

Abstract
We previously demonstrated that Transforming Growth Factor (TGF) β1 suppresses IgE-mediated signaling in human and mouse mast cells in vitro, an effect that correlated with decreased expression of the high affinity IgE receptor, FcεRI. The in vivo effects of TGFβ1 and the means by which it suppresses mast cells have been less clear. The current study shows that TGFβ1 suppresses FcεRI and c-Kit expression in vivo. By examining changes in cytokine production concurrent with FcεRI expression, we found that TGFβ1 suppresses TNF production independent of FcεRI levels. Rather, IgE-mediated signaling was altered. TGFβ1 significantly reduced expression of Fyn and Stat5, proteins critical for cytokine induction. These changes may partly explain the effects of TGFβ1, since Stat5B overexpression blocked TGF-mediated suppression of IgE-induced cytokine production. We also found that Stat5B is required for mast cell migration toward SCF, and that TGFβ1 reduced this migration. We found evidence that genetic background may alter TGF responses. TGFβ1 greatly reduced mast cell numbers in Th1-prone C57BL/6 but not Th2-prone 129/Sv mice. Furthermore, TGFβ1 did not suppress IgE-induced cytokine release, and increased c-Kit-mediated migration in 129/Sv mast cells. These data correlated with high basal Fyn and Stat5 expression in 129/Sv cells, which was not reduced by TGFβ1 treatment. Finally, primary human mast cell populations also showed variable sensitivity to TGFβ1-mediated changes in Stat5 and IgE-mediated IL-6 secretion. We propose that TGFβ1 regulates mast cell homeostasis, and that this feedback suppression may be dependent upon genetic context, predisposing some individuals to atopic disease.
Keywords: asthma, allergy, Stat5, Fyn, inflammation, IgE
Introduction
Atopic diseases, including asthma, atopic dermatitis, and allergic rhinitis are caused by environmental and genetic factors (1-3). During the last three decades, the prevalence of atopic disease has increased in developed nations, making them a major health concern (1-4). While their etiology is incompletely understood, a plausible factor promoting these chronic inflammatory diseases is failure of feedback mechanisms facilitating immune homeostasis. Understanding these regulatory circuits would provide insight for allergic and autoimmune diseases.
One means by which the immune system is regulated is through cytokine signaling. Of the many factors involved, members of the TGFβ family are ideal candidates. TGFβs are multipotent cytokines that control cell proliferation, tissue repair, inflammation, migration and angiogenesis (5, 6). The first observation of TGFβ-mediated immune regulation was made two decades ago (7). TGFβ1 is now linked to autoimmune diseases and cancer (5, 6, 8, 9), and has recently been shown to have a critical role in regulatory T cell and Th17 differentiation (10-12).
Mast cells are important effectors of atopic disease. Once activated by antigen-mediated IgE crosslinkage, mast cells link the innate and adaptive immune responses by altering vascular function and recruiting leukocytes to inflammatory sites. Mast cells can elicit TGFβ1-mediated paracrine signaling, since they secrete TGFβ1 as well as proteases that activate latent TGFβ1 present in blood and connective tissues (13-15).
We and others have found that TGFβ1 suppresses mast cell development, function and survival, offering homeostatic feedback regulation (16). TGFβ1 inhibited proliferation of mouse peritoneal mast cells and bone marrow–derived mast cells (BMMC) (17, 18), and prevented stem cell factor (SCF)-mediated rescue from apoptosis (25). TGFβ1 inhibitory effects on mast cell effector functions have also been shown. Bissonette et al (19) showed that Ag–induced histamine and TNF production from rat peritoneal mast cells were inhibited by TGFβ1, whereas Meade et al (20) showed that in vivo treatment with TGFβ1 inhibited IgE–mediated, mast cell–dependent, immediate hypersensitivity responses in mice. Despite studies supporting the theory that TGFβ1 inhibits mast cell function, contradictory evidence exists. This includes a report that reducing TGFβ1 levels diminished IgE–dependent cutaneous anaphylaxis (21). Moreover, these studies have not revealed the mechanisms by which TGFβ acts on mast cells, which could include decreased IgE receptor expression and/or altered signaling. Furthermore, why this potentially suppressive cascade fails to limit mast cell responses in some individuals is unknown.
We have found that Stat5 is critical for mast cell responses to both SCF via cKit and allergens via IgE-FcεRI (22-25). We now show that TGFβ1 suppresses Stat5 expression in mast cells derived from Th1-prone C57BL/6 mice. Intriguingly, mast cells from Th2-prone 129/Sv mice had much higher Stat5 expression that was unchanged by TGFβ1 treatment. While TGFβ1 suppressed IgE-mediated cytokine production and SCF-induced migration in C57BL/6 mast cells, it had no effect or even enhanced mast cell activation among 129/Sv mast cells. Similarly, TGFβ1 suppressed Stat5 expression and IgE-induced IL-6 secretion among approximately half of the human donor-derived mast cell populations tested. These data indicate specific pathways targeted for mast cell regulation, and suggest that genetic predisposition to atopy may include loss of homeostatic regulation by cytokines such as TGFβ1.
Materials and Methods
Animals
C57BL/6 and 129S1/SvImJ (hence referred to as 129/Sv) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). They were maintained in a specific pathogen-free facility at Virginia Commonwealth University (VCU). Protocols and studies involving animals were performed in accordance with the VCU Institutional Animal Care and Use Committee guidelines.
Cytokines and reagents
Purified dinitrophenol (DNP)-specific mouse IgE was purchased from BD Pharmingen (San Diego, CA). DNP-coupled human serum albumin (HSA) was purchased from Sigma Fine Chemicals (St. Louis, MO). Murine IL–3, SCF and human TGFβ1 were purchased from PeproTech (Rocky Hill, NJ). Antibodies recognizing actin were bought from Sigma–Aldrich (St. Louis, MO). Rat anti–mouse FcγRII/RIII (2.4G2), purified mouse IgE, purified anti–mouse IgE, FITC–conjugated rat IgG isotype control, and FITC conjugated anti–mouse CD117 (c-Kit) were purchased from BD Pharmingen. PE–conjugated rat IgG2b isotype control and PE conjugated anti–mouse IgE were purchased from eBioscience (San Diego, CA). Anti-Akt, Stat5, and Syk antibodies were purchased from Cell Signaling (Danvers, MA). Anti-Fyn antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Mouse mast cell cultures
Bone marrow derived mast cells (BMMC) were derived from mice by culture in complete RPMI (cRPMI) 1640 medium (Invitrogen Life Technologies, Carlsbad, CA) containing 10% FBS, 2 mM L–glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 mM sodium pyruvate, and 10 mM HEPES (Biofluids, Rockville, MD), supplemented with IL–3 containing supernatant from WEHI–3 cells and stem cell factor (SCF)–containing supernatant from BHK–MKL cells for 21 days. The final concentration of IL–3 and SCF was adjusted to 1ng/ml and 10ng/ml, respectively, as measured by ELISA. Peritoneal cells were extracted in phosphate buffered saline (PBS), then expanded in culture for 7 days as described for BMMC. For in vitro analyses, BMMC were washed and incubated at 37°C for 4 hours in cRPMI without cytokines to reduce background phosphorylation. Cells were then plated at 5×105 cells/ml and incubated at 37°C for 4 days in cRPMI with IL–3 (5ng/ml) +/- TGFβ1 (10ng/ml). Cells were activated by culturing overnight with 0.5μg/ml anti-DNP IgE, followed by crosslinking with 50ng/ml DNP-HSA for 16 hours, after which culture supernatants were harvested.
Human mast cell cultures
All protocols involving human tissues were approved by the human studies Internal Review Board at VCU or at NIAID/NIH. Surgical skin samples were obtained from the Cooperative Human Tissue Network of the National Cancer Institute or from the National Disease Research Interchange. Skin MCs were prepared and cultured as described (26) and were used after 6-10 weeks, at which time purity was essentially 100% mast cells, as determined by staining with toluidine blue. Peripheral blood human mast cells were developed from CD34+ pluripotent peripheral blood progenitor cells in StemPro-34 medium with supplement (Life Technologies) containing penicillin (100 U/ml), streptomycin (100 U/ml), L-glutamine (2 mM), IL-3 (30 ng/ml; first week only), IL-6 (100ng/ml) and SCF (100ng/ml) (PeproTech) for 8 weeks. Human mast cells were sensitized and activated as described for BMMC above, with the exception that culture media included soybean tryptase inhibitor (100μg/ml) to prevent cytokine degradation (27).
TGF–Beta 1 injection
Mice (BL/6 or 129/Sv, 8-12 weeks old, N=5/group) were injected intraperitoneally with 0.5μg TGFβ1 or PBS twice daily for 3 days, once on day 4, and harvested 4 hours later. Peritoneal lavage cells were counted with trypan blue and stained for flow analysis. Ear skin and small intestine sections were fixed in 4% paraformaldehyde and stained with chloracetate esterase before counting blinded samples with light microscopy. Mean mast cell numbers per 40X field were determined from 10 fields/mouse, after which the mean numbers were then averaged from all 5 mice. Mast cells from entire small intestine cross sections were counted from at least 3 sections/mouse, and the mean mast cell number from each mouse was then averaged from all 5 mice.
Flow cytometric analysis
Surface expression analysis was performed using a standard flow cytometry protocol described previously (28). To assess the percentage of peritoneal populations, B cells were identified as B220+ cells, neutrophils as CD11b-negative/Gr1+, high side-scatter cells, and macrophages as CD11b+/Gr-1-negative, high forward-scatter cells. Mast cells were identified as FcεRI+/Kit+. For cytokine measurements, cells were first activated for 90 minutes with IgE+antigen, then cultured for 5 hours in the presence of 5μM monensin at 37°C, prior to fixation in PBS/4% paraformaldehyde, and staining in the presence of 0.5% saponin with PE-conjugated anti-TNF. Stat5 was detected using rabbit-anti-Stat5 (Assay Biotech, clone C13341), with spectrum red-conjugated rat anti-rabbit IgG (Southern Biotechnology).
Enzyme–linked immunosorbent assay (ELISA)
Cytokine levels were detected by standard ELISA Kits purchased from PeproTech (Rocky Hill, NJ) using the manufacturer’s protocol.
Western Blot analysis
Western blotting was performed using 50μg of total cellular protein per sample. Protein was loaded and separated over 8–16% or 4–20% gradient SDS polyacrylamide gels (Bio–Rad, Hercules, CA). Proteins were transferred to nitrocellulose (Pall Corporation, Ann Arbor, MI), and blocked for 60 minutes in 5% nonfat dry milk (NFDM) in TBS plus 0.1% Tween 20 (TBST). Blots were incubated in 5% NFDM/TBST with a 1:1000 dilution of anti-Fyn, Syk, STAT5, Akt or β–actin overnight at 4°C with gentle rocking. Blots were washed six times for 10 minutes each in TBST, followed by incubation in Blotto B containing at 1:5000 dilution of HRP–linked anti–IgG (matched to the appropriate primary antibody source species, from Cell Signaling, Danvers, MA). Size estimates for proteins were obtained using molecular weight standards from Bio–Rad (Hercules, CA).
Nucleofection of siRNAs
C57BL/6 BMMC were transfected with a Dharmacon ON-TARGET SMARTpool of Stat5B or non-targeting siRNAs (Thermo Scientific). The Amaxa Nucleofector II and Cell Line Nucleofector Kit V (Lonza Cologne GmbH, Cologne, Germany) were utilized according to the manufacturer’s protocol for human monocytes. For the initial nucleofection step, a concentration of 2μM siRNA and three million cells were used. The cultures were allowed to rest for 72 hours in growth medium added to dilute the siRNAs to a final concentration of 57nM. Efficiency of knockdown was determined through immunoblot analyses.
Stat5B transfection
Stat5B, encoded by the plasmid pMSCV that bears the PGK promoter, was transfected in C57BL/6 BMMC using the Amaxa Nucleofector, with program Y-001 and Amaxa reagents. Cells were cultured for three days prior to assessing Stat5 expression levels and TGFβ1 responsiveness.
Mast cell migration
To measure migration, cells were cultured in IL-3+/-TGFβ1 for 72 hours, washed and re-plated at 2×106 cells/ml in cRPMI lacking fetal calf serum, and containing 10 mg/ml of bovine serum albumin (BSA). Cells (200μl) were added to the top chamber of an 8μm transwell insert, and assessed after a 16–hour incubation in response to SCF in the bottom chamber. Live cell migration was assessed using timed flow cytometry-based counting of quadruplicate samples harvested from the bottom chamber, using propidium iodide to exclude dead cells. Control wells were stimulated with media alone, and were used as the denominator to determine fold migration.
Statistics
Data shown in each figure are the mean and standard errors of the indicated number of samples. For comparisons of two samples, Student’s t–Test was used. For comparisons of multiple samples to a control group, one–way analysis of variance (ANOVA) with Dunnett’s post–hoc test was employed. Cluster analysis from the R statistical package was used to assign human mast cell populations into two groups based on TGF-mediated suppression of Stat5.
Results
TGFβ1 decreases mouse mast cell numbers, c–Kit and FcεRI expression in vivo
We previously found that TGFβ1 suppresses mast cell function and survival in vitro (29-31). In this work we investigated these effects in vivo, first by injecting C57BL/6 mice with TGFβ1 intraperitoneally for 4 days, and assessing changes in peritoneal mast cells. When gating on Kit-positive mast cells, we found that TGFβ1 significantly diminished FcεRI and c-Kit expression on peritoneal mast cells (Figure 1A,B). We also noted that mast cell IgE receptors were similarly diminished after only 2 days of injections, while c-Kit was unchanged (data not shown). FcεRI inhibition was not related to reduced circulating IgE during this short course of TGFβ1 treatment, since we found no difference in serum IgE levels (Figure 1C). These data matched the changes in mast cell surface antigens we have previously noted in vitro (29, 30, 32).
Figure 1. TGFβ1 decreases mouse mast cell number, c-Kit and FcεRI expression in vivo.

TGFβ1 or PBS was injected intraperitoneally twice daily for four days, and peritoneal lavage cells were assessed after a final injection on day 5 as described in Materials and Methods. (A and B) Peritoneal mast cell FcεRI and cKit expression, as determined by flow cytometry; (C) Serum IgE levels as measured by ELISA. (D) Changes in peritoneal mast cell numbers, based on FcεRI+/Kit+ gating and cell counting. Data shown in (A-D) are representative of two separate experiments that yielded similar results. (E) Plasma TGFβ1 levels, as determined by ELISA from 5 animals per group. Control received PBS. (F) Mast cell numbers in ear skin or small intestine (N=5/group), based on histological staining, as described in Materials and Methods. Data shown are mean and standard error.
We also found a 65% decrease in mast cell numbers (Figure 1D). Elevated TGFβ1 was detectable in plasma within 4 hours post-injection, and decreased to baseline levels approximately 24 hours later (Figure 1E). This suggested that TGFβ1 injection might alter mast cell migration, perhaps explaining the decline in peritoneal mast cells. However, mast cell numbers in the skin and intestine were unchanged (Figure 1F). Therefore, it seems unlikely that the loss of peritoneal mast cells was caused by emigration to other sites. This decline could be due to cell death. In support of this theory, TGFβ1 induces mast cell apoptosis in vitro after 4-6 days (31). Similarly, peritoneal mast cell numbers were unchanged after 2 days of injections (data not shown), but significantly decreased on day 4. These data demonstrated that TGFβ1-mediated suppression of the mast cell compartment can be translated to an in vivo setting.
TGFβ1 suppresses the Fyn-Stat5 pathway
The suppressive effects of TGFβ1 may stem from altered FcεRI signaling. We recently found that Stat5, activated by Fyn kinase, is critical for IgE-mediated signaling (22, 23). In addition to Fyn and Stat5, FcεRI activates Syk kinase, which, along with Fyn, induces Akt activation (33). We therefore investigated TGFβ1 suppression of these proteins. BMMC treated with TGFβ1 for four days showed no change in Syk or Akt expression, but had a significant reduction in Fyn and Stat5 levels (Figure 2A-D). There was no change in mRNA expression encoding any of these proteins, as judged by quantitative PCR (data not shown). As shown in Figure 2E, these data were not restricted to in vitro-derived BMMC, since peritoneal mast cells cultured for 4 days with TGFβ1 also had diminished Stat5 expression. These data indicated that TGFβ1 selectively altered FcεRI signaling, targeting the Fyn-Stat5 pathway through a post-transcriptional mechanism.
Figure 2. TGFβ1 selectively suppresses Fyn and Stat5 expression.

BMMC were cultured for four days in IL–3 (5ng/ml) with or without TGFβ1 (10ng/ml), after which the cells were lysed. Lysates were blotted for (A) Syk, (B) Fyn, (C) Akt, or (D) Stat5. Data are representative of three populations, with mean±SE, after normalizing to actin loading, shown in the bar charts below. (E) Peritoneal cells were cultured for 4 days in IL-3 and SCF, in the presence or absence of TGFβ1. Stat5 expression on c-Kit+ mast cells was determined by flow cytometry. Data shown are mean±SE from 4 mice.
TGFβ-mediated suppression varies with genetic background
We have previously found that mast cells from the Th2-prone 129/Sv background are resistant to IL-10 effects (34), and express Fyn at high levels (35). To determine if genetic background alters TGFβ1 responses, we compared Th1-prone C57BL/6 and Th2-prone 129/Sv mast cell populations. As shown in Figures 3A and B, TGFβ1 significantly reduced Fyn and Stat5 expression in C57BL/6 BMMC, as expected. In contrast, BMMC cultured from 129/Sv mice had Fyn and Stat levels that trended down after TGFβ1 culture, but did not reach statistical significance (p=.075 for Fyn and .15 for Stat5). More strikingly, 129/Sv BMMC expressed 2-3 times more Fyn and Stat5 protein than C57BL/6 cells (p<.01) – and post-TGFβ1 levels approached basal expression among C57BL/6 BMMC. These data suggest that genetic background may predict TGFβ1 effects.
Figure 3. C57BL/6 and 129/Sv BMMC show variation in TGFβ1 responsiveness.

(A and B) Fyn and Stat5 expression were measured by western blotting, using lysates from the indicated BMMC populations cultured ±TGFβ1 for 4 days as described in Figure 2. Fyn and Stat5 changes induced by TGFβ1 were quantified using 3-5 samples per point, with all points compared to control C57BL/6 samples, after normalization to actin loading. (C) Stat5B was transfected into C57BL/6 BMMC, and Stat5 expression relative to control transfection with empty vector was determined by western blotting. (D) Transfected BMMC populations described in (C) were cultured for 4 days ± TGFβ1, then activated with IgE+DNP-HSA for 16 hours. IL-6 concentration in supernatants was determined by ELISA. Data shown are mean±SE from quadruplicate samples.
To determine if altered Stat5 regulation is functionally important, Stat5B was overexpressed (approximately 2-fold) in C57BL/6 BMMC by transfection (Figure 3C). TGFβ1 reduced IgE-mediated IL-6 secretion in control vector-transfected cells as expected. By contrast, Stat5B-transfected cells showed a slight but significant increase in IL-6 (Figure 3D). Thus, increasing Stat5B can prevent suppression of the IgE response. Collectively, these data suggest that both Fyn/Stat5 expression and TGFβ1 responsiveness can vary with genetic background, which can contribute to loss of mast cell homeostasis.
We expanded the study of 129/Sv responses to TGFβ1 to include in vivo responses. 129/Sv mice were injected intraperitoneally twice daily for four days, as previously done with C57BL/6 mice. TGFβ1 suppressed mast cell FcεRI and c-Kit expression to a similar extent observed with C57BL/6 mice (Figure 4A,B). We also compared the relative change in several immune cell lineages in the peritoneum, which were diminished by TGFβ1 (Figure 4C). While the trends were similar, several differences were notable between the two mouse strains. C57BL/6 mice showed significantly diminshed peritoneal cells, especially B cells, CD4 T cells, and neutrophils. But the most overt difference was among mast cells, with 129/Sv mice showing less than 10% loss, while C57BL/6 mast cell numbers declined by more than 70%. Thus the in vivo response of 129/Sv mice to TGFβ1, especially within the mast cell lineage, differed from C57BL/6 animals.
Figure 4. Genetic background affects TGFβ1 effects in vivo.

(A and B) 129/Sv mice (N=5) were injected with TGFβ1 for 4 days as described in Figure 1. Anti-IgE and anti-c-Kit staining intensity among FcεRI+/c-Kit+ peritoneal mast cells was measured by flow cytometry. (C) The fold change in number of each peritoneal cell lineage was measured by flow cytometry, using mice of the indicated genotype injected with PBS as the reference sample. Cell populations were identified as described in Materials and Methods. N=5. The indicated p values were obtained by Student’s t Test.
These in vivo data partly confirmed the TGFβ1-resistant phenotype of 129/Sv mast cells. However, they also demonstrated that TGFβ1 reduced FcεRI and c-Kit expression, indicating that some TGF-mediated suppressive effects persist on the 129/Sv background. Why responses vary and how they affect mast cell function remained unclear. We found that BMMC derived from the two mouse strains had similar expression of TGFβ receptors I and II (Figure 5A), indicating that TGFβ1 resistance was not related to loss of TGFβ receptor expression. We next addressed the functional relevance of TGFβ1 effects, by examining IgE-mediated cytokine production. Each BMMC population was cultured for 4 days in the presence or absence of TGFβ1, then activated by IgE XL. As we and others have previously noted (19, 29, 32), TGFβ1 reduced IgE-induced cytokine secretion by at least 50% among C57BL/6 BMMC. In contrast, 129/Sv cells cultured with TGFβ1 showed no loss of TNF production, and actually secreted more IL-6 (Figure 5B). Hence 129/Sv mast cells were functionally resistant to TGFβ1-mediated suppression.
Figure 5. TGFβ1 effects on IgE-mediated cytokine production.

(A) TGFβ RI and RII surface expression on C57BL/6 and 129/Sv BMMC was measured by flow cytometry. Data shown are means ±SEM of 6-15 samples. (B) IgE-induced IL-6 and TNF production was assessed by ELISA after 4 days of culture in IL-3±TGFβ1. Data are means ±SEM of three populations. (C) BMMC were cultured ±TGFβ1 for 4 days, then activated with IgE XL, and assessed for TNF production by intracellular staining as described in Materials and Methods. FcεRI expression levels were determined using anti-IgE staining, after gating on TNF+ cells. Data shown are geometric mean fluorescence intensity (gMFI) of 7-9 samples/point. *p<.05 when using t Test to compare TNF staining of C57BL/6 cells to 129/Sv cells cultured in the absence of TGFβ1. (D and E) The TNF+ populations from (C) were used to determine the geometric (g) MFI of TNF staining among all TNF+ cells, and the fraction of cells producing TNF. Data shown are means ±SEM from 8-9 samples/point.
The ability of TGFβ1 to reduce FcεRI expression without suppressing cytokine secretion led us to investigate the importance of FcεRI suppression under these conditions. We examined the relationship between FcεRI and TNF levels, using intracellular staining to examine groups of cells with similar FcεRI expression levels. Among TNF-positive cells, those receiving TGFβ1 produced as much TNF as untreated cells at each FcεRI level measured (Figure 5C). While TNF levels initially increased in parallel with FcεRI expression, this curve quickly reached a plateau, indicating that a small number of receptors were needed for maximal cytokine production. This suggested that the 40-50% decrease in IgE receptor expression elicited by TGFβ1 was insufficient to reduce TNF production. We additionally noted that 129/Sv mast cells produced more TNF at any given FcεRI level than C57BL/6 BMMC (Figure 5C), suggesting that IgE signaling in 129/Sv cells is more potent. The effects of TGFβ1 treatment were evident when we determined the fraction of cells expressing TNF. As shown in Figures 5D and E, the average TNF-producing cell made only slightly less TNF when exposed to TGFβ1, whereas the fraction of TNF-producing cells decreased by 50% in C57BL/6 BMMC. In contrast, the proportion of TNF-positive cells was unchanged in the 129/Sv population. These data collectively support the hypothesis that TGFβ1 decreases mast cell responses to IgE not by downregulating its receptor, but by altering the outcome of FcεRI signaling. 129/Sv mast cells are resistant to this suppressive effect, showing no change in cytokine-producing cells.
SCF-induced migration requires Stat5B, and is blocked by TGFβ1: influence of genetic background
In addition to FcεRI, TGFβ1 also suppressed c-Kit expression. SCF, the ligand for c-Kit, is a potent mast cell survival and migration factor (36-39). We previously found that SCF activates Stat5 and requires this protein to elicit survival (24, 40). The role of Stat5 in SCF-induced migration is unknown. Since TGFβ1 suppressed Stat5 as well as c-Kit, we examined the importance of Stat5 in SCF-induced migration, and how TGFβ1 alters this response. Using siRNA, we demonstrated that SCF-mediated migration was reduced to background levels when Stat5B was suppressed (Figure 6A). Loss of Stat5B in this short-term assay had no impact on survival, which did not differ between control- and Stat5B-suppressed cells under the migration conditions (data not shown). This pathway, like SCF-mediated survival or IgE-induced cytokine secretion, appears to be dependent on Stat5B.
Figure 6. The role of Stat5 in SCF-mediated migration and its regulation by TGFβ1.

(A) C57BL/6 BMMC were transfected with non-targeting (NT) or Stat5B-targeting (siB) siRNAs prior to measuring SCF-induced migration as described in Materials and Methods. Data shown are mean ±SEM of N=4 from one of two representative experiments. Inset is western blotting to show efficacy and specificity of siRNA treatment. (B) BMMC of the indicated background were cultured in IL-3 +/- TGFβ1 for 3 days prior to measuring SCF-induced migration. Data shown are means ±SEM from 14 samples analyzed in 2 experiments.
We next examined the ability of TGFβ1 to suppress SCF-induced migration in C57BL/6 or 129/Sv BMMC. As shown in Figure 6B, there was a striking contrast between these populations. Prior culture in TGFβ1 for 3 days reduced SCF-mediated migration by more than 50% in C57BL/6 BMMC. However, 129/Sv mast cells exposed to TGFβ1 for 3 days showed at least 5-fold greater migration in response to SCF. These data showed that TGFβ1 effects can diverge greatly among genetically different mast cell populations, impacting the functional effects of both IgE and SCF.
Variable responsiveness to TGFβ1 is consistent in primary human mast cells
We extended our study of the Fyn-Stat5 cascade, assessing TGFβ1-mediated suppression in three preparations of primary human skin mast cells. Surprisingly, none of these isolates showed a change in Fyn expression when cultured with TGFβ1 for 4 days (data not shown). In contrast, Stat5 expression was diminished 50-60% in two mast cell isolates, while a third showed little change (Figure 7A). By expanding this study to a total of 9 donors, we found variable effects on Stat5 expression that fell into two groups upon cluster analysis. As shown in Figure 7B, mast cells from 3 donors (“Group A”) showed no decrease in Stat5 after culture in TGFβ1. By contrast, cells from 6 donors (“Group B”) decreased Stat5, with a significant difference between the two groups. These data demonstrated that the variable Stat5 suppression in C57BL/6 and 129/Sv mast cells may have a human correlate.
Figure 7. Variation in human mast cell responsiveness to TGFβ1.
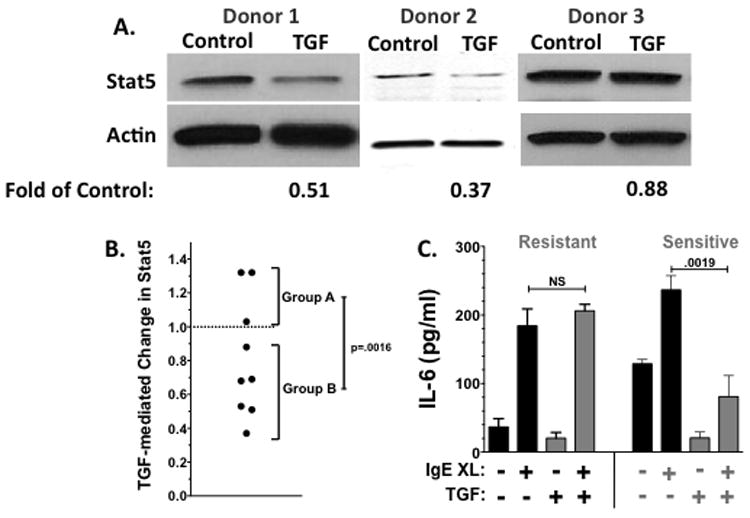
(A) Mast cells cultured from human skin donors were cultured ±TGFβ1 for 4 days as described in Materials and Methods, and Stat5 expression was assessed by Western blotting. The change in Stat5 expression was determined by comparison to the control lane, after normalizing to actin loading. Data are from 3 independent experiments. (B) TGFβ1-mediated change in Stat5 expression, as determined in part (A), from a total of 9 subjects. (C) Human mast cells were cultured for 4 days in the presence or absence of TGFβ1, prior to activation with IgE and antigen for 16 hours. Culture supernatants were analyzed for the production of IL-6. Data shown are mean±SE from 3 resistant and 2 sensitive populations, each analyzed in triplicate.
Due to low cell numbers, we were not able to test all human mast cell populations for both Stat5 expression and IgE responses following TGFβ1 stimulation. We did find that among 5 preparations from different donors, 3 showed resistance to TGFβ1, as demonstrated by continued IL-6 secretion in response to IgE crosslinkage. In contrast, 2 populations showed a significant decrease in IgE-mediated IL-6 production (Figure 7C). Two members of “Group A”, which showed no change in Stat5, were included in these experiments. One population was TGFβ1 resistant, while the other was sensitive; hence TGFβ1 may inhibit mast cell function even in the absence of Stat5 suppression.
Discussion
Mast cells are known as key initiators of the atopic response. We have sought to understand how factors such as TGFβ1 control mast cell homeostasis. Human and mouse mast cells secrete TGFβ1 along with proteases such as chymase that activate latent TGFβ1 (16). This has direct consequences for allergic disease, as mast cell-derived TGFβ1 has been associated with the induction of regulatory T cells (41), smooth muscle contractility (42), and fibroblast proliferation and collagen deposition (43-45). Moreover, TGFβ1 has many direct effects on mast cells, including increased mMCP-1, and -7 expression, enhanced adhesion and increased migration. However, we and others have also noted that TGFβ1 has suppressive activities on mast cells, including decreased proliferation, FcεRI expression, cytokine production, and reduced c-Kit expression, which some correlated with the induction of apoptosis (reviewed in (46) and (16). How these suppressive activities are carried out has not been determined.
Our data allow us to postulate a mechanism by which TGFβ1 acts on mast cells. We show that C57BL/6 BMMC exposed to TGFβ1 produce less TNF as a population – but that on a per cell basis, TNF-producing cells make similar amounts regardless of TGFβ1 exposure. Additionally, the relationship between FcεRI levels and TNF secretion was not altered by TGFβ1 (Figure 5). So, while TGFβ1 suppresses FcεRI expression, this is not causally linked to reduced cytokine production. These data are logical: since TGFβ1 reduces FcεRI surface expression 50-60% (29), and human mast cells express approximately 200,000 IgE receptors (47), an excess of receptors needed for activation would still be available. By comparison, the productive use of anti-IgE therapy (e.g. omaluzimab) reduces FcεRI levels by 95% or more (48, 49). Instead, the fraction of TNF-producing mast cells is halved by TGFβ1 exposure, suggesting that a subset of the population has defective IgE signaling.
Supporting this theory, we demonstrate that TGFβ1 reduces Fyn and Stat5 expression in mouse mast cells. Among peritoneal cells treated ex vivo (Figure 2A), we noted both a decrease in per cell Stat5 levels and in the percentage of the population expressing Stat5. As we have recently shown, Fyn associates with Stat5 and is required for Stat5 tyrosine phosphorylation (23). Since both Fyn and Stat5 are essential for IgE-induced cytokine secretion (22, 50), a blockade in this pathway could explain our observations. Unlike IL-10, which suppresses Syk and Akt in addition to Fyn and Stat5 (28), the effects of TGFβ1 were more selective. We also noted no change in Fyn or Stat5 mRNA expression (data not shown), suggesting alterations in protein production. We have previously found that TGFβ1 reduces the translational efficiency of FcεRI beta (29), and hypothesize that this mechanism may also be in place here.
TGFβ1 is one of the more enigmatic factors contributing to atopic disease. Several studies have noted polymorphisms in the TGFβ1 promoter that associate with asthma. TGFβ1 levels are increased in atopic dermatitis and in asthmatics, where TGFβ1 is associated with lung remodeling and collagen deposition. For a review of TGFβ1 effects on atopic disease, see (51-53). Work in mouse asthma models has implicated TGFβ1 as an exacerbating agent. For example, blocking TGFβ1 signaling through overexpression of the Smad7 antagonist reduced inflammation (54), as did treatment with anti-TGFβ1 immunoglobulin (55) in murine airway hyperresponsiveness (AHR) models. A recent study found that AHR treatment with the anti-malarial drug Mepacrine suppressed airway remodeling, and correlated with reduced TGFβ1 production (56). It has recently been suggested that targeting TGFβ1 may be a therapeutic intervention, based on data such as these (51-53).
In contrast, other studies find that transient increases in TGFβ1 levels may suppress AHR in mouse models. Fu and coworkers employed a TGFβ1–expressing plasmid to suppress AHR (57). A recent study by Nemeth and colleagues suggests that TGFβ1 secretion by bone marrow stromal cells correlates with reduced eosinophilia, mucus production, and cytokines in mouse AHR (58). Furthermore, genotypes associated with low TGFβ1 production have been linked with asthma (59). Separate from asthma, food allergy studies have demonstrated that oral TGF can suppress atopic inflammation (60). Finally, regulatory T cell function, including allergy suppression, has been postulated to occur partly via TGF production (61). A unifying hypothesis is that TGFβ1, while immunosuppressive and hence beneficial to asthma amelioration, also exacerbates the remodeling phase of chronic asthma – hence short term exposure to TGFβ1 is beneficial, while long term is detrimental.
Perhaps the most striking observation in this study was the importance of genetic background in TGFβ1 responsiveness. When human mast cell populations were exposed to TGFβ1, we noted two groups: one with significantly reduced Stat5 levels after incubation with TGFβ1, compared to another group resisted suppression. Similarly, we noted responsive and resistant groups when assessing TGFβ1-mediated effects on IL-6 secretion. Further work is needed to discern a link between reduced Stat5 and attenuated cytokine production. However, treatment of one preparation of human skin mast cells with TGFβ1 did not reduce Stat5, but still suppressed IL-6 secretion. Thus we conclude that Stat5 suppression may not be an absolute requirement for blocking mast cell function in human mast cells. Importantly, our interpretations are limited by access to a small number of human mast cell populations. This study demonstrates the plausibility of TGFβ1-sensitive and -resistant individuals; further work with a larger number of donors is needed to fully characterize these groups.
These data suggested that Stat5 is a target of TGFβ1, and that genetic variation might alter TGFβ1 effects. We reasoned that the question of genetic influence could be addressed using in-bred mouse strains with known Th1- or Th2-prone responses. Because Th2-prone 129/Sv mice were previously shown to have increased mast cell numbers and histamine release in vivo (35), we compared their TGFβ1 response to Th1-prone C57BL/6 mice, the source of BMMC for the majority of our studies. We found that 129/Sv and C57BL/6 BMMC expressed TGFβ1 receptors at comparable levels, but that 129/Sv mast cells were completely resistant to suppression of IgE-induced cytokines. In fact, IL-6 production increased among 129/Sv BMMC treated with TGFβ1. The explanation for this enhanced response is not known, but we postulate that TGFb1 could enhance activity of the Fyn-Stat5 cascade. TGFβ1 resistance correlated with 2-3 fold greater Fyn and Stat5 expression in 129/Sv BMMC – levels that were not statistically decreased by TGFβ1 treatment. Fyn was previously reported to be highly expressed in 129/Sv mast cells (35). Our corroboration of these data, along with enhanced Stat5 expression, fits their TGFβ1 resistance. These data suggest that genetic background contributes to the ultimate effects of TGFβ1 on mast cell homeostasis, and implicate the Fyn-Stat5 pathway as a contributing factor.
TGFβ1 resistance in the 129/Sv strain was not limited to in vitro IgE stimulation. When injected intraperitoneally, 129/Sv mice showed a reduction among all cell types except macrophages – like their C57BL/6 counterparts. However, these effects were generally mitigated on the 129/Sv background, and essentially absent when examining peritoneal mast cell numbers, which were greatly suppressed among C57BL/6 mice. Further, TGFβ1 inhibited c-Kit expression on both C57BL/6 and 129/Sv mast cells, but the effects on c-Kit-mediated migration were completely divergent. TGFβ1 reduced SCF-induced migration among C57BL/6 BMMC, while greatly enhancing 129/Sv mast cell migration. These differences might also be connected to Stat5. We demonstrate that c-Kit-mediated migration requires Stat5B expression. Importantly, our data do not exclude a role for Stat5A in migration, as we were unable to suppress Stat5A without diminshing Stat5B and hence could not clarify its function in these assays (data not shown). This is the first reported role for Stat5B in mast cell or c-Kit-induced migration. Our data concur with studies showing a role for Stat5 in cancer cell invasion and metastasis (62, 63). One of these prior studies found that Stat5B is selectively involved in glioblastoma invasion (64). Understanding how TGFβ1 alters c-Kit signaling and the importance of genetic background could impact multiple fields of study.
Our current hypothesis is that the Fyn-Stat5 pathway may be more robustly expressed on some genetic backgrounds, predisposing to stronger mast cell responses and greater resistance to suppression by factors such as TGFβ1. Importantly, other features of the Th2 response could operate separately or together with changes in Fyn-Stat5 to alter mast cell homeostasis. For example, T cells from asthmatic donors were recently shown to be TGFβ1-resistant perhaps due to signaling by IL-2 and IL-4 (65). Similarly, we found that prior exposure to IL-4 can block TGFβ1 effects on mast cells (66). Thus the true role of TGFβ1 may depend upon issues such as timing, other factors present in the microenvironment, and genetically determined levels of Fyn and Stat5. Resolving these variables will provide testable, quantifiable factors explaining predisposition to atopy and offering therapeutic targets.
Acknowledgments
The authors thank Dr. Michael Gurish, Harvard Medical School, for his kind assistance with histology samples.
Supported by grants from the National Institutes of Health (1R01AI59638 to JJR, U19AI077435 to JR, SS, and DC; 2R01DK059380 to KDB; K01AR053186 and 1R01AI095494 to CAO).
Footnotes
Disclosures: The authors have no conflicting financial interests.
References
- 1.Oettgen HC, Geha RS. IgE in asthma and atopy: cellular and molecular connections. J Clin Invest. 1999;104:829–835. doi: 10.1172/JCI8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Umetsu DT, McIntire JJ, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat Immunol. 2002;3:715–720. doi: 10.1038/ni0802-715. [DOI] [PubMed] [Google Scholar]
- 3.von Mutius E. Gene-environment interactions in asthma. J Allergy Clin Immunol. 2009;123:3–11. doi: 10.1016/j.jaci.2008.10.046. quiz 12-13. [DOI] [PubMed] [Google Scholar]
- 4.Wilson DH, Adams RJ, Tucker G, Appleton S, Taylor AW, Ruffin RE. Trends in asthma prevalence and population changes in South Australia, 1990-2003. Med J Aust. 2006;184:226–229. doi: 10.5694/j.1326-5377.2006.tb00207.x. [DOI] [PubMed] [Google Scholar]
- 5.Yoshimura A, Wakabayashi Y, Mori T. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J Biochem. 2010;147:781–792. doi: 10.1093/jb/mvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, Sporn MB, Fauci AS. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 9.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 10.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 11.Ziegler SF, Buckner JH. FOXP3 and the regulation of Treg/Th17 differentiation. Microbes Infect. 2009;11:594–598. doi: 10.1016/j.micinf.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee YK, Mukasa R, Hatton RD, Weaver CT. Developmental plasticity of Th17 and Treg cells. Curr Opin Immunol. 2009;21:274–280. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 13.Kanbe N, Kurosawa M, Nagata H, Saitoh H, Miyachi Y. Cord blood-derived human cultured mast cells produce transforming growth factor beta1. Clin Exp Allergy. 1999;29:105–113. doi: 10.1046/j.1365-2222.1999.00459.x. [DOI] [PubMed] [Google Scholar]
- 14.Lindstedt KA, Wang Y, Shiota N, Saarinen J, Hyytiainen M, Kokkonen JO, Keski-Oja J, Kovanen PT. Activation of paracrine TGF-beta1 signaling upon stimulation and degranulation of rat serosal mast cells: a novel function for chymase. FASEB J. 2001;15:1377–1388. doi: 10.1096/fj.00-0273com. [DOI] [PubMed] [Google Scholar]
- 15.Pennington DW, Lopez AR, Thomas PS, Peck C, Gold WM. Dog mastocytoma cells produce transforming growth factor beta 1. J Clin Invest. 1992;90:35–41. doi: 10.1172/JCI115853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan JJ, Kashyap M, Bailey D, Kennedy S, Speiran K, Brenzovich J, Barnstein B, Oskeritzian C, Gomez G. Mast cell homeostasis: a fundamental aspect of allergic disease. Crit Rev Immunol. 2007;27:15–32. doi: 10.1615/critrevimmunol.v27.i1.20. [DOI] [PubMed] [Google Scholar]
- 17.Toyota N, Hashimoto Y, Matsuo S, Iizuka H. Transforming growth factor beta 1 inhibits IL-3- and IL-4-dependent mouse connective tissue-type mast cell proliferation. Arch Dermatol Res. 1995;287:198–201. doi: 10.1007/BF01262332. [DOI] [PubMed] [Google Scholar]
- 18.Broide DH, Wasserman SI, Alvaro-Gracia J, Zvaifler NJ, Firestein GS. Transforming growth factor-beta 1 selectively inhibits IL-3-dependent mast cell proliferation without affecting mast cell function or differentiation. J Immunol. 1989;143:1591–1597. [PubMed] [Google Scholar]
- 19.Bissonnette EY, Enciso JA, Befus AD. TGF-beta1 inhibits the release of histamine and tumor necrosis factor-alpha from mast cells through an autocrine pathway. Am J Respir Cell Mol Biol. 1997;16:275–282. doi: 10.1165/ajrcmb.16.3.9070612. [DOI] [PubMed] [Google Scholar]
- 20.Meade R, Askenase PW, Geba GP, Neddermann K, Jacoby RO, Pasternak RD. Transforming growth factor-beta 1 inhibits murine immediate and delayed type hypersensitivity. J Immunol. 1992;149:521–528. [PubMed] [Google Scholar]
- 21.Kim HM, Lee YM. Role of TGF-beta 1 on the IgE-dependent anaphylaxis reaction. J Immunol. 1999;162:4960–4965. [PubMed] [Google Scholar]
- 22.Barnstein BO, Li G, Wang Z, Kennedy S, Chalfant C, Nakajima H, Bunting KD, Ryan JJ. Stat5 expression is required for IgE-mediated mast cell function. J Immunol. 2006;177:3421–3426. doi: 10.4049/jimmunol.177.5.3421. [DOI] [PubMed] [Google Scholar]
- 23.Pullen NA, Barnstein BO, Falanga YT, Wang Z, Suzuki R, Tamang TD, Khurana MC, Harry EA, Draber P, Bunting KD, Mizuno K, Wilson BS, Ryan JJ. Novel mechanism for Fc{epsilon}RI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J Biol Chem. 2012;287:2045–2054. doi: 10.1074/jbc.M111.311142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shelburne CP, McCoy ME, Piekorz R, Sexl V, Roh KH, Jacobs-Helber SM, Gillespie SR, Bailey DP, Mirmonsef P, Mann MN, Kashyap M, Wright HV, Chong HJ, Bouton LA, Barnstein B, Ramirez CD, Bunting KD, Sawyer S, Lantz CS, Ryan JJ. Stat5 expression is critical for mast cell development and survival. Blood. 2003;102:1290–1297. doi: 10.1182/blood-2002-11-3490. [DOI] [PubMed] [Google Scholar]
- 25.Shelburne CP, McCoy ME, Piekorz R, Sexl VV, Gillespie SR, Bailey DP, Gharse A, Mirmonsef P, Mann MN, Kashyap M, Wright HV, Chong HJ, Bouton LA, Ramirez CD, Lantz CS, Ryan JJ. Stat5: an essential regulator of mast cell biology. Mol Immunol. 2002;38:1187–1191. doi: 10.1016/s0161-5890(02)00061-5. [DOI] [PubMed] [Google Scholar]
- 26.Kambe N, Kambe M, Kochan JP, Schwartz LB. Human skin-derived mast cells can proliferate while retaining their characteristic functional and protease phenotypes. Blood. 2001;97:2045–2052. doi: 10.1182/blood.v97.7.2045. [DOI] [PubMed] [Google Scholar]
- 27.Zhao W, Oskeritzian CA, Pozez AL, Schwartz LB. Cytokine production by skin-derived mast cells: endogenous proteases are responsible for degradation of cytokines. J Immunol. 2005;175:2635–2642. doi: 10.4049/jimmunol.175.4.2635. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy Norton S, Barnstein B, Brenzovich J, Bailey DP, Kashyap M, Speiran K, Ford J, Conrad D, Watowich S, Moralle MR, Kepley CL, Murray PJ, Ryan JJ. IL-10 suppresses mast cell IgE receptor expression and signaling in vitro and in vivo. J Immunol. 2008;180:2848–2854. doi: 10.4049/jimmunol.180.5.2848. [DOI] [PubMed] [Google Scholar]
- 29.Gomez G, Ramirez CD, Rivera J, Patel M, Norozian F, Wright HV, Kashyap MV, Barnstein BO, Fischer-Stenger K, Schwartz LB, Kepley CL, Ryan JJ. TGF-beta 1 inhibits mast cell Fc epsilon RI expression. J Immunol. 2005;174:5987–5993. doi: 10.4049/jimmunol.174.10.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kashyap M, Bailey DP, Gomez G, Rivera J, Huff TF, Ryan JJ. TGF-beta1 inhibits late-stage mast cell maturation. Exp Hematol. 2005;33:1281–1291. doi: 10.1016/j.exphem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Norozian F, Kashyap M, Ramirez CD, Patel N, Kepley CL, Barnstein BO, Ryan JJ. TGF beta1 induces mast cell apoptosis. Exp Hematol. 2006;34:579–587. doi: 10.1016/j.exphem.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 32.Zhao W, Gomez G, Yu SH, Ryan JJ, Schwartz LB. TGF-beta1 attenuates mediator release and de novo Kit expression by human skin mast cells through a Smad-dependent pathway. J Immunol. 2008;181:7263–7272. doi: 10.4049/jimmunol.181.10.7263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilfillan AM, Rivera J. The tyrosine kinase network regulating mast cell activation. Immunol Rev. 2009;228:149–169. doi: 10.1111/j.1600-065X.2008.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Speiran K, Bailey DP, Fernando J, Macey M, Barnstein B, Kolawole M, Curley D, Watowich SS, Murray PJ, Oskeritzian C, Ryan JJ. Endogenous suppression of mast cell development and survival by IL-4 and IL-10. J Leukoc Biol. 2009;85:826–836. doi: 10.1189/jlb.0708448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamashita Y, Charles N, Furumoto Y, Odom S, Yamashita T, Gilfillan AM, Constant S, Bower MA, Ryan JJ, Rivera J. Cutting edge: genetic variation influences Fc epsilonRI-induced mast cell activation and allergic responses. J Immunol. 2007;179:740–743. doi: 10.4049/jimmunol.179.2.740. [DOI] [PubMed] [Google Scholar]
- 36.Galli SJ, Tsai M, Wershil BK, Tam SY, Costa JJ. Regulation of mouse and human mast cell development, survival and function by stem cell factor, the ligand for the c-kit receptor. Int Arch Allergy Immunol. 1995;107:51–53. doi: 10.1159/000236928. [DOI] [PubMed] [Google Scholar]
- 37.Iemura A, Tsai M, Ando A, Wershil BK, Galli SJ. The c-kit ligand, stem cell factor, promotes mast cell survival by suppressing apoptosis. Am J Pathol. 1994;144:321–328. [PMC free article] [PubMed] [Google Scholar]
- 38.Meininger CJ, Yano H, Rottapel R, Bernstein A, Zsebo KM, Zetter BR. The c-kit receptor ligand functions as a mast cell chemoattractant. Blood. 1992;79:958–963. [PubMed] [Google Scholar]
- 39.Sundstrom M, Alfredsson J, Olsson N, Nilsson G. Stem cell factor-induced migration of mast cells requires p38 mitogen-activated protein kinase activity. Exp Cell Res. 2001;267:144–151. doi: 10.1006/excr.2001.5239. [DOI] [PubMed] [Google Scholar]
- 40.Ryan JJ, Huang H, McReynolds LJ, Shelburne C, Hu-Li J, Huff TF, Paul WE. Stem cell factor activates STAT-5 DNA binding in IL-3-derived bone marrow mast cells. Exp Hematol. 1997;25:357–362. [PubMed] [Google Scholar]
- 41.Zhang W, Wu K, He W, Gao Y, Huang W, Lin X, Cai L, Fang Z, Zhou Q, Luo Z, Chen ZK, Zhou H. Transforming growth factor beta 1 plays an important role in inducing CD4(+)CD25(+)forhead box P3(+) regulatory T cells by mast cells. Clin Exp Immunol. 2010;161:490–496. doi: 10.1111/j.1365-2249.2010.04190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woodman L, Siddiqui S, Cruse G, Sutcliffe A, Saunders R, Kaur D, Bradding P, Brightling C. Mast cells promote airway smooth muscle cell differentiation via autocrine up-regulation of TGF-beta 1. J Immunol. 2008;181:5001–5007. doi: 10.4049/jimmunol.181.7.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao XY, Zhao LY, Zheng QS, Su JL, Guan H, Shang FJ, Niu XL, He YP, Lu XL. Chymase induces profibrotic response via transforming growth factor-beta 1/Smad activation in rat cardiac fibroblasts. Mol Cell Biochem. 2008;310:159–166. doi: 10.1007/s11010-007-9676-2. [DOI] [PubMed] [Google Scholar]
- 44.Kendall JC, Li XH, Galli SJ, Gordon JR. Promotion of mouse fibroblast proliferation by IgE-dependent activation of mouse mast cells: role for mast cell tumor necrosis factor-alpha and transforming growth factor-beta 1. J Allergy Clin Immunol. 1997;99:113–123. doi: 10.1016/s0091-6749(97)70308-7. [DOI] [PubMed] [Google Scholar]
- 45.Gordon JR, Galli SJ. Promotion of mouse fibroblast collagen gene expression by mast cells stimulated via the Fc epsilon RI. Role for mast cell-derived transforming growth factor beta and tumor necrosis factor alpha. J Exp Med. 1994;180:2027–2037. doi: 10.1084/jem.180.6.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pullen NA, Falanga YT, Morales JK, Ryan JJ. The Fyn-STAT5 Pathway: A New Frontier in IgE- and IgG-Mediated Mast Cell Signaling. Front Immunol. 3:117. doi: 10.3389/fimmu.2012.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffmann HJ, Frandsen PM, Christensen LH, Schiotz PO, Dahl R. Cultured human mast cells are heterogeneous for expression of the high-affinity IgE receptor FcepsilonRI. Int Arch Allergy Immunol. 2012;157:246–250. doi: 10.1159/000328756. [DOI] [PubMed] [Google Scholar]
- 48.Chang TW, Shiung YY. Anti-IgE as a mast cell-stabilizing therapeutic agent. J Allergy Clin Immunol. 2006;117:1203–1212. doi: 10.1016/j.jaci.2006.04.005. quiz 1213. [DOI] [PubMed] [Google Scholar]
- 49.Beck LA, Marcotte GV, MacGlashan D, Togias A, Saini S. Omalizumab-induced reductions in mast cell Fce psilon RI expression and function. J Allergy Clin Immunol. 2004;114:527–530. doi: 10.1016/j.jaci.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 50.Gomez G, Gonzalez-Espinosa C, Odom S, Baez G, Cid ME, Ryan JJ, Rivera J. Impaired FcepsilonRI-dependent gene expression and defective eicosanoid and cytokine production as a consequence of Fyn deficiency in mast cells. J Immunol. 2005;175:7602–7610. doi: 10.4049/jimmunol.175.11.7602. [DOI] [PubMed] [Google Scholar]
- 51.Bosse Y, Rola-Pleszczynski M. Controversy surrounding the increased expression of TGF beta 1 in asthma. Respir Res. 2007;8:66. doi: 10.1186/1465-9921-8-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Howell JE, McAnulty RJ. TGF-beta: its role in asthma and therapeutic potential. Curr Drug Targets. 2006;7:547–565. doi: 10.2174/138945006776818692. [DOI] [PubMed] [Google Scholar]
- 53.Makinde T, Murphy RF, Agrawal DK. The regulatory role of TGF-beta in airway remodeling in asthma. Immunol Cell Biol. 2007;85:348–356. doi: 10.1038/sj.icb.7100044. [DOI] [PubMed] [Google Scholar]
- 54.Luo X, Ding Q, Wang M, Li Z, Mao K, Sun B, Pan Y, Wang Z, Zang YQ, Chen Y. In vivo disruption of TGF-beta signaling by Smad7 in airway epithelium alleviates allergic asthma but aggravates lung carcinogenesis in mouse. PLoS One. 2010;5:e10149. doi: 10.1371/journal.pone.0010149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bottoms SE, Howell JE, Reinhardt AK, Evans IC, McAnulty RJ. Tgf-Beta isoform specific regulation of airway inflammation and remodelling in a murine model of asthma. PLoS One. 2010;5:e9674. doi: 10.1371/journal.pone.0009674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mabalirajan U, Aich J, Agrawal A, Ghosh B. Mepacrine inhibits subepithelial fibrosis by reducing the expression of arginase and TGF-beta1 in an extended subacute mouse model of allergic asthma. Am J Physiol Lung Cell Mol Physiol. 2009;297:L411–419. doi: 10.1152/ajplung.00138.2009. [DOI] [PubMed] [Google Scholar]
- 57.Fu CL, Ye YL, Lee YL, Chiang BL. Effects of overexpression of IL-10, IL-12, TGF-beta and IL-4 on allergen induced change in bronchial responsiveness. Respir Res. 2006;7:72. doi: 10.1186/1465-9921-7-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nemeth K, Keane-Myers A, Brown JM, Metcalfe DD, Gorham JD, Bundoc VG, Hodges MG, Jelinek I, Madala S, Karpati S, Mezey E. Bone marrow stromal cells use TGF-beta to suppress allergic responses in a mouse model of ragweed-induced asthma. Proc Natl Acad Sci U S A. 2010;107:5652–5657. doi: 10.1073/pnas.0910720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gentile DA, Doyle WJ, Zeevi A, Howe-Adams J, Trecki J, Skoner DP. Association between TNF-alpha and TGF-beta genotypes in infants and parental history of allergic rhinitis and asthma. Hum Immunol. 2004;65:347–351. doi: 10.1016/j.humimm.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 60.Noh G, Lee JH. Oral tolerance induction for human food allergy. Inflamm Allergy Drug Targets. 11:131–142. doi: 10.2174/187152812800392869. [DOI] [PubMed] [Google Scholar]
- 61.Ostroukhova M, Ray A. CD25+ T cells and regulation of allergen-induced responses. Curr Allergy Asthma Rep. 2005;5:35–41. doi: 10.1007/s11882-005-0052-6. [DOI] [PubMed] [Google Scholar]
- 62.Furth PA, Nakles RE, Millman S, Diaz-Cruz ES, Cabrera MC. Signal transducer and activator of transcription 5 as a key signaling pathway in normal mammary gland developmental biology and breast cancer. Breast Cancer Res. 13:220. doi: 10.1186/bcr2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Swiatek-Machado K, Kaminska B. STAT signaling in glioma cells. Adv Exp Med Biol. 986:189–208. doi: 10.1007/978-94-007-4719-7_10. [DOI] [PubMed] [Google Scholar]
- 64.Liang QC, Xiong H, Zhao ZW, Jia D, Li WX, Qin HZ, Deng JP, Gao L, Zhang H, Gao GD. Inhibition of transcription factor STAT5b suppresses proliferation, induces G1 cell cycle arrest and reduces tumor cell invasion in human glioblastoma multiforme cells. Cancer Lett. 2009;273:164–171. doi: 10.1016/j.canlet.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 65.Liang Q, Guo L, Gogate S, Karim Z, Hanifi A, Leung DY, Gorska MM, Alam R. IL-2 and IL-4 stimulate MEK1 expression and contribute to T cell resistance against suppression by TGF-beta and IL-10 in asthma. J Immunol. 2010;185:5704–5713. doi: 10.4049/jimmunol.1000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Macey MR, Sturgill JL, Morales JK, Falanga YT, Morales J, Norton SK, Yerram N, Shim H, Fernando J, Gifillan AM, Gomez G, Schwartz L, Oskeritzian C, Spiegel S, Conrad D, Ryan JJ. IL-4 and TGF-beta 1 counterbalance one another while regulating mast cell homeostasis. J Immunol. 2010;184:4688–4695. doi: 10.4049/jimmunol.0903477. [DOI] [PMC free article] [PubMed] [Google Scholar]