

Abstract
Lysine methylation is a key epigenetic mark, the dysregulation of which is linked to many diseases. Small molecule antagonism of methyl-lysine (Kme) binding proteins that recognize such epigenetic marks can improve our understanding of these regulatory mechanisms and potentially validate Kme binding proteins as drug discovery targets. We previously reported the discovery of 1 (UNC1215), the first potent and selective small molecule chemical probe of a methyl-lysine reader protein, L3MBTL3, which antagonizes the mono- and dimethyl-lysine reading function of L3MBTL3. The design, synthesis, and structure activity relationship studies that led to the discovery of 1 are described herein. These efforts established the requirements for potent L3MBTL3 binding and enabled the design of novel antagonists, such as compound 2 (UNC1679), that maintain in vitro and cellular potency with improved selectivity against other MBT-containing proteins. The antagonists described were also found to effectively interact with unlabeled endogenous L3MBTL3 in cells.
INTRODUCTION
Expression of the genetic code is largely regulated by cell-type specific transcription factors and chemical modifications to histone proteins and DNA. The intricate and dynamic set of post-translational modifications (PTMs), of which methylation is one of the most significant, control access of transcriptional machinery to DNA, in turn determining protein expression and cell function. The methylation state of lysine residues in histones is regulated by families of enzymes that can either ‘write’ (create a PTM) or ‘erase’ (chemically remove) such PTMs, known as methyltransferases and demethylases, respectively, while ‘readers’ recognize PTMs via proteinprotein interactions.
Lysine residues can be mono-, di-, or trimethylated. Both the location of this PTM within a histone and the degree of methylation dictate the transcriptional outcome (activation versus repression), as well as the recruitment of Kme modulators (readers, writers, and erasers) that work collectively to maintain an appropriate level of methylation within the cell. Methyl-lysine acts as docking site for specific reader proteins that can in turn alter chromatin structure and direct various cellular processes, often by attracting additional regulatory proteins in a highly coordinated manner.1 In addition, histone methylation is dynamic and has been shown to play an important role in cell-cycle regulation, DNA damage and stress response, and cell fate during development and differentiation.2
Aberrant methylation levels and ensuing changes in gene expression patterns due to the mutation or altered expression of Kme regulators is one mechanism by which such epigenetic factors can contribute to disease.3 There is increasing evidence that many epigenetic regulators are critical proteins dysregulated in cancer, as the levels of histone marks are often altered within cancer epigenomes.4–5 However, the underlying mechanisms of chromatin regulation in oncogenesis via miswriting, misreading, and/or miserasing methyl-lysine are not understood. One approach to increase our knowledge of these regulatory mechanisms is through small molecule perturbation. High-quality potent, selective, and cell-penetrant chemical probes serve as excellent tools for improving our understanding of their molecular targets and the broader biological and therapeutic consequences of modulating these targets.6 Accordingly, chemical biology efforts focused on deciphering the function of lysine methylation with small molecule tools have gained momentum, resulting in a number of freely available high-quality chemical probes.7–8 The methyltransferases were an initial focus of this effort which has resulted in chemical probes for enzymes including G9a/GLP,9 EZH2,10–16 and DOT1L,17–18 all of which have been implicated in tumorgenesis.
Kme readers have recently emerged as less precedented epigenetic targets,19–20 and antagonism of reader domains may result in cellular effects that are distinct from enzyme inhibitors. We recently reported a first-in-class chemical probe, 1, (UNC1215, Figure 1a), which selectively binds L3MBTL3, a member of the MBT (malignant brain tumor) family of methyl-lysine reader proteins, validating this class of proteins as tractable for probe discovery.21 Compound 1 provided valuable insight into the binding mechanism of L3MBTL3 and enabled the identification of a non-histone L3MBTL3 Kme substrate, BCLAF1. Although the role of L3MBTL3 in chromatin biology is largely unexplored to date, proteins containing MBT domains have generally been functionally associated with transcriptional repression, chromatin compaction, and significant developmental biology due to their presence in Polycomb complexes.22–23 Furthermore, it has been reported that L3MBTL3 knockout mice are embryonic lethal due to defects in myeloid lineage differentiation.24
Figure 1.
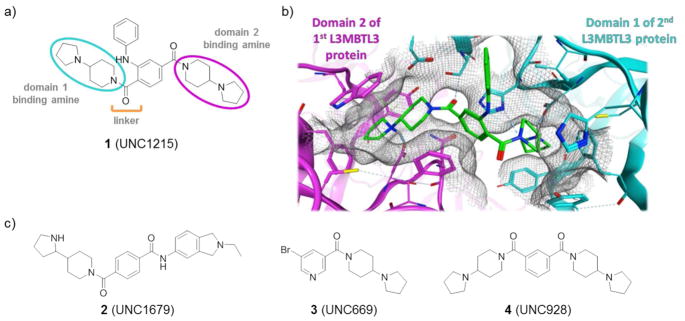
a) Structure of L3MBTL3 chemical probe, 1. b) Co-crystal structure of 1 (green) bound to two molecules of L3MBTL3. The amine meta to the aniline substituent (domain 2 amine) binds in the Kme binding pocket of MBT domain 2 of one L3MBTL3 molecule (magenta) and the amine ortho to the aniline substituent (domain 1 amine) binds to MBT domain 1 of a second L3MBTL3 molecule (cyan). c) Structures of other L3MBTL3 inhibitors.
Herein we summarize the structure activity relationship (SAR) studies that led to the discovery of compound 1. Binding affinities for L3MBTL3 were determined for multiple series of compounds by an AlphaScreen assay, and these binding trends were subsequently confirmed in an orthogonal LANCE time resolved fluorescence resonance energy transfer (TR-FRET) assay, which, to the best of our knowledge, is the first report of this assay in the evaluation of inhibitors of epigenetic protein-protein interactions. In order to assess the selectivity of these compounds and their affinity for other Kme reader targets, each ligand was screened against a small panel of Kme readers including related proteins from the MBT family as well as representative proteins from other Kme reader families including tudor domains, PHD fingers, and chromodomains. These studies established the requirements for potent L3MBTL3 binding and enabled the design of novel antagonists (such as 2, UNC1679; Figure 1c) that maintain in vitro and cellular potency with improved selectivity against other MBT-containing proteins. Lastly, we have demonstrated that the inhibitors described interact with unlabeled endogenous L3MBTL3 in the context of protein pull-down assays.
RESULTS AND DISCUSSION
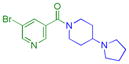
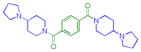
In an attempt to design potent small molecule inhibitors of the tudor domain-containing protein, p53-binding protein 1 (53BP1), that recognizes dimethyl-lysine (Kme2) in sequences with an adjacent basic residue (histone 4 R19K20me225 and p53 K381K382me226, for example), we initially synthesized compounds containing two basic amines, one of which we hypothesized would function as a specific Kme mimic and anchor the compound in the methyl-lysine pocket, and the other which would interact with the protein similarly to the adjacent unmethylated lysine or arginine in the native substrates. Based on prior work towards the discovery of an antagonist of L3MBTL1 (3, UNC669; Figure 1c), a reader of the H4K20me1–2 mark, we found that a 4-(pyrrolidin-1-yl)piperidine amine effectively filled the lysine binding cavity of this domain, enhancing the number of van der Waals contacts in the pocket as compared to Kme2 while also positioning the basic amine in close enough proximity to make a key hydrogen bond in the second MBT domain.27–28 Anticipating that this amine may be well suited to bind other H4K20me1–2 readers, compound 4 (UNC928; Figure 1c) was prepared, containing two meta-4-(pyrrolidin-1-yl)piperidine amines. While the affinity of 4 appeared to be weak for 53BP1, our cross-screening strategy20 revealed sub-micromolar inhibition of L3MBTL3 (Table 1). In comparison to our previous work with analogs of 3 containing only a single amine,27 it was apparent that the expansion of this scaffold to include a second basic site resulted in a significant boost in potency and selectivity for L3MBTL3. Intrigued by this result, we set out to optimize the inhibitory activity of this compound for L3MBTL3, with the hope that this early lead would evolve into a potent and selective L3MBTL3 antagonist.
Table 1.
Effect of varying the substitution pattern of the 4-(pyrrolidin-1-yl)piperidine aminesa
| ID | R | LANCE IC50 (μM) | AlphaScreen IC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L3MBTL3 | L3MBTL3 | L3MBTL1 | L3MBTL4 | SFMBT1 | MBTD1 | CBX7 | 53BP1 | UHRF1 | PHF23 | JARID1A | ||
| 3 |
|
4.7 ± 1.3 (8) | 3.1 ± 0.77 (6) | 4.2 ± 0.87 (6) | >10 (6) | >10 (10) | >10 (17) | >10 (8) | >10 (19) | >10 (6) | >10 (6) | >10 (6) |
| 4 |
|
0.29 ± 0.14 (4) | 0.36 ± 0.021 (3) | 2.8 ± 0.91 (3) | >10 (3) | >10 (4) | >10 (11) | >10 (4) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 5 |
|
0.048 ± 0.0025 (3) | 0.071 ± 0.0023 (3) | 2.9 ± 1.3 (3) | >10 (3) | >10 (10) | >10b (3) | >10 (3) | >10b (3) | >10 (3) | >10 (3) | >10 (3) |
| 6 |
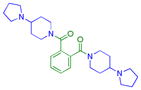
|
0.35 ± 0.025 (3) | 1.1± 0.1 (3) | 3.0 ± 0.91 (3) | >10 (3) | >10 (9) | >10b (3) | >10 (3) | 4.7 ± 1.7 (8) | >10 (3) | >10 (3) | >10 (3) |
| 7 |
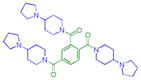
|
0.11 ± 0.044 (4) | 0.12 ± 0.0058 (3) | 2.2 ± 0.3 (3) | >10 (3) | >10 (5) | >10b (10) | >10 (4) | 7.9 ± 1.1b (10) | >10 (3) | >10 (3) | >10 (3) |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
In the course of these studies, we solved the co-crystal structure of 1 and L3MBTL3 (Figure 1b, PDB code 4FL6) which helped to elucidate the compound’s mechanism of action and rationalize many of the SAR trends observed.21 Surprisingly, a unique 2:2 polyvalent mode of interaction was identified whereby two different L3MBTL3 molecules interact with two molecules of 1. L3MBTL3 is a multi-domain protein containing three MBT domains, of which MBT domain 1 and MBT domain 2 were found to participate in binding to 1. The pyrrolidine amine meta to the aniline substituent of 1 binds in the Kme reader pocket of MBT domain 2 while the pyrrolidine amine ortho to the aniline bridges the two L3MBTL3 proteins by binding more shallowly in MBT domain 1 of a second L3MBTL3 molecule. Site directed mutagenesis demonstrated that domain 2 interactions were dominant in the binding of 1, while binding to MBT domain 1 was essential for dimerization and the observed boost in potency versus monovalent binding. This is in contrast to the binding of compounds 1 and 3 to L3MBTL1, which are monovalent interactions involving only the second MBT domain Kme pocket as confirmed by crystallography (PDB codes 3P8H and 3UWN).28 While our understanding of this binding mode was not contemporaneous with all of the SAR studies we present herein, this binding mode provides a rational explanation for the trends observed, and based on this structural model, the amines described will generally be referred to as “domain 1 binding” or “domain 2 binding” (Figure 1a).
Compounds were initially designed to explore the SAR of the domain 1 binding amine, the position of the two amines, the linker between the domain 1 amine and the aromatic core, and modifications to the aromatic core. Significant efforts had previously been made to optimize the amine that binds in the dominant Kme binding pocket (domain 2 binding amine) in the context of monobasic scaffolds; however, little improvement was achieved over 3 for L3MBTL3.27 As a result, the domain 2 binding amine was held constant as 4-(pyrrolidin-1-yl)piperidine throughout most of these SAR studies.
The inhibitory activity of analogs of 4 versus histone peptide binding to L3MBTL3, as well as a small panel of various other Kme reader proteins, was evaluated using an AlphaScreen competition assay reported previously.29 The direct binding affinity of compounds of particular interest was also confirmed by isothermal titration calorimetry (ITC). Although signal amplification and savings on protein supplies are an advantage of the AlphaScreen assay,29 we observed variation in the results from day to day. Therefore, in order to most accurately evaluate the SAR of these analogs and their relative activities, about 100 analogs of compound 4 that demonstrate the most important trends were prepared in serial dilution screening plates and tested simultaneously. This allowed for a more precise comparison of the compounds’ activities and an accurate depiction of the observed binding trends. The serial dilution screening was performed up to 10 μM, and therefore the SAR trends were most closely evaluated for those compounds that were active within the concentration range explored. In addition, a counterscreen assay was conducted in the absence of protein with a His6-biotin fusion peptide substrate to ensure the compounds did not interfere with the assay by serving as metal chelators, biotin mimetics, or singlet-oxygen quenchers.29
To further validate the SAR trends observed by AlphaScreen, a secondary L3MBTL3 assay was developed, comparable to AlphaScreen in throughput and efficiency. This LANCE TR-FRET assay is based on the interaction of a biotinylated H4K20me2 peptide bound to a streptavidin-dye conjugate (ULight streptavidin) with a His-tagged L3MBTL3 molecule recognized by an anti-6× His antibody labeled with a europium donor fluorophore. When L3MBTL3 binds the H4K20me2 peptide, the donor and acceptor fluorophores are in close enough proximity to produce a FRET signal, which in turn can be disrupted by small molecule inhibitors. ULight is a PerkinElmer proprietary dye, chosen for its small size, brightness, and reduced steric hindrance. Similarly to AlphaScreen, this assay was easily miniaturized and automated in a 384-well format and proved to be equally efficient and cost effective. Due to some of the shortcomings in day-to-day reproducibility that we have encountered with the AlphaScreen assay, we are currently investigating expansion of TR-FRET based assays for screening Kme reader proteins more broadly. In order to compare the screening results of the AlphaScreen and LANCE TR-FRET assays following at least three biological replicates in each format, a scatter plot was generated comparing the two data sets (Figure 2). This revealed an overall linear regression with an R2 value of 0.8 which, when combined with ITC confirmations of key compounds, gave us high confidence in interpreting the reported SAR trends.
Figure 2.

Scatter plot showing the excellent, linear correlation between L3MBTL3 AlphaScreen data and L3MBTL3 TR-FRET data.
Our first step towards the optimization of compound 4 involved the preparation of various structural isomers to determine whether the position of the two amines influenced potency. The synthesis of these compounds was straightforward due to their symmetry, and they were easily prepared in one step from the appropriate phthalic acids. All three dibasic compounds (4, 5, and 6) showed improved potency for L3MBTL3 over compound 3, while maintaining a comparable potency to 3 against L3MBTL1 (Table 1). This revealed that this series of compounds had the potential to be developed as selective L3MBTL3 inhibitors. The 1,4-bis-benzamide compound, 5 (UNC1021),21 demonstrated about a 5-fold improvement in potency relative to 4 (1,3-bisbenzamide), whereas the 1,2-bis-benzamide (6) showed weaker in vitro potency. These geometrical preferences led us to believe that the improved inhibition of L3MBTL3 relative to L3MBTL1 was not purely a non-specific interaction resulting from the addition of a second basic amine or a change in the compound’s physical properties. Furthermore, the addition of a third 4-(pyrrolidin-1-yl)piperidine amine in compound 7 did not result in any additional improvement in potency against L3MBTL3. Compounds 4 – 7 all are weakly potent against L3MBTL1, which was anticipated; however, compound 5 demonstrated more selective inhibition of L3MBTL3 over other panel members, whereas 6 and 7 show modest inhibition of 53BP1. The affinity of 5 for L3MBTL3 was confirmed by ITC, resulting in an average Kd of 390 nM, which is about 7-fold more potent than compound 4 by ITC. This ITC data closely agrees with the activity trends observed by both high-throughput assays.
To explore the nature of the linker between the domain 1 amine and the aromatic core, one amide of compound 5 was varied to generate asymmetric compounds 8, 9, and 10 (Table 2). While conversion to a sulfonamide linker in compound 8 was well tolerated, this modification did not further improve potency against L3MBTL3. Similarly, attaching the amine to the aromatic core directly in compound 9 to give a more rigid structure and replacing the amide with a methylene group in compound 10 to increase flexibility resulted in a 2–3-fold decrease in potency in each case relative to 5. Compound 5 is also the only inhibitor from this series demonstrating IC50’s greater than 10 μM versus all other panel members (except L3MBTL1), making it the preferred candidate for future optimization. Interestingly, compound 9 is the only analog screened that shows weak affinity for L3MBTL4, another member of the MBT family, suggesting that the phenyl-4-(pyrrolidin-1-yl)piperidine moiety may be a good starting point for L3MBTL4 ligand development.
Table 2.
Effect of varying the linker between the domain 1 amine and the phenyl corea
| ID | R | LANCE IC50 (μM) | AlphaScreen IC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L3MBTL3 | L3MBTL3 | L3MBTL1 | L3MBTL4 | SFMBT1 | MBTD1 | CBX7 | 53BP1 | UHRF1 | PHF23 | JARID1A | ||
| 8 |
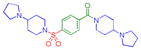
|
0.097 ± 0.049 (4) | 0.099 ± 0.02 (3) | 3.3 ± 1.0 (3) | >10 (3) | >10 (5) | 5.4 ± 2.2 (11) | >10 (4) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 9 |
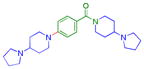
|
0.15 ± 0.017 (3) | 0.17 ± 0.01 (3) | 1.5 ± 0.61 (3) | 8.4 ± 1.1 (3) | >10 (10) | 8.2 ± 1.2b (3) | >10 (3) | 7.4 ± 1.1b (9) | >10 (3) | >10 (3) | >10 (3) |
| 10 |
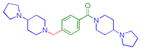
|
0.13 ± 0.041 (4) | 0.14 ± 0.038 (3) | 3.8 ± 1.3 (3) | >10 (3) | >10 (5) | 8.1 ± 1.1b (5) | >10 (3) | >10 (5) | >10 (3) | >10 (3) | >10 (3) |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
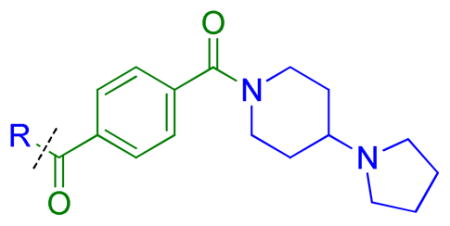
As compound 5 showed the most promising activity versus L3MBTL3, we next desymmetrized the molecule to explore the domain 1 binding amine (Table 3). This series of compounds was prepared by reacting 4-(tert-butoxycarbonyl)benzoic acid with 4-(1-pyrrolidinyl)piperidine, followed by deprotection with TFA and coupling with the desired amine. Expansion of the domain 1 amine to either a piperidine or azepane in compounds 11 and 12, respectively, decreased potency by about 3–4-fold indicating that increasing the size of this ring is not beneficial. Converting the piperidine amine in compound 11 to a morpholine (13) or cyclohexyl (14) group where the basicity of the ring is either diminished or eliminated decreases the potency greater than 20-fold, demonstrating that two basic amines are required for nanomolar in vitro potency. In contrast, a smaller dimethyl amino group in compound 15 results in only about a 4-fold loss in potency relative to compound 5. This suggests that although the presence of a basic nitrogen in the domain 1 amine is essential for high potency, a variety of substitutents or ring systems are well tolerated.
Table 3.
Effect of varying the domain 1 aminea
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | R | LANCE IC50 (μM) | AlphaScreen IC50 (μM) | |||||||||
| L3MBTL3 | L3MBTL3 | L3MBTL1 | L3MBTL4 | SFMBT1 | MBTD1 | CBX7 | 53BP1 | UHRF1 | PHF23 | JARID1A | ||
| 11 |
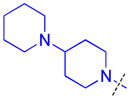
|
0.16 ± 0.0071 (3) | 0.23 ± 0.017 (3) | 6.8 ± 1.3b (3) | >10 (3) | >10 (10) | 10.0 ± 1.3b (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 12 |
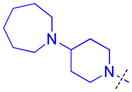
|
0.21 ± 0.012 (3) | 0.27 ± 0.015 (3) | 4.2 ± 1.2 (3) | >10 (3) | >10 (10) | 9.7 ± 1.4b (3) | >10 (3) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 13 |
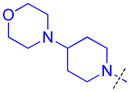
|
1.1 ± 0.071 (3) | 1.5 ± 0.15 (3) | 7.1 ± 1.1b (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 14 |
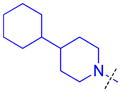
|
1.6 ± 0.10 (3) | 2.4 ± 0.21 (3) | >10 (3) | >10 (3) | >10 (10) | 8.1 ± 0.68 (3) | >10 (3) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 15 |
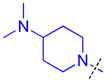
|
0.18 ± 0.0071 (3) | 0.31 ± 0.046 (3) | 7.2 ± 2.9 (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 16 |
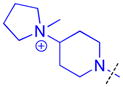
|
0.081 ± 0.0041 (3) | 0.14 ± 0.010 (3) | 7.5 ± 1.1b (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 17 |
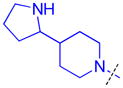
|
0.092 ± 0.027 (4) | 0.12 ± 0.017 (3) | 5.2 ± 2.0 (3) | >10 (3) | >10 (5) | 5.5 ± 1.0 (5) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
| 18 |
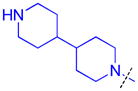
|
0.80 ± 0.41 (4) | 0.74 ± 0.060 (3) | 3.5 ± 1.1 (3) | >10 (3) | >10 (5) | >10 (11) | >10 (4) | >10 (10) | >10 (3) | >10 (3) | >10 (3) |
| 19 |
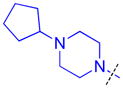
|
3.2 ± 0.16 (3) | 3.8 ± 0.49 (3) | 7.6 ± 1.2b (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (7) | >10 (3) | >10 (3) | >10 (3) |
| 20 |

|
0.77 ± 0.44 (4) | 0.83 ± 0.24 (3) | 3.3 ± 0.87 (3) | >10 (3) | >10 (5) | >10 (11) | >10 (4) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 21 |
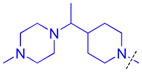
|
0.25 ± 0.092 (4) | 0.30 ± 0.075 (3) | 4.4 ± 1.6 (3) | >10 (3) | >10 (5) | >10b (11) | >10 (4) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 22 |
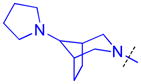
|
0.092 ± 0.0074 (3) | 0.16 ± 0.015 (3) | 6.6 ± 0.68 (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 23 |
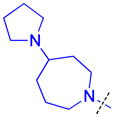
|
0.074 ± 0.026 (3) | 0.096 ± 0.0032 (3) | 5.9 ± 2.4 (3) | >10 (3) | >10 (4) | >10 (5) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
| 24 |
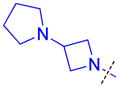
|
0.30 ± 0.11 (4) | 0.33 ± 0.021 (3) | 9.5 ± 1.1b (3) | >10 (3) | >10 (5) | >10 (5) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
| 25 |
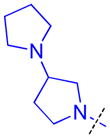
|
0.22 ± 0.098 (4) | 0.25 ± 0.035 (3) | 6.5 ± 3.1 (3) | >10 (3) | >10 (5) | >10 (5) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
| 26 |
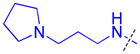
|
0.34 ± 0.0071 (3) | 0.68 ± 0.053 (3) | 5.5 ± 2.1 (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 27 |
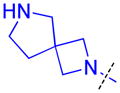
|
2.7 ± 0.78 (4) | 3.6 ± 0.29 (3) | 5.6 ± 1.8 (3) | >10 (3) | >10 (5) | >10 (4) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
| 28 |
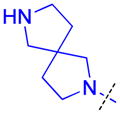
|
2.2 ± 0.79 (4) | 1.8 ± 0.36 (3) | 7.1 ± 1.1b (3) | >10 (3) | >10 (5) | >10 (4) | >10 (3) | >10 (5) | >10 (3) | >10 (3) | >10 (3) |
| 29 |
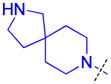
|
0.62 ± 0.23 (4) | 0.56 ± 0.15 (3) | 5.0 ± 1.7 (3) | >10 (3) | >10 (5) | >10 (5) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
| 30 |
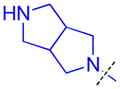
|
3.5 ± 1.3 (4) | 3.1 ± 0.87 (3) | 6.9 ± 3.0 (3) | >10 (3) | >10 (5) | >10 (5) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
| 31 |
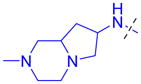
|
4.5 ± 0.49 (3) | 4.6 ± 0.70 (3) | 8.4 ± 1.7 (3) | >10 (3) | >10 (5) | >10 (11) | >10 (4) | >10 (10) | >10 (3) | >10 (3) | >10 (3) |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
Methylation of the pyrrolidine nitrogen to form a quaternary amine in the domain 1 amine position of compound 16 is well tolerated, as is the installation of 4-(pyrrolidin-2-yl)piperidine in compound 17, resulting in a secondary amine slightly further removed from the aromatic core. Neither compound shows improved potency over compound 5 for L3MBTL3, but compound 17 does show modest affinity for MBTD1 (IC50 = 5.5 μM). Increasing the distance between the secondary amine and the aromatic ring further in compound 18 additionally decreases L3MBTL3 potency by greater than 10-fold. Compound 19 demonstrated a more than a 50-fold loss in affinity by simply swapping the position of the pyrrolidine nitrogen with the adjacent tertiary carbon. This suggests that the nitrogen must be positioned appropriately to make a key interaction(s) and facilitate binding to the first MBT domain of L3MBTL3. Compounds such as 20 and 21 containing additional basic amines also did not improve potency.
The role of the piperidine ring of the domain 1 binding amine was also investigated. While replacement with a larger 3-azabicyclo[3.2.1]octane (22) or azepane (23) only resulted in a minimal decrease in L3MBTL3 in vitro potency, smaller azetidine (24) and pyrrolidine (25) ring systems at this position resulted in a decrease in activity. Increasing the flexibility of the molecule by linking the pyrrolidine to the phenyl core via an alkyl amine as in compound 26 was also not favorable. We last investigated various spirocycles (27 – 29) or fused bicyclic amines (30 and 31). While compound 29 containing the largest spirocycle amine binds L3MBTL3 about 10-fold weaker than 5, the remaining compounds were micromolar ligands, indicating that they are likely not forming favorable interactions with the first MBT domain and therefore not facilitating L3MBTL3 dimerization.
To determine whether the symmetric nature of compound 5 enhanced binding to L3MBTL3 relative to the asymmetric compounds in Table 3, a number of other symmetric 1,4-bisbenzamide compounds were synthesized (Table 4). In agreement with the trends from Table 3, L3MBTL3 binding is affected more significantly by modification of the pyrrolidine rings than by changing the nature of the piperidine rings in compound 5. Consequently, compound 32 with symmetric 4-(pyrrolidin-1-yl)azepane amines shows diminished affinity for L3MBTL3 by about 2-fold, while compound 33 with (1R,5S)-8-(pyrrolidin-1-yl)-3-azabicyclo[3.2.1]octane amines binds about 6-fold weaker. Both of these compounds also show somewhat diminished affinity for L3MBTL1, indicating that the larger rings are not ideally suited for binding in the domain 2 pocket of L3MBTL1, yet this can be overcome to some extent with L3MBTL3 due to the added interactions of the second amine binding to domain 1. Compounds 34 and 35 where the pyrrolidines are replaced by smaller azetidine rings or the pyrrolidine nitrogens are moved to the 2 position of the ring, respectively, both show reduced activity against L3MBTL3 and L3MBTL1, while methylation of compound 35 to give tertiary amines (36) binds L3MBTL3 with micromolar affinity. The most significant changes are seen with compounds 37 and 38, as although the 1,4′-bipiperidine and N,N-dimethylpiperidin-4-amine are reasonably well tolerated in the domain 1 amine position, these amines are not well suited to bind the Kme pocket of domain 2, resulting in affinities of ca. 10 μM or greater. Since compound 37 is structurally similar to 1 but substantially less potent as a L3MBTL3 antagonist it was used as a negative control compound in prior cellular studies.21 None of the compounds in Table 4 were equipotent to 5, indicating that the specific nature of the 4-(pyrrolidin-1-yl)piperidine amines contributes more significantly to L3MBTL3 binding than the inherent symmetry of the molecule. Symmetric compounds 35 and 37 also are weak antagonists of MBTD1, which correlates well with the observed MBTD1 binding of their asymmetric analogs in Table 3 (compounds 17 and 11, respectively).
Table 4.
Potency of symmetric dibasic compoundsa
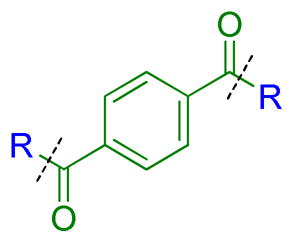
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | R | LANCE IC50 (μM) | AlphaScreen IC50 (μM) | |||||||||
| L3MBTL3 | L3MBTL3 | L3MBTL1 | L3MBTL4 | SFMBT1 | MBTD1 | CBX7 | 53BP1 | UHRF1 | PHF23 | JARID1A | ||
| 32 |
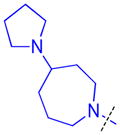
|
0.11 ± 0.031 (4) | 0.11 ± 0.045 (3) | 8.1 ± 1.1b (3) | >10 (3) | >10 (5) | >10b (5) | >10 (3) | >10 (5) | >10 (3) | >10 (3) | >10 (3) |
| 33 |
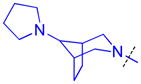
|
0.28 ± 0.082 (4) | 0.40 ± 0.042 (3) | 10.7 ± 1.4b (3) | >10 (3) | >10 (5) | >10 (5) | >10 (3) | >10b (7) | >10 (3) | >10 (3) | >10 (3) |
| 34 |
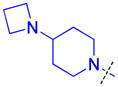
|
0.35 ± 0.099 (4) | 0.4 ± 0.012 (3) | 7.4 >10 (2) | >10 (3) | >10 (5) | >10b (5) | >10 (3) | >10 (5) | >10 (3) | >10 (3) | >10 (3) |
| 35 |
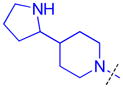
|
0.80 ± 0.47 (4) | 0.64 ± 0.040 (3) | >10 (3) | >10 (3) | >10 (5) | 4.0 ± 1.1b (11) | >10 (4) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 36 |
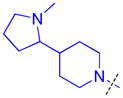
|
1.9 ± 1.1 (4) | 1.7 ± 0.15 (3) | >10 (3) | >10 (3) | >10 (5) | >10 (9) | >10 (4) | >10 (10) | >10 (3) | >10 (3) | >10 (3) |
| 37 |
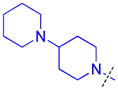
|
>10b (3) | >10 (3) | >10 (3) | >10 (3) | >10 (9) | 4.9 ± 0.21 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 38 |
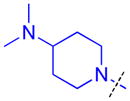
|
8.0 ± 1.2b (4) | >10b (3) | >10 (3) | >10 (3) | >10 (5) | >10 (5) | >10 (3) | >10 (6) | >10 (3) | >10 (3) | >10 (3) |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
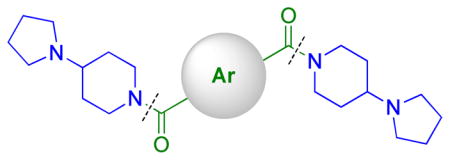
Because extensive exploration of the amine groups did not result in an L3MBTL3 ligand more potent than compound 5, we next evaluated whether modification to the phenyl core would serve to further improve L3MBTL3 affinity (Table 5). Overall, a variety of modifications to compound 5 were reasonably well tolerated, as all ligands retained sub-micromolar affinity for L3MBTL3, while their affinity for L3MBTL1 was virtually unaffected. The introduction of a pyridine (39), bromophenyl (40), or nitrophenyl (41) core yielded IC50 values less than 2-fold weaker than 5, whereas the aniline (42), tetrafluorophenyl (43), and 2,5-dimethylphenyl (44) analogs were only slightly less potent with IC50 values ranging from 3–5-fold weaker. This indicates that neither the presence of electron donating nor electron withdrawing substituents serve to enhance affinity for L3MBTL3. Furthermore, expansion of the phenyl core to a 1,4-substituted naphthalene (45) results in comparable potency to 5, while demonstrating slightly enhanced affinity for MBTD1 and 53BP1. In contrast, a 2,6-substituted naphthalene core (46), which increases the distance between the two amines, is less favorable yielding a 10-fold decrease in affinity for L3MBTL3. Lastly, substitution of the phenyl ring for a smaller 2,5-substituted furan ring (47) is less potent by about 7-fold, while a more isosteric replacement by a thiophene core (48) is virtually equipotent to compound 5.
Table 5.
Effect of modifications to the aromatic corea
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Ar | LANCE IC50 (μM) | AlphaScreen IC50 (μM) | |||||||||
| L3MBTL3 | L3MBTL3 | L3MBTL1 | L3MBTL4 | SFMBT1 | MBTD1 | CBX7 | 53BP1 | UHRF1 | PHF23 | JARID1A | ||
| 39 |
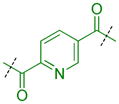
|
0.061 ± 0.0031 (3) | 0.11 ± 0.0058 (3) | 4.5 ± 1.6 (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 40 |
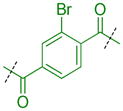
|
0.074 ± 0.0096 (3) | 0.12 ± 0.040 (3) | 2.5 ± 1.1 (3) | >10 (3) | >10 (10) | 7.8 ± 1.2 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 41 |
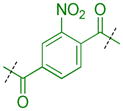
|
0.088 ± 0.0057 (3) | 0.16 ± 0.010 (3) | 2.7 ± 0.85 (2) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 42 |
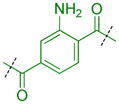
|
0.22 ± 0.035 (3) | 0.23 ± 0.049 (3) | 6.8 ± 3.1 (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 43 |
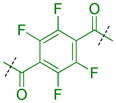
|
0.13 ± 0.012 (3) | 0.21 ± 0.012 (3) | 2.9 ± 1.0 (3) | >10 (3) | >10 (10) | >10b (3) | >10 (3) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 44 |
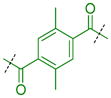
|
0.24 ± 0.13 (4) | 0.21 ± 0.046 (3) | 1.2 ± 0.35 (3) | >10 (3) | >10 (5) | >10b (11) | >10 (4) | >10b (10) | >10 (3) | >10 (3) | >10 (3) |
| 45 |
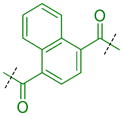
|
0.043 ± 0.007(3) | 0.072 ± 0.0042 (3) | 3.1 ± 0.71 (3) | >10 (3) | >10 (10) | 5.7 ± 2.4 (3) | >10 (3) | 10.7 ± 1.1b (8) | >10 (3) | >10 (3) | >10 (3) |
| 46 |
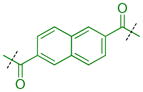
|
0.45 ± 0.016 (3) | 0.69 ± 0.17 (3) | 5.0 ± 2.2 (3) | >10b (3) | >10 (9) | >10 (3) | >10 (3) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 47 |
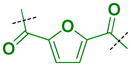
|
0.31 ± 0.021 (3) | 0.53 ± 0.056 (3) | 2.3 ± 0.98 (3) | >10 (3) | >10 (10) | >10 (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 48 |
|
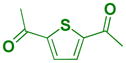
0.068 ± 0.0035 (3) | 0.096 ± 0.0059 (3) | 3.7 ± 1.4 (3) | >10 (3) | >10 (10) | >10b (3) | >10 (3) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
Compounds were next synthesized with bulkier substituents on the aromatic core (Table 6) under Buchwald, Suzuki, or reductive amination conditions from compounds 40 or 42. Both compound 1 with a 2-phenylamino substituent and compound 49 with a 2-benzylamino substituent bind L3MBTL3 virtually identically to compound 5 in vitro, albeit with a decrease in ligand efficiency. Interestingly, 49 also displays sub-micromolar potency against L3MBTL1 as determined by AlphaScreen. However, follow-up ITC studies revealed that 49 binds L3MBTL1 with micromolar affinity, comparable to the Kd values determined previously for compounds 121 and 3.28 The 2-benzyl (50) and 2-phenoxy (51) analogs of compound 1 were also found to be equipotent, as was the biphenyl derivative (52). The increased hydrophobicity of this series of inhibitors also resulted in a modest increase in affinity for MBTD1. Although compound 1 did not show improved potency over compound 5 in vitro, simultaneous cellular studies revealed that 1 was a more potent L3MBTL3 inhibitor in cells. As reported previously, compound 1 showed potent effects on the subnuclear localization of a GFP fusion protein containing the three MBT domains of L3MBTL3 (GFP-3MBT), with an EC50 of approximately 500 nM, relative to 3 μM for compound 5.21 One reason for this difference in cellular activities may be due to the increased lipophilicity of compound 1, which has a cLogP value of 2.4 relative to compound 5 which has a cLogP of 0.55.
Table 6.
Effect of phenyl substituents on the aromatic corea
| ID | Ar | LANCE IC50 (μM) | AlphaScreen IC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L3MBTL3 | L3MBTL3 | L3MBTL1 | L3MBTL4 | SFMBT1 | MBTD1 | CBX7 | 53BP1 | UHRF1 | PHF23 | JARID1A | ||
| 1 |
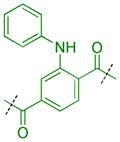
|
0.049 ± 0.025 (11) | 0.064 ± 0.02 (9) | 2.3 ± 0.85 (8) | >10 (9) | >10 (20) | 9.0 ± 1.1b (16) | >10 (10) | >10b (24) | >10 (9) | >10 (9) | >10 (9) |
| 49 |
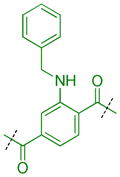
|
0.050 ± 0.025 (4) | 0.039 ± 0.0021 (3) | 0.74 ± 0.25 (3) | >10 (3) | >10 (5) | >10b (11) | >10 (4) | 8.7 ± 1.1b (10) | >10 (3) | >10 (3) | >10 (3) |
| 50 |
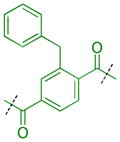
|
0.051 ± 0.029 (4) | 0.048 ± 0.0095 (3) | 1.7 ± 0.66 (3) | >10 (3) | >10 (5) | 6.8 ± 1.1b (11) | >10 (4) | >10b (10) | >10 (3) | >10 (3) | >10 (3) |
| 51 |
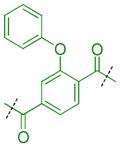
|
0.053 ± 0.026 (4) | 0.058 ± 0.0050 (3) | 3.0 ± 1.0 (3) | >10 (3) | >10 (5) | 9.3 ± 1.2b (8) | >10 (4) | >10b (9) | >10 (3) | >10 (3) | >10 (3) |
| 52 |
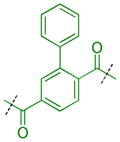
|
0.061 ± 0.026 (4) | 0.066 ± 0.011 (3) | 2.0 ± 0.72 (3) | >10 (3) | >10 (5) | >10b (11) | >10 (4) | >10b (10) | >10 (3) | >10 (3) | >10 (3) |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
Despite the fact that compound 1 demonstrated potent cellular activity and selectivity against more than 250 chromatin-associated effector proteins,21 we were interested in designing potent L3MBTL3 inhibitors with enhanced selectivity over L3MBTL1. Based on the SAR presented and the binding mode shown in Figure 1b, this should be achievable by replacing each 4-(pyrrolidin-1-yl)piperidine amine of 1 with amines that bind L3MBTL1 more weakly than compound 3. We first sought to identify a monobasic ligand (therefore limiting binding to domain 2 of each MBT protein) that selectivity antagonizes histone peptide binding to L3MBTL3 versus L3MBTL1 (Figure 3). Extensive screening of compounds that could effectively serve as methyl-lysine mimics led to the identification of compound 58, which binds L3MBTL3 with an IC50 of 1.2 μM yet has an IC50 of greater than 30 μM for L3MBTL1. This is in contrast to compound 57 which binds L3MBTL3 and L3MBTL1 equally well. Accordingly, this 2-ethylisoindoline scaffold seemed promising as a Kme mimic to bind the second domain of L3MBTL3 while enhancing selectivity by decreasing binding to the second domain of L3MBTL1. In choosing an amine to interact with the first domain of L3MBTL3, we started with those compounds in Table 3 that still have L3MBTL3 activity within 3-fold of compound 5, but that display diminished potency for L3MBTL1 as monobasic species relative to 3. For example, compound 22 is only about 2-fold weaker than 5 against L3MBTL3, while monobasic compound 59 binds L3MBTL1 weakly with an IC50 of 20 μM, suggesting 3-azabicyclo[3.2.1]octane may be a good choice for the domain 1 amine position. Similarly, 4-(pyrrolidin-2-yl)piperidine seemed promising, as 17 binds L3MBTL3 potently yet compound 60 binds both L3MBTL1 and L3MBTL3 with an IC50 greater than 30 μM.
Figure 3.
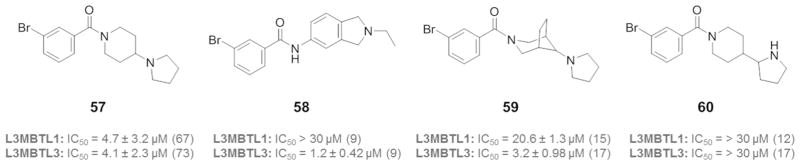
Structures of monobasic MBT domain inhibitors.
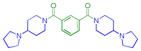
In the event, compound 53 was synthesized, and not surprisingly, binds L3MBTL1 with an IC50 of 4.0 μM (Table 7). When the 4-(pyrrolidin-1-yl)piperidine amine was replaced with 3-azabicyclo[3.2.1]octane as in compound 54, weak potency for L3MBTL1 was maintained, whereas compound 2 containing 4-(pyrrolidin-2-yl)piperidine showed an IC50 of greater than 10 μM, as presumably neither amine in compound 2 binds the domain 2 Kme pocket of L3MBTL1 very well, as intended. However, both compounds 54 and 2 retained nanomolar potency versus L3MBTL3, within 3-fold of 5. Furthermore, they both demonstrated IC50’s greater than 10 μM against the remaining Kme reader proteins in the panel, maintaining the overall selectivity of compound 5. As compound 2 appeared to be the weakest L3MBTL1 antagonist, we next prepared aniline derivatives analogous to 1, with the aniline substituent both ortho and meta to the 2-ethylisoindoline Kme mimic (55 and 56). While both of these compounds were equipotent to 2 against L3MBTL3, they gained some activity against L3MBTL1, MBTD1, and 53BP1 in the case of compound 55. Overall this suggests that the aniline substituent may increase nonspecific binding and that its location relative to the 2-ethylisoindoline is not significant in L3MBTL3 binding. While neither 55 nor 56 showed improved in vitro selectivity properties over compound 2, we expect that similarly to 1, these compounds could have a higher potency in cells.
Table 7.
Efficacy of compounds containing novel methyl-lysine mimicsa
| ID | R | LANCE IC50 (μM) | AlphaScreen IC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L3MBTL3 | L3MBTL3 | L3MBTL1 | L3MBTL4 | SFMBT1 | MBTD1 | CBX7 | 53BP1 | UHRF1 | PHF23 | JARID1A | ||
| 53 |
|
0.35 ± 0.15 (4) | 0.37 ± 0.053 (3) | 4.0 ± 1.3 (3) | >10 (3) | >10 (5) | >10 (10) | >10 (4) | >10 (8) | >10 (3) | >10 (3) | >10 (3) |
| 54 |
|
0.15 ± 0.059 (4) | 0.24 ± 0.0058 (3) | 6.8 ± 2.4 (3) | >10 (3) | >10 (5) | >10b (11) | >10 (4) | >10b (10) | >10 (3) | >10 (3) | >10 (3) |
| 2 |
|
0.16 ± 0.075 (4) | 0.17 ± 0.010 (3) | >10b (3) | >10 (3) | >10 (5) | >10b (10) | >10 (4) | >10 (9) | >10 (3) | >10 (3) | >10 (3) |
| 55 |
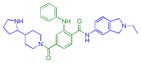
|
0.16 ± 0.035 (4) | 0.17 ± 0.023 (3) | 6.6 ± 2.0 (3) | >10 (3) | >10 (5) | 3.5 ± 1.1b (11) | >10 (4) | 3.5 ± 1.1b (10) | >10 (3) | >10 (3) | >10 (3) |
| 56 |
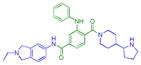
|
0.14 ± 0.045 (4) | 0.13 ± 0.025 (3) | 9.6 ± 1.2b (3) | >10 (3) | >10 (5) | 5.1 ± 1.1b (11) | >10 (4) | >10b (10) | >10 (3) | >10 (3) | >10 (3) |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
Since compounds 2 and 56 showed promise as more selective L3MBTL3 inhibitors, we sought to further quantify the level of selectivity by ITC (Table 8). Compound 2 binds L3MBTL3 with a Kd of 0.47 ± 0.14 μM and L3MBTL1 with a Kd of 68 ± 18 μM resulting in ca. 150-fold selectivity, whereas compound 56 is even more selective for L3MBTL3 with approximately 400-fold selectivity by ITC. In comparison, ITC data for compound 5 revealed that the compound was 16-fold selective for L3MBTL3 over L3MBTL1, while compound 1 is 78-fold selective for L3MBTL3 over L3MBTL1 due to its slightly higher L3MBTL3 potency by ITC.21 These data suggest that it is possible to improve the selectivity of potent L3MBTL3 inhibitors by varying the amines that bind to both the first and second MBT domain in the dimeric binding mode (Fig. 1b), and highlights the feasibility of generating novel amine scaffolds that function as Kme mimetics.
Table 8.
Inhibitor selectivity for L3MBTL3 over L3MBTL1 as determined by AlphaScreen and ITCa
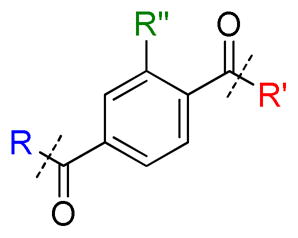
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ID | R | R′ | R″ | AlphaScreen IC50 (μM) | AlphaScreen Selectivity Ratio (L3MBTL1/L3MBTL3) | ITC Kd (μM) | ITC Selectivity Ratio (L3MBTL1/L3MBTL3) | ||
| L3MBTL3 | L3MBTL1 | L3MBTL3 | L3MBTL1 | ||||||
| 5 |
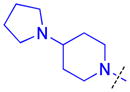
|
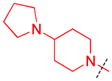
|
H | 0.071 ± 0.0023 (3) | 2.9 ± 1.3 (3) | 41 | 0.39 ± 0.044 (3) | 6.2 ± 1.9 (3) | 16 |
| 1 |
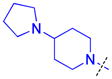
|
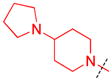
|
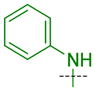
|
0.064 ± 0.02 (9) | 2.3 ± 0.85 (8) | 35.4 | 0.12 ± 0.11 (5) | 9.4 ± 1.7 (3) | 78 |
| 53 |
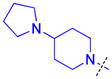
|
|
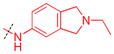
H | 0.37 ± 0.053 (3) | 4.0 ± 1.3 (3) | 11 | ND | ND | ND |
| 54 |
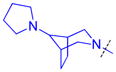
|
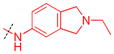
|
H | 0.24 ± 0.0058 (3) | 6.8 ± 2.4 (3) | 28 | ND | ND | ND |
| 2 |
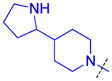
|
|
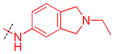
H | 0.17 ± 0.010 (3) | >10 (3)b | > 59 | 0.47 ± 0.14 (3) | 68 ± 18 (2) | 145 |
| 55 |
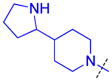
|
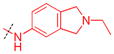
|
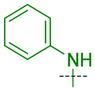
|
0.17 ± 0.023 (3) | 6.6 ± 2.0 (3) | 39 | ND | ND | ND |
| 56 |
|
|
|
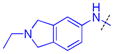
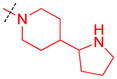
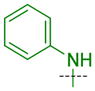
0.13 ± 0.025 (3) | 9.6 ± 1.2 (3)b | 74 | 0.35 ± 0.087 (3) | 132 ± 32 (2) | 380 |
The data for the average IC50 values was calculated from the number of replicate runs indicated in parentheses.
IC50 values were calculated by averaging data points for each compound concentration and plotted using 4-parameter curve fitting when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
In attempt to better understand the preference of the 2-ethylisoindoline Kme mimic for L3MBTL3, we performed a structural comparison of the Kme binding pockets of L3MBTL1 and L3MBTL3. Both proteins are highly similar with the major exception being a phenylalanine near the L3MBTL3 Kme reader pocket (F387) that is a leucine in L3MBTL1 (L361). Compound 58 (Figure 3) was docked into L3MBTL1 by superimposing it with compound 3 in the co-crystal structure of L3MBTL1 and 3 (pdb 3P8H).28 The docked ligand was then transferred into L3MBTL3 by protein-protein alignment. Subsequent energy optimizations of compound 58 docked to both proteins suggested that there were favorable interactions between F387 of L3MBTL3 and the 2-ethylisoindoline group of 58, stabilized by either π-π or CH-π interactions. To experimentally evaluate the contribution of F387 to binding, an L3MBTL1 L361F mutant was prepared in attempt to engineer a more ‘L3MBTL3-like’ binding pocket and the binding of the mutant protein to compounds 1, 2, and 57 was evaluated by ITC. While compounds 1 and 57 were virtually unaffected by the mutation, binding L3MBTL1 L361F within about 2-fold of wildtype L3MBTL1, compound 2 demonstrated a 12-fold increase in affinity for L3MBTL1 L361F relative to wildtype. This data supports the notion that the enhanced selectivity of 2-ethylisoindoline containing compounds is due to specific interactions with F387 in L3MBTL3 which are absent in L3MBTL1 binding. In contrast, the binding of 4-(pyrrolidin-1-yl)piperidine Kme mimics does not seem to be significantly influenced by the presence of a phenylalanine at this position.
The cytotoxicity of compound 2 was also evaluated in comparison to 1 and 5. All compounds showed no observable level of cytotoxicity up to 100 μM as measured by a CellTiter-Glo luminescent cell viability assay. Furthermore, while compound 1 was previously reported to have weak antagonist activity against the M1 muscarinic receptor (IC50 = 3.5 μM),21 compound 2 had no appreciable antagonist activity against M1 (less than 20% inhibition at 30 μM).
We next wanted to confirm the cellular activity of compounds 2 and 56. In order to demonstrate the ability of 1 to bind full length L3MBTL3 in cells, we previously reported the use of a fluorescent conjugate, mero76-1, to visualize the co-localization of the fluorescent antagonist with an N-terminal GFP fusion of the full-length L3MBTL3 protein (GFP-FLMBT).21 We therefore treated HEK293 cells containing GFP-FLMBT and mero76-1 with 1 or 5 equivalents (relative to mero76-1) of untagged competitor and observed the displacement of mero76-1 from L3MBTL3 (Figure 4). As expected, treatment with compound 1 largely displaces mero76-1 from GFP-FLMBT, eliminating mero76-1 foci at both concentrations tested. Compounds 2 and 56 were also found to be potent cellular L3MBTL3 antagonists, abolishing mero76-1 foci when the cells were treated with 5 equivalents of compound. When treating the cells with only 1 equivalent of compound, both 1 and 56 appear slightly more effective than 2 at displacing mero76-1 from L3MBTL3. Furthermore, mero76 alone (not conjugated to 1) does not co-localize with GFP-FLMBT, confirming that foci formation is dependent on binding to compound 1, and mero76-1 also does not form nuclear foci in the absence of GFP-FLMBT.
Figure 4.

Mero76-1 co-localizes with GFP-L3MBTL3 in the nucleus in HEK293 cells (row 1), and compounds 1, 2, and 56 (1 equivalent relative to mero76-1; rows 2 – 4) all displace mero76-1 from GFP-L3MBTL3 (red is mero76-1; green is GFP-L3MBTL3; blue is Hoechst). Scale bar, 10 μm.
Encouraged by the observed cellular potency of these compounds, we sought to demonstrate for the first time that L3MBTL3 antagonists bind endogenous L3MBTL3. Due to the generally low levels of endogenous L3MBTL3 expression, we investigated L3MBTL3 mRNA levels in a variety of cell lines. G-401 cells were identified as having higher levels (by 5-fold or greater) of endogenous L3MBTL3 than other cell lines, including HEK293, U2OS, and MCF10A. G-401 cells are derived from a rhabdoid tumor of the kidney, however the relevance of this higher expression level is not known. Through traditional pull-down assays we explored the ability of a biotinylated analog of 121 to affinity purify L3MBTL3 from G-401 cell lysates (Figure 5). Indeed, endogenous L3MBTL3 could be efficiently purified from whole cell lysates with this biotinylated reagent as well as competed off with soluble 1. In addition, compound 56 also demonstrated potent binding to endogenous L3MBTL3 by effectively competing with biotin-1, while the negative control compound (37) failed to compete for binding to endogenous L3MBTL3. These results together strongly suggest that compounds 1 and 56 are potent cellular inhibitors of the Kme binding function of L3MBTL3 and that neither artificial tagging of L3MBTL3 or L3MBTL3 overexpression are required for this interaction.
Figure 5.
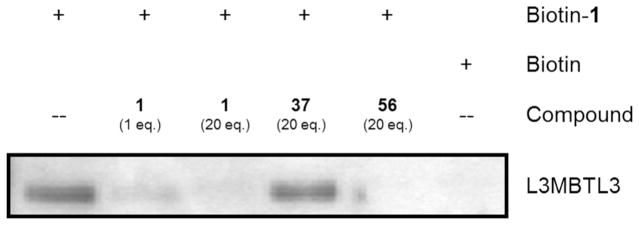
Western analysis of biotin-1 pull-down experiments in G-401 cell lysates. Biotin-1 effectively binds endogenous L3MBTL3. Compounds 1 and 56 inhibit this interaction, whereas the inactive control compound, 37, cannot successfully block L3MBTL3 binding.
CONCLUSIONS
To improve the in vitro potency of a preliminary sub-micromolar inhibitor of L3MBTL3 (4) and subsequently explore the mechanism of action of our L3MBTL3 chemical probe (1), we designed several series of novel analogs and evaluated their SARs in order to define the properties required to achieve potent L3MBTL3 antagonism. All compounds were screened against a panel of 11 Kme reader proteins by AlphaScreen, and an orthogonal L3MBTL3 TR-FRET assay was developed which further validated the observed SAR trends. Through the knowledge gained in these studies we were able to design novel antagonists (2) that maintain in vitro and cellular potency with improved selectivity against the closely related MBT-containing protein, L3MBTL1. The ability of subtle modifications, steric and electronic in nature, to the amine groups that occupy the Kme pockets of L3MBTL3 to significantly modulate affinity bodes well for the design of selective ligands for other Kme domains. It was also demonstrated that compounds 1 and 56 interact with endogenous L3MBTL3 in the context of protein pulldown assays, which is essential for the validation of tools to study the function of L3MBTL3 endogenously. Having demonstrated the chemical tractability of this target class, increased efforts towards the development of small molecule tools for additional Kme reader domains will provide new opportunities to investigate their role in chromatin biology and drug discovery.30
EXPERIMENTAL SECTION
General Chemistry Procedures
Analytical LCMS data for all compounds were acquired using an Agilent 6110 series system with the UV detector set to 220 and 254 nm. Samples were injected (<10 μL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 μm, C18 column at room temperature. A mobile phase of A (H2O + 0.1% acetic acid) and B (MeOH + 0.1% acetic acid) was used with a linear gradient from 10% to 100% B in 5.0 min, followed by a flush at 100% B for another 2 minutes with a flow rate of 1.0 mL/min. Mass spectra data were acquired in positive ion mode using an Agilent 6110 single quadrupole mass spectrometer with an electrospray ionization source. Nuclear Magnetic Resonance (NMR) spectra were recorded on a Varian Mercury spectrometer at 400 MHz for proton (1H NMR) and 100 MHz for carbon (13C NMR); chemical shifts are reported in ppm (δ). Preparative HPLC was performed using an Agilent Prep 1200 series with the UV detector set to 220 nm and 254 nm. Samples were injected onto a Phenomenex Luna 250 × 30 mm, 5 μm, C18 column at room temperature. Mobile phases of A (H2O + 0.1% TFA) and B (MeOH) were used with a flow rate of 40 mL/min. A general gradient of 0–5 minutes at 5% B, 5–20 minutes increasing from 5 to 40% B, and 20–25 minutes increasing from 40 to 80% B was used, followed by a 100% B flush for another 3 minutes. Small variations in this purification method were made as needed to achieve ideal separation for each compound. All compounds that were evaluated in biochemical and biophysical assays had >95% purity as determined by 1HNMR and LCMS.
AlphaScreen
The AlphaScreen assay was generally performed as previously described.29 In brief, compound plates (1 μL at 10 or 30 mM highest concentration; 3-fold, 10-point dilutions in DMSO) were diluted in 1× assay buffer (20 mM TRIS pH 8.0, 25 mM NaCl, 2 mM DTT and 0.05% Tween-20) over two steps to 100 or 300 μM using a Multimek robotic pipettor (Nanoscreen) and 1 μL was spotted into the wells of 384-well low-volume Proxiplates (PerkinElmer). To these plates 9 μL of protein-peptide mix in 1× assay buffer was added by Multidrop (Thermo) and incubated for 30 min at room temperature. Next, 2 μL of a 1:1 mixture of streptavidin-conjugate donor and nickel-chelate acceptor beads (45 Gg/mL in 1× assay buffer) were added and the plates were allowed to incubate for an additional 30 min in the dark at room temperature. After incubation, the plates were read on an EnVision multi-label reader equipped with an HTS AlphaScreen laser (Perkin Elmer). The screens reported were performed at concentrations up to either 10 μM or 30 μM as indicated, and therefore it should be noted that those compounds referred to as inactive are indeed inactive only within the concentration range tested. PHF23 and JARID1A are GST tagged and, consequently, GST-acceptor beads were used for these assays.
The IC50 values reported are the average of at least 3 values ± the standard deviation. When specified in Tables 1 – 7, the IC50 values were calculated using 4-paramter curve fitting (GraphPad Prism 5) from replicate runs using averaged response values for each compound concentration. This was required only when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
LANCE TR-FRET
Compound plates were treated as described for the AlphaScreen experiments. Compound plates (1 μL at 10 mM highest concentration; 3-fold, 10-point dilutions in DMSO) were diluted in 1× assay buffer (20 mM TRIS pH 8.0, 25 mM NaCl, 2 mM DTT and 0.05% Tween-20) over two steps to 100 μM using a Multimek robotic pipettor (Nanoscreen) and 1 μL was spotted into the wells of 384-well low-volume Proxiplates (PerkinElmer). To these plates 4.5 μL of a 1:1 L3MBTL3:H4K20me2 mixture (555 nM each) in 1× assay buffer was added by Multidrop (Thermo) and incubated for 30 min at room temperature. Next, 4.5 μL of a mixture of Eu-W1024-anti6×His (2.2 nM) and Ulight-streptavidin (137.5 nM) were added and the plates were allowed to incubate for an additional 30 min in the dark at room temperature. After incubation, the plates were read on an EnVision multi-label reader (Perkin Elmer) equipped with an LANCE/DELFIA D400/630 dual mirror, UV2(TRF) 320 nm excitation filter, and two emission filters(APC 665 nm and Europium 615 nM).
The IC50 values reported are the average of at least 3 values ± the standard deviation. When specified in Tables 1 – 7, the IC50 values were calculated using 4-paramter curve fitting (GraphPad Prism 5) from replicate runs using averaged response values for each compound concentration. This was required only when IC50 values for a single compound were not all active (< 10 μM) or inactive (> 10 μM).
Isothermal Titration Calorimetry
All ITC measurements were recorded at 25 °C with an AutoITC200 microcalorimeter (MicroCal Inc.). All protein and compound stock samples were in the target buffer (25 mM Tris-HCl, pH 8, 75–150 mM NaCl, and 2 mM β-mercaptoethanol), and then diluted in the same buffer to achieve the desired concentrations: 30 – 90 μM protein and 0.3 – 2 mM compound depending on the expected dissociation constant. The concentration of protein stock solutions were established using the Edelhoch method, whereas 10 mM compound stock solutions were prepared gravimetrically based on molecular weight. A typical experiment included a single 0.2 μL compound injection into a 200 μL cell filled with protein, followed by 26 subsequent 1.5 μL injections of compound. Injections were performed with a spacing of 180 seconds and a reference power of 8 Gcal/sec. Control experiments were performed titrating each compound into buffer under identical conditions to determine the heat signals, if any, that arise from diluting the compound. If applicable, the heats of dilution generated were then subtracted from the protein-compound binding curves. The titration data was analyzed using Origin Software (MicroCal Inc.) by non-linear least squares, fitting the heats of binding as a function of the compound:protein ratio. The data were fit based on a one set of sites model. L3MBTL3 binding was also fit based on one site due to the fact that two binding events were not observed, and therefore the data could not be fit based on two independent or sequential binding events.
Immunofluorescence Experiments
150,000 HEK293 cells were seeded on coverslips in 12-well plates. After 24 hours, cells were transfected with GFP-L3MBTL3, and after another 16 hours they were treated with mero76-1 (5 μM). After a 3 hour incubation period, cells were treated with 5 μM or 25 μM of compound 1, 2, or 56 for an additional 3 hours. Prior to imaging, cells were stained with Hoechst 33342 dye, washed with PBS, and incubated with DMEM/F-12 media without phenol red (11039). Images were acquired using a Quorum Spinning Disk Confocal microscope equipped with 405, 491, 561, and 642 nm lasers. Image analysis was performed using Volocity 5.4.1.
Affinity Purification Experiments
Frozen G-401 cell pellets were lysed in lysis buffer containing 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, and complete protease inhibitor cocktail (Roche). After vortexing and centrifugation at 13,000 rpm, 4 °C, 5 min to pellet the DNA, the supernatant was diluted to 150 mM NaCl (while keeping other reagents’ concentrations constant). The lysate concentration was determined by a BCA colorimetric assay (Pierce). Approximately 1 mg of cell lysate was used per immunoprecipitation. Lysate was incubated in the absence or presence of compound (1 or 20 equivalents relative to biotin-1; 1.75 nmol or 35 nmol respectively) at 4 °C for 1 hour. Magnetic streptavidin beads (50 μL, binding capacity is 1.75 nmol; Pierce) pre-washed with IP buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and protease inhibitor cocktail (Roche)) were saturated with biotin-1 (or biotin as a negative control). The beads were incubated with biotin-1 or biotin (35 nmol) in 200 μL IP buffer for 30 min at room temperature. Three washes were performed to remove all excess biotin-1 (or biotin), and then the beads were resuspended in the corresponding lysate and rotated overnight at 4 °C. Three washes were performed with IP buffer, followed by elution with 1× NuPAGE Sample Reducing Agent (Invitrogen) and 1× NuPAGE LDS Sample Buffer (Invitrogen), analysis by SDS-PAGE, and immunoblotting.
Supplementary Material
Acknowledgments
We thank Emily Hull-Ryde for support with the CellTiter-Glo cell viability assay, Chris MacNevin and Klaus Hahn for providing mero76, Xi-Ping for support with the M1 muscarinic receptor assay, Greg Wang for providing PHF23 and JARID1 proteins, and Timothy Willson and John Moore for helpful discussions and access to resources for the endogenous L3MBTL3 pulldown assays. The research described here was supported by the National Institute of General Medical Sciences, US National Institutes of Health (NIH, grant RC1GM090732 and R01GM100919), the Carolina Partnership and the University Cancer Research Fund, University of North Carolina at Chapel Hill, the Ontario Research Fund (grant ORF-GL2), the Natural Sciences and Engineering Research Council of Canada, and the Structural Genomics Consortium which is a registered charity (number 1097737) that receives funds from AbbVie, Boehringer Ingelheim, Canada Foundation for Innovation, the Canadian Institutes for Health Research (CIHR), Genome Canada through the Ontario Genomics Institute [OGI-055], GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]. L.K. holds a Frederick Banting and Charles Best Canada Graduate Scholarship from the CIHR and C.H.A. holds a Canada Research Chair in Structural Genomics.
ABBREVIATIONS USED
- Kme
methyl-lysine
- Kme2
dimethyl-lysine
- SAR
structure activity relationships
- PTM
post-translational modification
- MBT
malignant brain tumor
- L3MBTL3
Lethal(3)malignant brain tumor-like protein 3
- L3MBTL1
Lethal(3)malignant brain tumor-like protein 1
- TR-FRET
time resolved fluorescence resonance energy transfer
- H4K20me1-2
mono- or dimethylation of lysine 20 on histone 4
- ITC
isothermal titration calorimetry
- GFP
green fluorescent protein
Footnotes
Supporting Information. The synthesis of compounds 1 – 60, protein purification and expression details, experimental details of other in vitro results, and supplementary figures (ITC, CellTiter-Glo, M1 and M2 functional assays, cellular localization results, L3MBTL3 mRNA levels, and 1H NMR spectra) are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007;8(12):983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13(5):343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 4.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frye SV. The art of the chemical probe. Nat Chem Biol. 2010;6(3):159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- 7.Structural Genomics Consortium. http://www.thesgc.org/scientists/chemical_probes.
- 8.Itoh Y, Suzuki T, Miyata N. Small-molecular modulators of cancer-associated epigenetic mechanisms. Mol Biosyst. 2013;9(5):873–896. doi: 10.1039/c3mb25410k. [DOI] [PubMed] [Google Scholar]
- 9.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7(8):566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8(11):890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 11.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian XR, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 12.Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Aller GSV, Machutta CA, Diaz E, Jiang Y, Johnson NW, Knight SD, Kruger RG, McCabe MT, Dhanak D, Tummino PJ, Creasy CL, Miller WH. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Medicinal Chemistry Letters. 2012;3(12):1091–1096. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang JH, Yan X, Fang J, Mi Y, Zhang M, Zhou T, Feng G, Chen Z, Li G, Yang T, Zhao K, Liu X, Yu Z, Lu CX, Atadja P, Li E. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109(52):21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M, Jin J. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8(6):1324–1334. doi: 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Pollock RM, Kuntz KW, Keilhack H. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez-Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, Van Aller GS, Kruger RG, Liu Y, McHugh CF, Scott DW, Chung YR, Kelleher N, Shaknovich R, Creasy CL, Gascoyne RD, Wong KK, Cerchietti L, Levine RL, Abdel-Wahab O, Licht JD, Elemento O, Melnick AM. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell. 2013;23(5):677–692. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu W, Chory EJ, Wernimont AK, Tempel W, Scopton A, Federation A, Marineau JJ, Qi J, Barsyte-Lovejoy D, Yi J, Marcellus R, Iacob RE, Engen JR, Griffin C, Aman A, Wienholds E, Li F, Pineda J, Estiu G, Shatseva T, Hajian T, Al-Awar R, Dick JE, Vedadi M, Brown PJ, Arrowsmith CH, Bradner JE, Schapira M. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 18.Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, Jin L, Kuntz KW, Chesworth R, Moyer MP, Bernt KM, Tseng JC, Kung AL, Armstrong SA, Copeland RA, Richon VM, Pollock RM. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herold JM, Ingerman LA, Gao C, Frye SV. Drug discovery toward antagonists of methyl-lysine binding proteins. Curr Chem Genomics. 2011;5:51–61. doi: 10.2174/1875397301005010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.James LI, Frye SV. Targeting chromatin readers. Clin Pharmacol Ther. 2013;93(4):312–314. doi: 10.1038/clpt.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.James LI, Barsyte-Lovejoy D, Zhong N, Krichevsky L, Korboukh VK, Herold JM, Macnevin CJ, Norris JL, Sagum CA, Tempel W, Marcon E, Guo H, Gao C, Huang XP, Duan S, Emili A, Greenblatt JF, Kireev DB, Jin J, Janzen WP, Brown PJ, Bedford MT, Arrowsmith CH, Frye SV. Discovery of a chemical probe for the L3MBTL3 methyllysine reader domain. Nat Chem Biol. 2013;9(3):184–191. doi: 10.1038/nchembio.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonasio R, Lecona E, Reinberg D. MBT domain proteins in development and disease. Semin Cell Dev Biol. 2010;21(2):221–230. doi: 10.1016/j.semcdb.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trojer P, Li G, Sims RJ, 3rd, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, Wang YH, Reinberg D. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129(5):915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 24.Arai S, Miyazaki T. Impaired maturation of myeloid progenitors in mice lacking novel Polycomb group protein MBT-1. EMBO J. 2005;24(10):1863–1873. doi: 10.1038/sj.emboj.7600654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy S, Musselman CA, Kachirskaia I, Hayashi R, Glass KC, Nix JC, Gozani O, Appella E, Kutateladze TG. Structural insight into p53 recognition by the 53BP1 tandem Tudor domain. J Mol Biol. 2010;398(4):489–496. doi: 10.1016/j.jmb.2010.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herold JM, James LI, Korboukh VK, Gao C, Coil KE, Bua DJ, Norris JL, Kireev DB, Brown PJ, Jin J, Janzen WP, Gozani O, Frye SV. Structure–activity relationships of methyl-lysine reader antagonists. MedChemComm. 2012;3(45):45–51. [Google Scholar]
- 28.Herold JM, Wigle TJ, Norris JL, Lam R, Korboukh VK, Gao C, Ingerman LA, Kireev DB, Senisterra G, Vedadi M, Tripathy A, Brown PJ, Arrowsmith CH, Jin J, Janzen WP, Frye SV. Small-molecule ligands of methyl-lysine binding proteins. J Med Chem. 2011;54(7):2504–2511. doi: 10.1021/jm200045v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wigle TJ, Herold JM, Senisterra GA, Vedadi M, Kireev DB, Arrowsmith CH, Frye SV, Janzen WP. Screening for inhibitors of low-affinity epigenetic peptide-protein interactions: an AlphaScreen-based assay for antagonists of methyl-lysine binding proteins. J Biomol Screen. 2010;15(1):62–71. doi: 10.1177/1087057109352902. [DOI] [PubMed] [Google Scholar]
- 30.Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo JL, Patel DJ, Allis CD. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459(7248):847–851. doi: 10.1038/nature08036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.