developed by M. A. Saunders at Stanford University.
All the expression estimates in this paper are in units compatible with RPKM(Reads Per Kilobase of exon model per Million mapped reads) (see Mortazavi et al. (2008))
Contributor Information
Julia Salzman, Email: julia.salzman@stanford.edu, Research Associate in the Department of Statistics and Biochemistry, Stanford University, Stanford, California 94305, USA.
Hui Jiang, Email: jiangh@stanford.edu, Postdoctoral Scholar in the Department of Statistics and Stanford Genome Technology Center, Stanford University, Stanford, California 94305, USA.
Wing Hung Wong, Email: whwong@stanford.edu, Professor of Statistics and of Health Research and Policy, Stanford University, Stanford, California 94305, USA.
References
- Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mrna-seq experiments. BMC Bioinformatics. 2010 doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casella G, Berger R. Statistical inference. 2. Thomson Learning; 2002. [Google Scholar]
- Chi KR. The year of sequencing. Nature Methods. 2008;5:11–14. doi: 10.1038/nmeth1154. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Brenner SE, Dudoit S. Biases in illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010 Apr; doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Lareau LF, Blanchette M, Green RE, Meng Q, Rehwinkel J, Gallusser FL, Izaurralde E, Sandrine Dudoit DCR, Brenner SE. Genome-wide identification of alternative splice forms down-regulated by nonsense-mediated mrna decay in drosophila. PLoS Genetics. 2009:6. doi: 10.1371/journal.pgen.1000525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller D, Jiang H, Xu W, Wong WH. Identifiability of isoform deconvolution from junction arrays and rna-seq. Bioinformatics. 2009 Dec;25(23):3056–3059. doi: 10.1093/bioinformatics/btp544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wang F, Dyer NP, Wong WH. Cisgenome browser: A flexible tool for genomic data visualization. Bioinformatics. 2010 May; doi: 10.1093/bioinformatics/btq286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wong WH. Seqmap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008 Aug;24(20):2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wong WH. Statistical inferences for isoform expression in rna-seq. Bioinformatics. 2009 Feb; doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg S. Ultrafast and memory-efficient alignment of short dna sequences to the human genome. Genome Biol. 2009 Mar;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann EL. Theory of point estimation. 2. Springer; 1998. [Google Scholar]
- Li J, Jiang H, Wong WH. Modeling non-uniformity in short-read rates in rna-seq data. Genome Biol. 2010 May;11(5):R50. doi: 10.1186/gb-2010-11-5-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh P, Nelder J. Generalized Linear Models. 2. Boca Raton: Chapman and Hall/CRC; 1989. [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by rna-seq. Nat Methods. 2008 Jul;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008 Dec;40(12):1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. Ncbi reference sequence (refseq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005 Jan;33(Database issue):D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ., 1 A large genome center’s improvements to the illumina sequencing system. Nat Methods. 2008:1005–1010. doi: 10.1038/nmeth.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She Y, Hubbell E, Wang H. Resolving deconvolution ambiguity in gene alternative splicing. BMC Bioinformatics. 2009 doi: 10.1186/1471-2105-10-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, Schmidt D, O’Keeffe S, Haas S, Vingron M, Lehrach H, Yaspo ML. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008 Aug;321(5891):956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. Tophat: discovering splice junctions with rna-seq. Bioinformatics. 2009 May;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010 May;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega VB, Cheung E, Palanisamy N, Sung W-K. Inherent signals in sequencing- based chromatin-immunoprecipitation control libraries. PLoS ONE. 2009:4. doi: 10.1371/journal.pone.0005241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstedt H, Daniel C, Enstero M, Ohman M. Large-scale mrna sequencing determines global regulation of rna editing during brain development. Genome Res. 2009;(19):978–986. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue tran- scriptomes. Nature. 2008 Nov;456(7221):470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Duan S, Bleibel WK, Wisel SA, Huang RS, Wu X, He L, Clark TA, Chen TX, Schweitzer AC, Blume JE, Dolan ME, Cox NJ. Identification of common genetic variants that account for transcript isoform variation between human populations. Journal Human Genetics. 2009;(1):81–93. doi: 10.1007/s00439-008-0601-x. [DOI] [PMC free article] [PubMed] [Google Scholar]