

Abstract
Purpose
Children with von Hippel-Lindau syndrome are at an increased risk for developing bilateral pheochromocytomas. In an effort to illustrate the advantage of partial adrenalectomy (PA) over total adrenalectomy in children with VHL, we report the largest single series on PA for pediatric VHL patients, demonstrating a balance between tumor removal and preservation of adrenocortical function.
Methods
From 1994 to 2011, a prospectively maintained database was reviewed to evaluate 10 pediatric patients with hereditary pheochromocytoma for PA. Surgery was performed if there was clinical evidence of pheochromocytoma and normal adrenocortical tissue was evident on preoperative imaging and/or intraoperative ultrasonography. Perioperative data were collected and patients were followed for postoperative steroid use and tumor recurrence.
Results
Ten pediatric patients with a diagnosis of VHL underwent 18 successful partial adrenalectomies (4 open, 14 laparoscopic). The median tumor size removed was 2.6 centimeters (range 1.2–6.5). Over a median follow up of 7.2 years (range 2.6–15.8) additional tumors in the ipsilateral adrenal gland were found in two patients. One patient underwent completion adrenalectomy and one underwent a salvage PA with resection of the ipsilateral lesion. One patient required short term steroid replacement therapy. At last follow up, 7 patients had no radiographic or laboratory evidence of pheochromocytoma.
Conclusion
At our institution, partial adrenalectomy is the preferred form of management for pheochromocytoma in the (VHL) pediatric population. This surgical approach allows for removal of tumor while preserving adrenocortical function and minimizing the side effects of long term steroid replacement on puberty and quality of life.
Keywords: adrenalectomy, partial adrenalectomy, pediatric VHL, pheochromocytoma
Background
Hereditary pheochromocytoma genetic disorders such as von Hippel-Lindau syndrome (VHL) are associated with an increased risk for the development of multiple, recurrent, and bilateral adrenal tumors 1,2. Historically, total adrenalectomy (TA) was utilized for the management of adrenal masses in the hereditary setting. Several series have reported on the oncologic efficacy of total adrenalectomy in hereditary tumor syndromes, with local recurrence rates reported to be 14% to 22% 3,4. However, total adrenalectomy places patients at a high risk of developing adrenal insufficiency and lifelong exogenous steroid dependence. Adrenal insufficiency is associated with a significant depreciation in quality of life and health including osteoporosis, weight gain, loss of libido, and risk of Addisonian crisis5,6. These problems have a much more significant impact on quality of life in the pediatric population.
Partial adrenalectomy has been previously underutilized but is gaining popularity worldwide7. At our institution, we have reported performing both laparoscopic and robotic PA with excellent outcomes8,9. Indications for PA include hereditary adrenal tumors, bilateral tumors, and tumors in a solitary adrenal gland. The safety and feasibility of PA, particularly by laparoscopic techniques, has been demonstrated by several groups10–17. Much like in adults, preservation of functional adrenal tissue is important in pediatric patients to maintain the physiologic hormonal surges required for homeostatic maintenance18,19. We advocate for partial adrenalectomy (PA) whenever possible in the pediatric population.
Despite multiple smaller studies and case reports, there are no large single center reports on PA in the pediatric hereditary population to our knowledge. We report our single institution contemporary series of PA in the pediatric VHL population.
MATERIALS AND METHODS
Ten patients (18 years of age or younger) with adrenal masses presented to our institution from December of 1994 to July of 2008 and underwent a total of 18 partial adrenalectomy (PA) procedures under an IRB protocol (IRB protocol number 97-c-0147). Germline mutation analysis was performed on all patients with parent or guardian consent. All patients had hereditary adrenal pheochromocytomas associated with von Hippel-Lindau syndrome (VHL). Patients were screened preoperatively with plasma and urine catecholamines, as well as either computed tomography (CT) or magnetic resonance imaging (MRI).
In preparation for surgery, patients underwent a two week preoperative blockade with oral metyrosine 250mg three times day, phenoxybenzamine 10mg twice a day, and atenolol 25mg once a day if needed29. They were admitted one week prior to surgery for inpatient observation and titration of medications in conjunction with the pediatric endocrinology service. Blockade was considered sufficient when blood pressure was normalized and the symptoms of pheochromocytoma abated. Postoperatively, patients were transferred to the intensive care unit to monitor for hemodynamic instability and for pain control.
Patients with pheochromocytomas were treated with either an open or minimally invasive (laparoscopic or robotic-assisted) PA. Three of the authors (GB, AM, PP) have experience operating on VHL patients with multiple tumors and performed PA in this study. Minimally invasive PA was performed in 14 (12 lap, 2 robotic) of the 18 procedures. Open surgery was performed in 4 procedures The decision to perform open surgery was dependent on the minimally invasive skill set of the surgeon and was also preferred in one patient (case 7) undergoing concomitant distal pancreatectomy. Bilateral operations performed synchronously were considered to be two distinct partial adrenalectomies in this study.
Minimally invasive PA was performed via transperitoneal access using a three or four-port technique with the patient in a modified flank position. Intraoperative ultrasonography and ultrasonic shears (Harmonic scalpel, Ethicon, Cincinnati, Ohio) were used in all minimally invasive cases. Specific operative techniques were previously described by Rogers et al.20 Tumors were resected with the help of intraoperative ultrasound which aided the surgeon in delineating a plane between normal and involved tissue. This allowed for the maximal preservation of normal adrenal tissue and its vascular supply. Open partial adrenalectomy was done through a chevron or flank approach. Tumors in the central portion of the gland were resected while dissection of the normal adrenal tissue and its pedicle were avoided to preserve vascular supply. Patients with a small adrenal remnant and sustained low blood pressure were considered at risk for Addisonian crisis after surgery and were given cortisol supplementation until ACTH stimulation testing at approximately two months demonstrated adequate adrenocortical function.
Data collection included tumor size (radiographic and pathologic), postoperative adrenal function (based on the need for adrenal hormone replacement), and tumor recurrence (based on postoperative plasma and 24-hour urine catecholamine levels and abdominal CT or MRI imaging). Elevated blood pressures, headaches, palpitations, dizziness, anxiety or any other possible symptoms of pheochromocytoma were recorded.
RESULTS
A total of 10 patients in VHL families (8 males, 2 females, Table 1) presented with adrenal masses. VHL germline missense mutations were confirmed in all patients; the most common resulted in a substitution of glutamine for arginine at the 167th codon. The procedures were done at a mean age of 14.3 years (range 6–18). Two patients (Case 3 & 9, Table 1) underwent partial adrenalectomy on a solitary adrenal gland. One patient (Case 8) had bilateral open partial adrenalectomies at another center before coming to our institution.
Table 1.
Clinical Characteristics and Outcomes
| Case No | Age* | Gender | Mutation | Largest lesion size (cm) | PA Operationsa | Recurrence site/time | Path | Follow up (yrs) | Steroids |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 17 | M | 167: Arg>Gln | 3.0 | lap-RPA | None | Pheo | 15.63 | No |
| 2 | 13 | M | 167: Arg>Gln | 2.7 | lap-LPA, lap-BPA | None | Pheo | 15.77 | No† |
| 3 | 13 | M | 128: Leu>Phe | 2.5 | open-LPA | None | Pheo | 12.74 | No |
| 4 | 13 | M | 167: Leu>Gln | 4.1 | lap-LPA, lap-RPA | None | Composite | 9.41 | No |
| 5 | 15 | F | 161: Leu>Gln | 1.4 | lap-LPA, lap-RPA | None | Pheo | 2.56 | No |
| 6 | 6 | M | 124: Thr>Ile | 3.5 | lap-LPA, lap-RPA | Right/5mos | Composite | 4.58 | No |
| 7 | 17 | F | 149: Ala>Ser | 2.0 | open-BPA | None | Pheo | ** | ** |
| 8 | 13 | M | 167: Leu>Gln | 1.5 | lap-RPA | None | Pheo | 5.00 | No |
| 9 | 16 | M | 167: Arg>Gln | 6.5 | open-RPA, lap-RPA | Right/1yr | Pheo | 4.06 | No |
| 10 | 15 | M | 129: Leu>Pro | 5.5 | robo-LPA, robo-RPA | None | Pheo | 4.42 | Yes x2 years post-op |
Age at first operation
Post-operative death
Operations are in chronological order and do not include total adrenalectomies.
Patient was steroid independent for 10 years post operatively. He has recently begun steroid supplementation after removal of an ACTH secreting ectopic tumor in the lung.
lap - laparoscopic
robo - robotic
LPA – left partial adrenalectomy
RPA – right partial adrenalectomy
BPA – bilateral partial adrenalectomy
Post-operative analysis (Table 2) of 18 PA procedures included a median tumor size of 2.6 cm (range 1.2–6.5cm). There was a median operative time in open cases of 353 minutes (range 255–450), laparoscopic 300 minutes (range 170–600), and robotic 273 minutes (range 245–300). There was a median estimated blood loss in open cases of 725 cc (range 700–750), 125 cc in laparoscopic cases (range 50–700), and 100 cc in robotic cases (range 50–150). One patient (Case 7) was not included in the analysis of operative parameters because her anesthesia record could not be found. Final pathology confirmed pheochromocytoma in all but 2 patients; they were found to have composite pheochromocytomas. We did not evaluate statistical significance due to our limited sample size.
Table 2.
Operative Characteristics
| Total Number (n) | ||
|---|---|---|
| Total adrenal units operated on | 18 | |
| Open | 4 | |
| Lap | 12 | |
| Robotic | 2 | |
| Median | Range | |
| Gross tumor size (cm) | 2.6 | 1.2 – 6.5 |
| Operative time (min) | 300 | 170 – 600 |
| Open | 353 | 255 – 450 |
| Lap | 300 | 170 – 600 |
| Robotic | 273 | 245 – 300 |
| Median | Range | |
| Estimated blood loss | 150 | 50 – 750 |
| Open | 725 | 700 – 750 |
| Lap | 125 | 50 – 700 |
| Robotic | 100 | 50 – 150 |
Case 7 not included in this analysis
During intraoperative manipulation of his tumor one patient (Case 6) had a rise in blood pressure that required a nitroprusside drip. This patient stabilized postoperatively. Two patients (Cases 4 & 10) had postoperative hypotension that required corticosteroids until discharge. Both patients had previous partial adrenalectomies on the contralateral gland. One patient (Case 4) was tapered off of steroids before discharge. The other (Case 10) remained on oral steroids for 2 years after discharge until he was weaned off by his local endocrinologist.
There was one death in our series (Case 7). This patient received open bilateral partial adrenalectomies with a simultaneous distal pancreatectomy. Ten days postoperatively, this patient developed a small bowel obstruction and underwent an exploratory laparotomy which revealed an incarcerated segment of small intestine. This segment was repaired and the patient was transferred to the ICU post-operatively where she experienced asystole and was unable to be resuscitated.
Tumor recurrence was assessed over a median follow-up of 7.2 years (range 2.6–15.8, Table 1). An additional tumor occurred in the ipsilateral adrenal gland of two patients (Cases 6 & 9). Case 6 underwent a completion total adrenalectomy on that side and Case 9 underwent repeat PA with resection of the recurrent ipsilateral lesion. One patient (Case 2) was steroid independent for ten years after bilateral PA. However, an ectopic pulmonary ACTH producing tumor was removed in 2010 and he subsequently required daily steroid supplementation.
At last follow up, 7 of 9 living patients had no radiographic or laboratory evidence of pheochromocytoma. One patient has increased plasma norepinephrine but no symptoms or other evidence of pheochromocytoma. The other patient has had episodic heart palpitations but also does not have further evidence of recurrent disease.
DISCUSSION
Adrenal hormones are critical for normal physiologic development and maintenance of the hypothalamic-pituitary adrenocortical axis in all ages. Total bilateral adrenalectomy creates a surgical Addisonian state that requires lifelong steroid replacement in the pediatric population. Partial adrenalectomy was described by Albanese et al. in non-hereditary pediatric patients with bilateral tumors21. In our series, we explored the use of partial adrenalectomy in a hereditary pediatric population. These children are more likely to present with bilateral disease and develop additional primary tumors over time. TA in this population is problematic due to the increased risk of developing new primary tumors within the contra-lateral gland, which when treated with total adrenalectomy, would render these patients adrenally insufficient for life.
Adrenal insufficiency results in a lifelong medical risk of Addisonian crisis and is associated with a lower quality of life in many cases.28 Pediatric patients bear a disproportionate burden of this risk due to substantially longer life expectancy than their adult counterparts5,25. PA has evolved as a strategy to balance the risk of local tumor recurrence with operative morbidity and long-term effects of steroid replacement.
Diner et al. described favorable outcomes after PA in a 33 patient cohort, most of whom had hereditary adrenal tumors. They demonstrated good preservation of adrenal function at a mean follow-up of 3 years (range 0.4–10 years)10. Adrenal hormone replacement was initially required in 5 patients, but four of these patients were weaned off of steroids within two months. Similarly, in our series one patient was steroid dependent at two months postoperatively (Case 10). This patient received a PA on a solitary adrenal gland and the size of this lesion was approximately 3 cm. This patient was followed by an endocrinologist and was successfully weaned off of steroids two years post-operatively. At last follow up, this patient is still steroid independent.
Rates of pheochromocytoma recurrence after PA in the hereditary population vary from 0–33% 22–25. This variability may be related to the duration of follow-up as well as the relative definition of recurrence, with de novo development of tumor being impossible to distinguish from true recurrence in the hereditary population. Recurrences may be seen as late as 10 years or longer23. The risk of recurrent tumors in the adrenal remnant is theoretically due to medullary cells that inevitably remain in the spared cortex. Such “recurrences” are thought to be derived from the formation of new tumors due to genetic predisposition rather than recurrence from incomplete resection of the original tumor24. One patient in this cohort (Case 1) likely had such a de novo tumor, although not included in our analysis because he was no longer a pediatric patient at the time of the original operation. He received a contralateral PA at age 19 and another operation on the same side at age 31 – the 12 year lapse is consistent the development of a new tumor.
We report additional tumors in 2 of 10 patients and do believe that these are true recurrences. Case 6 presented with rapid recurrence and received the subsequent operation just five months following the primary surgery, suggesting an incomplete resection. This patient had a 3.5cm tumor and final pathology demonstrated a composite pheochromocytoma. This pathological description is rarely addressed in the literature and some reports that have associated this histology with larger tumors at presentation26. It is possible that the size at surgery and the unique behavior may have contributed to the rapid recurrence in this patient. The recurrence in Case 9 occurred just one year following the primary PA, again suggesting true recurrence. He had a large 6.5cm tumor which likely made complete evacuation technically difficult and resulted in a positive margin. This patient also had a previous TA making his subsequent re-operation a PA on a solitary adrenal gland. In the instances of local tumor recurrence, previous groups have demonstrated the feasibility of repeat PA using laparoscopic techniques in the adult hereditary pheochromocytoma population24,27. Our experience supports this data as most patients can still safely undergo laparoscopic or robot-assisted salvage partial adrenalectomy.
Though treatment with PA may spare children with VHL from a lifetime of hormone replacement, the nature of the disease makes continued screening of utmost importance. We recommend that children be seen at least every 6 months for 1 to 2 years postoperatively and then annually after that to evaluate for signs and symptoms of recurrent pheochromocytoma. In addition to a history and physical and blood pressure screening at these intervals, we test for elevations in urinary or plasma catecholamines/metanephrines.
Furthermore, our patients with VHL undergo yearly imaging. In pediatric patients we opt for screening with MRI whenever possible to avoid the cumulative exposure to ionizing radiation and the increased risk of secondary malignancies later in life. However, VHL patients have a multitude of other systemic manifestations aside from pheochromocytoma. As a result, many of the patients in this study underwent CT imaging as part of a screening or follow up protocol to evaluate for tumors in other organ systems, especially for pancreatic and kidney lesions. While the frequency of follow up may seem to be a disadvantage over TA, patients with VHL must be continually screened regardless, as they are at risk for development of benign and malignant lesions throughout the body.
CONCLUSIONS
At our institution, partial adrenalectomy is the preferred surgical management for the pediatric population with hereditary pheochromocytomas. Due to the genetic predisposition for developing recurrent tumors, complete surgical tumor eradication may never be possible with one operative procedure. Our surgical experience has demonstrated that PA provides adequate long-term preservation of adrenal cortical function without compromising local tumor control. Partial adrenalectomy preserves adrenal function for subsequent operations and protects many of these patients from the risks associated with long-term steroid replacement.
Figure 1.
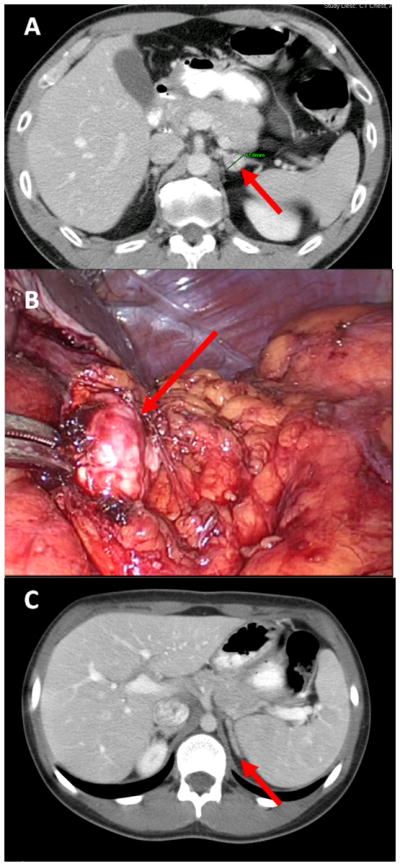
A. Pre-operative imaging of left adrenal pheochromocytoma.
B. Intra-operative image of the left adrenal pheochromocytoma during laparoscopic partial adrenalectomy.
C. Post-operative image (28 months) of left adrenal gland after partial adrenalectomy
Acknowledgments
This research was supported [in part] by the Intramural Research Program of the National Cancer Institute, NIH
References
- 1.Walther MM, Keiser HR, Choyke PL, Rayford W, Lyne JC, Linehan WM. Management of hereditary pheochromocytoma in von Hippel-Lindau kindreds with partial adrenalectomy. J Urol. 1999 Feb;161(2):395–398. [PubMed] [Google Scholar]
- 2.Neumann HP, Berger DP, Sigmund G, et al. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med. 1993 Nov 18;329(21):1531–1538. doi: 10.1056/NEJM199311183292103. [DOI] [PubMed] [Google Scholar]
- 3.Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997 May;29(5):1133–1139. doi: 10.1161/01.hyp.29.5.1133. [DOI] [PubMed] [Google Scholar]
- 4.Neumann HP, Reincke M, Bender BU, Elsner R, Janetschek G. Preserved adrenocortical function after laparoscopic bilateral adrenal sparing surgery for hereditary pheochromocytoma. J Clin Endocrinol Metab. 1999 Aug;84(8):2608–2610. doi: 10.1210/jcem.84.8.5872. [DOI] [PubMed] [Google Scholar]
- 5.Telenius-Berg M, Ponder MA, Berg B, Ponder BA, Werner S. Quality of life after bilateral adrenalectomy in MEN 2. Henry Ford Hosp Med J. 1989;37(3–4):160–163. [PubMed] [Google Scholar]
- 6.Hahner S, Loeffler M, Fassnacht M, et al. Impaired subjective health status in 256 patients with adrenal insufficiency on standard therapy based on cross-sectional analysis. J Clin Endocrinol Metab. 2007 Oct;92(10):3912–3922. doi: 10.1210/jc.2007-0685. [DOI] [PubMed] [Google Scholar]
- 7.Kaye DR, Storey BB, Pacak K, Pinto PA, Linehan WM, Bratslavsky G. Partial adrenalectomy: underused first line therapy for small adrenal tumors. J Urol. 2010 Jul;184(1):18–25. doi: 10.1016/j.juro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boris RS, Gupta G, Linehan WM, Pinto PA, Bratslavsky G. Robot-assisted laparoscopic partial adrenalectomy: initial experience. Urology. 2011 Apr;77(4):775–780. doi: 10.1016/j.urology.2010.07.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asher KP, Gupta GN, Boris RS, Pinto PA, Linehan WM, Bratslavsky G. Robot-assisted laparoscopic partial adrenalectomy for pheochromocytoma: the National Cancer Institute technique. Eur Urol. 2011 Jul;60(1):118–124. doi: 10.1016/j.eururo.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diner EK, Franks ME, Behari A, Linehan WM, Walther MM. Partial adrenalectomy: the National Cancer Institute experience. Urology. 2005 Jul;66(1):19–23. doi: 10.1016/j.urology.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Janetschek G, Finkenstedt G, Gasser R, et al. Laparoscopic surgery for pheochromocytoma: adrenalectomy, partial resection, excision of paragangliomas. J Urol. 1998 Aug;160(2):330–334. doi: 10.1016/s0022-5347(01)62886-6. [DOI] [PubMed] [Google Scholar]
- 12.Walther MM, Herring J, Choyke PL, Linehan WM. Laparoscopic partial adrenalectomy in patients with hereditary forms of pheochromocytoma. J Urol. 2000 Jul;164(1):14–17. [PubMed] [Google Scholar]
- 13.Al-Sobhi S, Peschel R, Zihak C, Bartsch G, Neumann H, Janetschek G. Laparoscopic partial adrenalectomy for recurrent pheochromocytoma after open partial adrenalectomy in von Hippel-Lindau disease. J Endourol. 2002 Apr;16(3):171–174. doi: 10.1089/089277902753716142. [DOI] [PubMed] [Google Scholar]
- 14.Sasagawa I, Suzuki Y, Itoh K, et al. Posterior retroperitoneoscopic partial adrenalectomy: clinical experience in 47 procedures. Eur Urol. 2003 Apr;43(4):381–385. doi: 10.1016/s0302-2838(03)00087-3. [DOI] [PubMed] [Google Scholar]
- 15.Walz MK, Peitgen K, Diesing D, et al. Partial versus total adrenalectomy by the posterior retroperitoneoscopic approach: early and long-term results of 325 consecutive procedures in primary adrenal neoplasias. World J Surg. 2004 Dec;28(12):1323–1329. doi: 10.1007/s00268-004-7667-y. [DOI] [PubMed] [Google Scholar]
- 16.Al-Sobhi S, Peschel R, Bartsch G, Gasser R, Finkenstedt G, Janetschek G. Partial laparoscopic adrenalectomy for aldosterone-producing adenoma: short-and long-term results. J Endourol. 2000 Aug;14(6):497–499. doi: 10.1089/end.2000.14.497. [DOI] [PubMed] [Google Scholar]
- 17.Jeschke K, Janetschek G, Peschel R, Schellander L, Bartsch G, Henning K. Laparoscopic partial adrenalectomy in patients with aldosterone-producing adenomas: indications, technique, and results. Urology. 2003 Jan;61(1):69–72. doi: 10.1016/s0090-4295(02)02240-9. discussion 72. [DOI] [PubMed] [Google Scholar]
- 18.Beltsevich DG, Kuznetsov NS, Kazaryan AM, Lysenko MA. Pheochromocytoma surgery: epidemiologic peculiarities in children. World J Surg. 2004 Jun;28(6):592–596. doi: 10.1007/s00268-004-7134-9. [DOI] [PubMed] [Google Scholar]
- 19.Bleicken B, Hahner S, Loeffler M, et al. Influence of hydrocortisone dosage scheme on health-related quality of life in patients with adrenal insufficiency. Clin Endocrinol (Oxf) 2010 Mar;72(3):297–304. doi: 10.1111/j.1365-2265.2009.03596.x. [DOI] [PubMed] [Google Scholar]
- 20.Rogers CG, Blatt AM, Miles GE, Linehan WM, Pinto PA. Concurrent robotic partial adrenalectomy and extra-adrenal pheochromocytoma resection in a pediatric patient with von Hippel-Lindau disease. J Endourol. 2008 Jul;22(7):1501–1503. doi: 10.1089/end.2007.0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Albanese CT, Wiener ES. Routine total bilateral adrenalectomy is not warranted in childhood familial pheochromocytoma. J Pediatr Surg. 1993 Oct;28(10):1248–1251. doi: 10.1016/s0022-3468(05)80307-0. discussion 1251–1242. [DOI] [PubMed] [Google Scholar]
- 22.Inabnet WB, Caragliano P, Pertsemlidis D. Pheochromocytoma: inherited associations, bilaterality, and cortex preservation. Surgery. 2000 Dec;128(6):1007–1011. doi: 10.1067/msy.2000.110846. discussion 1011–1002. [DOI] [PubMed] [Google Scholar]
- 23.Lee JE, Curley SA, Gagel RF, Evans DB, Hickey RC. Cortical-sparing adrenalectomy for patients with bilateral pheochromocytoma. Surgery. 1996 Dec;120(6):1064–1070. doi: 10.1016/s0039-6060(96)80056-0. discussion 1070–1061. [DOI] [PubMed] [Google Scholar]
- 24.Nambirajan T, Leeb K, Neumann HP, Graubner UB, Janetschek G. Laparoscopic adrenal surgery for recurrent tumours in patients with hereditary phaeochromocytoma. Eur Urol. 2005 May;47(5):622–626. doi: 10.1016/j.eururo.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Benhammou JN, Boris RS, Pacak K, Pinto PA, Linehan WM, Bratslavsky G. Functional and oncologic outcomes of partial adrenalectomy for pheochromocytoma in patients with von Hippel-Lindau syndrome after at least 5 years of followup. J Urol. 2010 Nov;184(5):1855–1859. doi: 10.1016/j.juro.2010.06.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lam KY, Lo CY. Composite Pheochromocytoma-Ganglioneuroma of the Adrenal Gland: An Uncommon Entity with Distinctive Clinicopathologic Features. Endocr Pathol. 1999 Winter;10(4):343–352. doi: 10.1007/BF02739777. [DOI] [PubMed] [Google Scholar]
- 27.Janetschek G, Neumann HP. Laparoscopic surgery for pheochromocytoma. Urol Clin North Am. 2001 Feb;28(1):97–105. doi: 10.1016/s0094-0143(01)80011-2. [DOI] [PubMed] [Google Scholar]
- 28.Hawn MT, Cook D, Deveney C, Sheppard BC. Quality of life after laparascopic bilateral adrenalectomy for Cushing’s disease. Surgery. 2002;132:1064. doi: 10.1067/msy.2002.128482. [DOI] [PubMed] [Google Scholar]
- 29.Pacak K. Preoperative Management of the Pheochromocytoma Patient. J Clin Endocrinol Metab. 2007 Nov;92(11):4069–4079. doi: 10.1210/jc.2007-1720. [DOI] [PubMed] [Google Scholar]