

Abstract
Signaling of the receptor for advanced glycation end products (RAGE) has been implicated in the development of injury-elicited vascular complications. Soluble RAGE (sRAGE) acts as a decoy of RAGE, and has been used to treat pathological vascular conditions in animal models. However, previous studies using sRAGE produced in insect Sf9 cells (sRAGESf9) used a high dose and multiple injections to achieve the therapeutic outcome. Here, we explore whether modulation of sRAGE N-glycoform impacts its bioactivity and augments its therapeutic efficacy. We first profiled carbohydrate components of sRAGECHO to show that a majority of its N-glycans belong to sialylated complex-types that are not shared by sRAGESf9. In cell-based NF-κB activation and vascular smooth muscle cell (VSMC) migration assays, sRAGECHO exhibited a significantly higher bioactivity relative to sRAGESf9 to inhibit RAGE alarmin ligand-induced NF-κB activation and VSMC migration. We next studied whether this N-glycoform-associated bioactivity of sRAGECHO is translated to higher in vivo therapeutic efficacy in a rat carotid artery balloon injury model. Consistent with the observed higher bioactivity in cell assays, sRAGECHO significantly reduced injury-induced neointimal growth and the expression of inflammatory markers in injured vasculature. Specifically, a single dose of 3 ng/g of sRAGECHO reduced neointimal hyperplasia by over 70%, whereas the same dose of sRAGESf9 showed no effect. The administered sRAGECHO is rapidly and specifically recruited to the injured arterial locus, suggesting that early intervention of arterial injury with sRAGECHO may offset an inflammatory circuit and reduce the ensuing tissue remodeling. Our findings showed that the N-glycoform of sRAGE is the key determinant underlying its bioactivity, and thus is an important glycobioengineering target to develop a highly potent therapeutic sRAGE for future clinical applications.
Keywords: sRAGE, N-glycoform, arterial injury, arterial inflammation, neointimal hyperplasia, therapeutic window
Introduction
It is well-established that inflammation, an innate response to vasculature injury, often results in excessive proliferation of VSMCs within the vessel wall and the subsequent formation of the neointima, often leading to the eventual occlusion of the vessel [1–3]. RAGE signaling has long been implicated in arterial inflammation, maladapdation, and atherogenesis [4–7]. RAGE- mediated vascular cell proliferation is due to the activation of pro-mitotic and pro-inflammatory cellular signaling programs, including NF-κB, by its diverse, endogenous ligands [8–10]. It has been demonstrated that a subset of RAGE ligands, especially those act as alarmins, function as chemoattractants for RAGE-mediated cell migration [11–13]. In addition to cellular inflammatory effects, the activation of these signaling cascades also triggers the RAGE promoter, which in turn, enhances RAGE expression, further augmenting the receptor’s signaling capacity in the vessel [14–16]. Vascular endothelial RAGE also functions as a receptor for Mac-1 integrin on leukocytes, promoting the recruitment of these inflammatory cells to the arterial wall [17]. Thus, arterial injury initiates these concurrent signaling and adhesion events that lead to chronic inflammation and tissue remodeling.
sRAGE is generated in vivo via the alternative splicing of the AGER gene, and by protease cleavage of RAGE on the cell surface, although the regulatory mechanisms of each event remains unclear [18, 19]. sRAGE functions as a decoy, competing with the plasma membrane-anchored counterparts for ligands, thus diminishing the signaling capacity of the latter [5]. Recombinant sRAGE produced in insect Sf9 cells (sRAGESf9) has been used to treat pathological vascular conditions in animal models, and a universal dose, i.e.100 μg/mouse/day, has been used in these studies in mice [4–6], and 0.5 mg/rat/day in rats [7]. A recent report has also shown that sRAGE binds monocytes and mediates inflammatory responses when RAGE ligands are not in excess [20]. Because insect and mammalian cells possess different glycosylation pathways, glycoproteins expressed in the two systems contain glycans of different structures [21]. In addition to the impact on the efficacy and in vivo half-life of a glycoprotein, glycoform modifications are also a potential source for immunogenicity. Currently, biosafety rules set by major regulatory authorities (FDA, EMEA) require human therapeutic glycoproteins to be produced from mammalian sources [22]. Therefore, there is a need to explore sRAGE produced in mammalian cells as a therapeutic candidate.
sRAGE encompasses the entire extracellular portion of RAGE including the N-glycosylation sites and the ligand binding regions. Prior studies have shown that polymorphisms that enhance N-glycosylation of RAGE increase its signaling [23], and that RAGE from tissues enriched by anti-carboxylated glycan antibodies has a higher ligand binding capacity to the high mobility group box 1 (HMGB1) and s100A family of RAGE ligands[24–26]. These observations imply that glycoform modifications play a role in RAGE-ligand interactions and signaling.
Based on these observations, we hypothesized that mammalian cell-produced sRAGE may have enhanced bioactivity. To test the hypothesis, we profiled the carbohydrate components of sRAGECHO and sRAGESf9, and performed both in vitro and in vivo studies using sRAGE produced in the two cell systems. Our results showed that sRAGECHO carrying the complex-type of N-glycoform with multivalent sialic acids has higher inhibitory activity to block RAGE-ligand-induced NF-κB activation and VSMC migration than that of sRAGESf9. In a rat carotid artery balloon injury model, sRAGECHO, in contrast to sRAGESf9, potently inhibited vascular inflammation in a single bolus injection of 3 ng/g dose, and reduces 71% neointimal hyperplasia. In addition, we also found that the administered sRAGECHO was rapidly and specifically recruited to the injured arterial locus, suggesting that an immediate treatment of arterial injury with sRAGE may offset inflammatory circuit to achieve a beneficial outcome. The greater efficacy of sRAGECHO to reduce injury-induced inflammation not only presents a proof of concept for realistic clinical applications, including treatment of arterial injury and inflammation, but also provides a molecular basis for further enhancement of sRAGE bioactivity via glyco-bioengineering in the future.
Materials and methods
Construction of sRAGE expression vector and recombinant Baculovirus
Human RAGE (NM_001136) cDNA sequence was used to construct the sRAGE expression vector. The coding sequence from amino acid 23–340 of RAGE was amplified with PCR and subcloned to a membrane-targeting expression vector that contains RAGE signal peptide and T7 epitope tag [27]. The same construct was used for establishing stable T7-sRAGE CHO-CD14 cell lines. Baculovirus carrying human T7-sRAGE cDNA was constructed with a Baculovirus Expression System with Gateway® Technology kit from Invitrogen, using pENTR1 vector. The constructed recombinant virus was used to infect Sf9 cells for the expression of sRAGESf9.
Affinity purification of sRAGE
Cell culture medium was collected daily from transient or stably-transfected CHO cells, or sRAGE-Baculovirus infected Sf9 cells, and centrifuged with low speed to remove dead cells. A Novagen T7 tag affinity purification kit was used to purify sRAGE, according to the manufacturer’s instruction. Total 5 fractions (1 ml of each) were collected, and fractions 2–4 were pooled. To monitor the purification steps, the input medium and individual fraction (15 μl of each) were resolved with 4–12% gradient SDS-polyacrylamide gel electrophoresis (PAGE) and probed with anti-T7 antibodies (Fig. S1a and b). A silver stain kit (Thermo Scientific) was used to stain the resolved samples to monitor the purity of the fractions (Fig. S1c). The concentration of purified sRAGE was assayed using a RAGE ELISA kit from R & D Systems, and the protein was stored in aliquot at −80°C. The purity of sRAGE, after affinity purification, is estimated to be about 80–85%. Prior to injection into animals, the sRAGE preparations were tested for endotoxin level using a ToxinSensor™ Chromogenic LAL Endotoxin Assay Kit from Genscript to ensure that the endotoxin level is below 0.5 EU/ml, and free of mycoplasma (tested with a Mycoscope™ mycoplasma PCR detection kit from Genlatis).
Western blotting and enzymatic treatments of sRAGE
Cell culture medium (10 μg of total protein) containing sRAGE were resolved with SDS 4–12% PAGE and western blotted with anti-T7 monoclonal antibodies (Novagen). Enzymatic treatments of sRAGE were performed according to the manufacturers’ instruction using cell culture medium (both PNGase F and neuraminidase were from New England Biolabs).
Carbohydrate Profiling of sRAGECHO and sRAGESf9 with high-performance anion-exchange chromatography (HPAEC)
Purified sRAGE was hydrolyzed and analyzed with HPAEC, using a Carbopac PA1 column with pulsed amperometric detection as described previously[28]. Briefly, PNGase F was used to release N-glycans from sRAGE. The released N-glycans were purified with a Sepharose CL-4B column, and resolved with HPAEC on a Carbopac PA-100 column (Dionex). The elution positions of N-glycans were compared to N-glycan standards. For chemical desialylation, N-glycans from sRAGECHO were treated with 20 mM HCl at 55°C for 30 min prior to HPAEC analysis.
NF-κB-luciferase reporter assay
A dual-luciferase reporter assay system (Promega) was used to monitor NF-κB activity, according to manufacturer’s instruction. Briefly, expression vectors harboring firefly luciferase gene controlled by NF-κB cis-element (pNF-κB-luciferase) and Renilla luciferase (pRL-SV40) were transfected to HEK293 or HEK293-RAGE cells in 24-well cell culture plates with a ratio of 1:7. The transfected cells were incubated in 0.1% FBS-supplemented medium for 24 h, and were then treated with RAGE ligands (100 nM) ± sRAGEs (100 nM) for 2 h in serum-free medium followed with luciferase activity assays. NF-κB activity was measured in triplicate and normalized with Renilla luciferase activity, and inhibition was calculated as percentage relative to no addition of sRAGE.
VSMC migration assay
Cell migration assays were performed as previously described [3]. Briefly, VSMCs were harvested from thoracic aortae of 24-months old male Fisher 344 rats and cultured in DMEM medium according to the protocol. The cells (< 5 passages) were serum-starved in 0.1% FBS-supplemented medium for 24 hours prior to the experiments, and then re-suspended in 0.1% BSA supplemented DMEM. VSMCs (106/assay) were placed on the upper Boyden chambers with 8 μm pore and Matrigel-coated polycarbonate filters (BD Biosciences), and RAGE ligands (100 nM) ± sRAGE (100nM) were added to the lower chamber. The chambers were incubated at 37°C for 4 h, and cells in the lower chambers were fixed and stained using HEMA3 system (Curtin Matheson Scientific Inc.). Each condition was measured in triplicate and migrated cells were counted in 4 random fields of each filter.
Rat carotid artery balloon denudation injury model and administration of sRAGE
Male Wistar rats (400–450g) were administered with 3% isoflurane for anesthesia, and a neck midline incision was made to expose the left carotid artery. A Fogarty balloon catheter (Edwards Lifesciences) was inserted into the external carotid branch to the aortic arch and inflated to produce slight resistance. The catheter was then withdrawn 3 times to produce sufficient injury on the endothelium. The investigator performing the surgical procedure was blinded with respect to the subsequent injections and other analyses. After the surgery, depending upon specific experiment, purified sRAGE ranging 0.5 – 6 ng /g body weight were administered via intraperitoneal injection with saline as placebo. The procedure was conducted according to the Institutional Animal Care and Use Committee and complied with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 3040-2, revised 1999).
Histomorphological analyses and tissue collection
Rats were euthanized 2 weeks post-surgery. Immediately following the sacrifice, the thorax cavity was opened, and the vasculature was flushed with saline and replenished with 10% neutral buffered formalin (NBF). The organs and aortae were then harvested as described [2]. The carotid aorta was dissected free from the surrounding connective tissue and further fixed in 10% NBF. Cross-sections of the aorta, 2–3 mm in length, were taken from aortic-arch to bifurcation region (internal and external carotid artery), processed, and embedded in paraffin to be further cut to 7μm thin sections for hematoxylin-eosin staining and immunohistochemistry. Morphological analyses of carotid artery segments were performed with a digital imaging analysis system (MCID, GE Healthcare). Investigators performing the injections and histomorphological analyses were blinded with respect to doses and preparations of sRAGE and placebo in the studies to obtain unbiased assessments.
Immunohistochemistry
A standard avidin biotin complex method was used. Briefly, vessel sections were deparaffinized with xylene and rehydrated through gradient ethanol immersion. Antigen retrieval was performed by microwaving the sections for 5 min in citric acid buffer (2 mM citric acid and 9 mM trisodium citrate dehydrate, pH 6.0). After washing with PBS, the slides were treated with peroxidase blocking buffer for 5 min to quench endogenous peroxidase activity and blocked with 2% BSA at room temperature for 1 h followed by washing and incubation with primary antibodies. Rabbit polyclonal anti-RAGE, anti-s100, and anti-HMGB1 (all with 1: 1,000 dilution), and mouse monoclonal anti-ICAM-1 antibodies (1: 100) were from Abcam, rat monoclonal anti-VCAM-1 antibodies (1: 100) were from Covance. The specimens were then incubated with biotinylated secondary antibodies, washed with PBS containing 0.02% Tween-20, and reincubated with streptavidin-horseradish peroxidase for detection. The sections were counterstained with hematoxylin, rinsed, dehydrated and mounted with mounting medium. The staining was visualized with a diaminobenzidine liquid substrate system (Dako).
Monitor tissue-targeting of sRAGECHO in the vessel
Immediately after the balloon injury procedure, the rat was administered with a dose of sRAGECHO (6 ng/g) via i.p. injection. The rat was euthanized 1 h post-injection, and both left and right carotid arteries (injured and non-injured, respectively) were isolated and embedded to paraffin blocks following the standard procedure. The vessel cross-sections were then deparaffinized and immunostained with Novagen mouse monoclonal anti-T7 antibodies (1:100 dilution) as described in the last section.
Statistical analyses
Data were reported as mean ± SEM. All histomorphological data were first subjected to normality tests (Shapiro-Wilk) prior to one-way analysis of variance (ANOVA) followed with the Student-Newman-Keuls post hoc multiple comparison procedure. SAS 9.2 was used to perform the analyses. Statistical significance is determined when p ≤ 0.05.
Results
The majority of sRAGECHO is modified with the complex type of N-glycoforms
RAGE contains two sequons, or consensus tripeptide sequence for N-linked glycosylation, starting at N residues 25 and 81[29]. To confirm that both sequons are indeed glycosylated in sRAGECHO, we mutated both N25 and N81 to T, and tested migration pattern of the wild-type sRAGE, or sRAGE carrying N25T, N81T, and N25T/N81T composite mutations on SDS-PAGE. sRAGECHO(N25T/N81T) migrates at the same position as sRAGECHO treated with peptide-N-glycosidase F (PNGase F), suggesting that sRAGECHO is N-glycoform modified at both sequons (Fig. S2). While it is known that insect cells generate only paucimannose-type N-glycans, mammalian cells may produce more diverse types of N-glycans, including pauci-/high-mannose-, hybrid-, and more often, complex-type of N-glycans [21]. To examine whether sRAGECHO is modified differently than sRAGESf9, we first compared the migration of untreated and PNGase F-treated sRAGE produced from the two cell systems on SDS-PAGE. The untreated sRAGESf9 migrates faster than that of sRAGECHO (Fig. 1, lanes 1 and 2); whereas sRAGE from the two sources migrate similarly after PNGase F treatment that removes the entire N-glycan (Fig. 1, lanes 7–10). These observations suggest that sRAGE produced from the two cell systems are both N-glycosylated but are modified with different N-glycoforms.
Fig. 1.
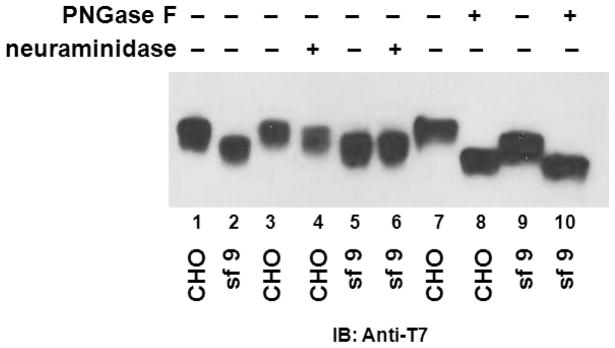
sRAGECHO and sRAGESf9 are modified with different N-glycoforms. Cell culture media of transfected CHO cells, or recombinant baculovirus infected Sf9, were collected and 10 μg of total protein were resolved on SDS 4–12% gel followed with anti-T7 antibodies blotting. Lane 1–2: untreated samples; lane 3–6: neuraminidase and mock-treated samples; lane 7–10: PNGase F and mock-treated samples.
We next analyzed the N-glycoform of sRAGECHO and sRAGESf9, using high-performance anion-exchange chromatography (HPAEC). Resolving PNGase F-released N-glycans from sRAGECHO and sRAGESf9 with HPAEC and comparing their elution positions with standard glycan types, we showed that N-glycans from these two cell origins exhibited different chromatographic patterns: while majority of N-glycans from sRAGESf9 are neutral species (Fig. 2a lower panel), majority of N-glycans (about 70%) from sRAGECHO are anionic species (Fig. 2a, upper panel). Insect cells modify glycoproteins with paucimannose glycans, whereas mammalian cells can modify glycoproteins with paucimannose, hybrid, and most often, complex-type of glycans [21]. Complex-type glycans contain a 3-mannose (Man) core structure linked to a variable number of N-acetylglucosamine (GlcNAc), galactose (Gal), fucose (Fuc) and sialic acid (also termed N-acetylneuraminic acid, Neu5Ac) residues, while high-mannose-type glycans have a 2–3-fold higher mannose content and no Gal, GlcNAc and sialic acid. Hybrid-type glycans have one Gal-GlcNAc sequence in one branch and variable numbers of Man on the other branch. Our carbohydrate profiling data (Table S1) suggests that sRAGECHO may contain all three types of N-glycans. Desialylation of N-glycans with mild acid almost completely eliminated anionic glycans and produced free aslialoglycans (Fig. 2b), suggesting that the majority of the observed anionic species from sRAGECHO are sialylated glycans, and hence belong to the complex-type. Treatment of sRAGECHO with neurominidase, which removes sialic acids from the N-glycan, also affects its migration on SDS-PAGE, whereas the same treatment does not affect sRAGESf9(Fig. 1, lanes 3–6), confirming that the majority of sRAGECHO, but not sRAGESf9, are indeed modified by sialylated complex-type N-glycoforms.
Fig. 2.

HPAEC analyses of N-glycan profile of sRAGECHO and sRAGESf9. a The HPAEC elution profile of PNGase F released sRAGECHO (upper panel), and sRAGESf9 (lower panel), N-glycans. b The profile of desilalylated (lower panel) versus untreated (upper panel) sRAGECHON-glycans.
The activity of sRAGE to inhibit NF-κB activation is associated with its N-glycoform modifications
RAGE signaling activates NF-κB transcription program that leads to transcription of inflammatory molecules and to tissue inflammation [9]. To test whether the N-glycoform of sRAGE is indeed associated with its bioactivity to inhibit RAGE signaling, we determined how differentially modified sRAGEs from Sf9 and CHO cells inhibit NF-κB activation, using NF-κB-driven luciferase reporter assays. RAGE alarmin ligands, HMGB1 and s100B, specifically activate NF-κB in human embryonic kidney (HEK293) cells that stably express human RAGE (Fig. 3a and b). Adding sRAGE with the RAGE ligand (in 1:1 molar ratio) dampens NF-κB activation (Fig. 3c and d): sRAGECHO blocks 58.6% of HMGB1-induced, and 40.2% of s100B-induced NF-κB activity respectively, whereas unglycosylated sRAGECHO(N25T/N81T) barely inhibits NF-κB activity (0.3% of HMGB1-induced and 4.1% of s100B-induced NF-κB activity, respectively). sRAGESf9 also blocks NF-κB activation but with reduced activity (19.7 % of HMGB1-induced, and 20.4% of s100B-induced, respectively). Together, these results demonstrated that sRAGE inhibition of NF-κB is associated not only with its N-glycosylation, but also with the specific N-glycoforms attached to the proteins.
Fig. 3.

Inhibition of RAGE ligand-activated NF-κB activity by different N-glycoform-modified sRAGE. For sRAGE blocking, RAGE ligand (100 nM) and sRAGE (100 nM) were added to cells for 2 h at 37 °C before lysis and luciferase assays. a NF-κB activation by RAGE ligand HMGB1; b NF-κB activation by RAGE ligand s100B. c Inhibition of HMGB-induced NF-κB activation by different N-glycoform-modified sRAGE. d Inhibition of s100B-induced NF-κB activation by different N-glycoform-modified sRAGE. *: p < 0.05; **: p < 0.01; ***: p < 0.0001, n=9 for each group.
The activity of sRAGE to block RAGE ligand-induced VSMC migration is associated with its N-glycoform modifications
VSMC migration is a critical step during injury-induced neointimal growth in the vessel [7], and it has been demonstrated that alarmin ligands HMGB1 and s100B mediate RAGE-dependent cell migration [11–13]. We thus tested whether N-glycoform-associated sRAGE bioactivities are also effecting at blocking VSMC migration. Adding differentially modified sRAGEs with HMGB1 to rat VSMCs demonstrated that a reduction of ligand-induced chemotaxis is associated with sRAGE N-glycoform: sRAGECHO has a more potent inhibitory effect than that of sRAGESf9 or of sRAGECHO(N25T/N81T) (Fig. 4a and b). Similar results were obtained in migration assays using s100B as the chemoattractant (Fig. 4c and d). In both cases, sRAGECHO appears to specifically block RAGE ligand-induced VSMC migration, as adding sRAGECHO does not affect platelet-derived growth factor (PDGF)-induced VSMC migration (Fig. 4). These results suggest that sRAGE bioactivity to block RAGE alarmin ligand-induced VSMC migration is also associated with specific N-glycoform modifications.
Fig. 4.

Inhibition of RAGE-ligand induced VSMC migration by different N-glycoform-modified sRAGE. PDGF was used as a position control. For inhibition, sRAGE was added to RAGE ligand with a molar ratio of 1:1. a Inhibition of HMGB1-induced VSMC migration by different N-glycoform-modified sRAGE (n=12). b Representative fields from a. c Inhibition of s100B-induced VSMC migration by different N-glycoform-modified sRAGE (n=12). d Representative fields from c. The scale bar = 100 μm. *: p < 0.05; **: p < 0.01; ***: p < 0.000, NS: not significant.
A single dose of administered sRAGECHO significantly reduced neointimal growth in a rat carotid artery balloon injury model
Previous studies showed that administration of sRAGESf9 reduces neointimal growth after arterial injury in mice and rat models [6, 7]. To study N-glycoform-associated sRAGE bioactivities in vivo, we employed a well-established rat carotid balloon injury model to monitor injury-elicited neointimal hyperplasia. Our initial tests showed that a much-lowered dose of sRAGECHO was effective (Fig. S3). Because tissue injury triggers the release of RAGE alarmin ligands, we first tested whether scavenging of RAGE alarmin ligands with sRAGECHO immediately after the injury will be more effective, i.e. to reduce the initial 3 regimens to a single bolus injection. Our results showed that a single bolus intraperitoneal (i.p.) injection of sRAGECHO immediately after the balloon-injury procedure was as effective as 3 injections of the same dose (Fig. 5), suggesting that blocking RAGE signaling immediately after arterial injury is beneficial. The effective dose of sRAGECHO was then determined with a single injection of varied dosages administered immediately after the balloon injury procedure in rats. As shown in Fig. 6 and Table 1, a single dose of 3 ng/g body weight blocked 71% neointimal growth, suggesting that sRAGECHO is highly effective in vivo. Doubling of the injected dosage (6 ng/g) did not render a significantly higher percentage of the blockade (Table 1). Examination of major organs of rats that received the injections found no apparent changes (Table S1).
Fig. 5.
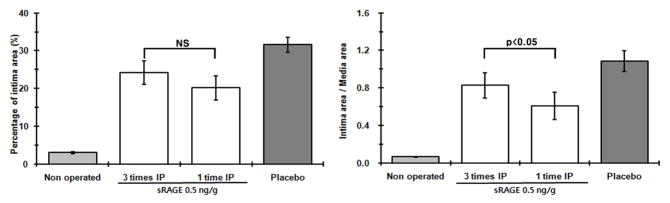
A single bolus injection of sRAGECHO suppresses neointimal hyperplasia in the vessel. sRAGECHO (0.5 ng/g) administered 3 times, or one time immediately after balloon injuries were compared. The left panel shows the percentage of neointima area in the vessel wall, and the right panel shows I/M (intima-to-media) ratio (n = 8–10, p< 0.05).
Fig. 6.

Dose-dependent suppression of neointimal growth by sRAGECHO. a Dose-dependent suppression of neointimal growth by sRAGECHO. A single dose of 0.5, 1, 1.5, 3, and 6 ng/g of sRAGECHO was injected via i.p. immediately after the balloon injury, and histomorphological analyses of vessel sections were made 2-week post-surgery. Both the percentage of neointimal area in the vessel wall (left panel) and the I/M ratio (right panel) were shown. (n = 8–10, p <0.05). b–g Representative histological vessel sections from each treatment group. b– g Non-operated, and balloon-injured vessel sections with the placebo, 0.5, 1, 1.5, 3, and 6 ng/g sRAGECHO treatment, respectively.
Table 1.
Effects of sRAGE dose in vessels of balloon-injured rats
| Intima area (μm2) | Reduced intima area (%) a | Intima/media | Reduced intima/media (%) b | ||
|---|---|---|---|---|---|
| Placebo | 120,204.15 | 0 | 1.08 | 0 | |
| sRAGE | 0.5ng/g | 66417.22 | 35.8 | 0.61 | 43.3 |
| 1.0ng/g | 52827.58 | 47.3 | 0.45 | 58.2 | |
| 1.5ng/g | 37672.84 | 60.2 | 0.34 | 68.3 | |
| 3.0ng/g | 22850.26 | 71.6 | 0.23 | 78.8 | |
| 6.0ng/g | 20889.66 | 73.2 | 0.20 | 81.4 | |
Prior pharmacokinetic studies in rats showed that i. p.-administered sRAGE peaks in the bloodstream shortly after the injection, and the plasma level of sRAGE then sharply declines to a low level that sustains for a few days [30]. To confirm that the administered sRAGECHO indeed reaches the injured locus in the vessel wall, we isolated injured (left) and non-injured (right) carotid vessels from a rat 1 h post-injection of sRAGECHO, and performed immunostaining of the vessel sections using anti-T7 antibodies that recognize the epitope tag of sRAGECHO. The injured carotid vessel section stained with more sRAGECHO than the non-injured vessel from the same animal (Fig. 7), suggesting that administered sRAGECHO not only quickly reaches the vessel wall, but also is specifically recruited to the injured locus. These observations establish an effective therapeutic window that maximizes a beneficial outcome.
Fig. 7.
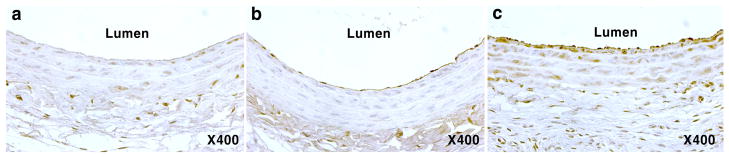
sRAGECHO is specifically recruited to the injured arterial locus. Carotid artery cross-sections from rats that was not administered with sRAGECHO (a), and administered with sRAGECHO (b and c) were immunostained with anti-T7 antibodies. b, the non-injured artery; c the injured artery.
Treatment of sRAGECHO reduces inflammatory markers in the vessel
Previous studies showed that sRAGESf9 reduces neointimal expansion by scavenging RAGE ligands, leading to attenuation of vascular inflammation and restriction of the expression of adhesion molecules as well as RAGE on the cell surface [6]. We next tested whether sRAGECHO also attenuates vascular inflammation by assessing the expression of inflammatory markers, ICAM-1 and VCAM-1, as well as RAGE, on sRAGECHO (3 ng/g) and placebo-treated vessel sections using immunostaining (Fig. 8a–c). Compared to placebo, the sRAGECHO significantly reduced expression of these markers, suggesting a down-regulation of inflammatory signaling and monocyte adhesion mediated by these markers. We also examined the expression of RAGE alarmin ligands, HMGB1 and s100B, on the vessel sections. As shown in Fig. 8d and e, HMGB1 and s100B immunolabeling of vessel sections is significantly reduced by sRAGECHO compared to placebo, suggesting that sRAGECHO exerts the therapeutic effects via restriction of RAGE alarmin ligand-induced inflammation.
Fig. 8.

Immunohistochemical assessments of inflammatory markers in sRAGECHO-treated vessels. Vessel sections from sRAGECHO treatment (3 ng/g) and placebo were stained with indicated primary antibodies. Left panel: representative immunohintochemical staining of vessel section; right panel: overall assessment (n = 8–10, p <0.05). a anti-ICAM-1 antibodies staining; b anti-VCAM-1 antibodies staining; c anti-RAGE antibodies staining; d anti-HMGB1 antibodies staining; e anti-s100B antibodies staining.
sRAGECHO exhibits significantly higher efficacy than sRAGESf9 to reduce neointimal growth
To compare the in vivo efficacy of sRAGECHO and sRAGESf9 in balloon-injured rats, we injected a single dose of 3 ng/g of sRAGECHO in parallel with the same dose of sRAGESf9 or sRAGECHO(N25T/N81T). Compared to placebo, neointimal growth was significantly reduced (i.e. 71% reduction) by a single dose of sRAGECHO, whereas the reduction of neointimal growth by the same dose of sRAGESf9 or sRAGECHO(N25T/N81T) was insignificant (Fig. 9). These results confirmed that the N-glycoform in sRAGE also significantly impacts its therapeutic efficacy in vivo.
Fig. 9.

N-glycoform of sRAGE contributes to its high therapeutic efficacy. A single dose of 3 ng/g sRAGECHO, sRAGESf9, and sRAGECHO(N25T/N81T) was injected to rats (n =8–12/group) immediately after surgery, and histomorphological analysis on vessel sections were performed 2-week post-surgery. a The percentage of neointimal area in the vessel wall. b The I/M (intima-to-media) ratio of the vessels. NS: not significant. (c–g). Representative histological vessel sections from each treatment group: c Non-operated; d sRAGECHO-treated; e sRAGESf9-treated; f sRAGECHO(N25T/N81T)-treated; g Placebo-treated. Paired arrows mark neointimal areas.
Discussion
Although the therapeutic functions of sRAGE have been well recognized in several disease models [4–7], and N-glycoform modifications have been implicated in RAGE signaling capacity or sRAGE bioactivity [13, 23, 25], how post-translational modifications impact the in vivo therapeutic efficacy of sRAGE, or the specific N-glycoform associated with sRAGE bioactivity, have not been studied. Our current studies addressed these aspects.
Complex-type N-glycoform modifications of sRAGE are critical for its bioactivity in vitro and therapeutic efficacy in vivo
Our N-glycan profiling results demonstrated that a majority of N-glycans from sRAGECHO belongs to the complex-type with multivalent sialylations (Fig. 2 and Table S1). Although the two sequons in sRAGE are both N-glycosylated in Sf9 and CHO cells, the specific glycoforms attached to sRAGE produced in the two systems are quite different (Figs.1 and 2, S1). In studies performed at cellular level, we showed that sRAGECHO are more potent than sRAGESf9 or sRAGECHO(N25T/N81T) to inhibit RAGE-ligand induced NF-κB activation and VSMC migration (Fig. 3 and 4). Our results suggest that the observed in vitro bioactivities of sRAGE not only require N-glycosylation of the protein, but also are associated with the complex type N-glycans that can only be generated in mammalian cells.
The enhanced efficacy of N-glycoform-specific bioactivities of sRAGECHO also reflected in our result thatdemonstrated a single low dose significantly blocks injury-elicited neointimal growth (Fig 5 and 6, S3, Table 1) and inflammation (Fig. 8). Previous studies in a mouse femoral artery injury model employed a dose of 100 μg/mouse/day of sRAGESf9 for one week to block neointimal growth [6]. Assuming that an average lab mouse weighs 20 g, this dose is equivalent to 5 μg/g body weight/day. In the same model, administration of 20 μg/mouse/day of sRAGESf9 did not block neointimal growth, suggesting that the use of a high dose was necessary to achieve the therapeutic outcome[6]. In a rat balloon injury model similar to the one used in our studies, Zhou and colleagues showed that a daily sRAGESf9 regimen of 0.5 mg/rat for 6 days suppresses neointimal growth [7]. This dose is equivalent to 1.25 μg/g body weight/day, as rats used in the study weighed 400 g in average. In contrast, our studies showed that only 3 ng/g of sRAGECHO was required to block over 70% of neointimal growth resulted from the balloon injury (Fig 6 and Table 1). In addition, we also showed that a single administration of sRAGECHO, rather than daily, multiple injections, was sufficient to attain a beneficial outcome (Fig. 5). Critically, in a direct comparison between sRAGESf9 and sRAGECHO in the rat carotid balloon injury model, we demonstrated that a dose of 3 ng/g sRAGESf9, like that of sRAGECHO(N25T/N81T), was ineffective in restricting neointimal growth (Fig. 9). The low efficacy of sRAGESf9 shown in our in vivo studies appears not due to an abnormality of the preparation such as protein denaturation, as sRAGESf9 was prepared with the same procedure as the CHO counterpart, and was able to inhibit NF-κB activation and VSMC migration albeit with lowered activities (Fig. 3 and 4).
Insect cell-originated paucimannose glycoforms are immunogenic in mammals, and human therapeutic glycoproteins therefore must be produced in mammalian cell lines or sources [22]. In addition to N-glycoform-associated bioactivities, potential immunogenicity of sRAGESf9 may also contribute to the low efficacy of sRAGESf9 observed in previous studies [6, 7]. The sRAGECHO protein is tagged with a T7 tag for affinity purification (Fig. S1), and epitope tags are highly immunogenic. Since our model deals with acute injuries, and sRAGECHO was administered with a single low dose, immunogenicity of the epitope tag appeared not to impact the therapeutic efficacy. A recent report showed that sRAGESf9 binds murine monocytes, and enhances their survival and differentiation when administered in a high dose (i.e. 5 μg/g) [20], it is unclear whether such inflammatory reactions are due to glycan immunogenicity, or due to use a high dose.
How N-glycoform modifications of sRAGE contribute to its therapeutic efficacy?
Although it is known that N-glycoform modifications increase efficacy of glycoproteins by enhancing their bioactivities including target binding capacity and in vivo duration, how N-glycoforms contribute to sRAGE bioactivity and efficacy require additional study. Prior studies found that HMGB1 interacts with the V domain of RAGE and that this interaction is glycosylation dependent [24]; the binding of s100B, however, does not depend upon glycosylation of RAGE[31]. Our surface plasmon resonance binding studies of these two ligands seem to agree with these early observations (Fig. S4 and Table S2). In our experimental setting, HMGB1 binds sRAGECHO only, whereas s100B appears to have higher affinity to the unglycosylated sRAGECHO(N25T/N81T). It is possible that sRAGECHO with a higher binding capacity for HMGB1 and an albeit compromised capacity for s100B, still possesses overall higher in vivo efficacy for treating arterial injury. Whether sRAGECHO is also more potent than sRAGESf9 for treating neuronal injury is unknown, as s100B is a dominant alarmin ligand released by injured astrocytes [32].
RAGE is a pattern recognition receptor, and its interactions with ligands may not follow a kinetic model used for a classic receptor. RAGE ligands such as AGE are of heterogeneous nature, or are oligomeric. Previous studies have found that either RAGE/sRAGE, or RAGE ligands form oligomers with undefined molecular masses [25, 33]. If the dissociation constant (KD) is calculated based on a molecular mass of monomeric RAGE or RAGE ligands, the resultant binding affinity index (KD−1) may not reflect the actual RAGE-ligand interactions or efficacy observed in vivo. Further, the participation of glycoforms in RAGE-ligand interactions may also preclude the process from a classic model, as geometric arrangement of carbohydrates of the glycans in a receptor may significantly enhance its clustering [34], and such clustering or polymerization of a receptor may dramatically influences entropic effects of ligand binding and signaling events in vivo[35, 36]. Our observations that RAGE/sRAGE form dimer-based oligomers [33] and that N-glycoforms influence sRAGE bioactivities appear to agree with the theory proposed by Srikrishna and colleagues, i.e. RAGE-ligand interactions may follow a complex, multivalent kinetic model that concerns the N-glycoform of the receptor [25], rather than a simple kinetic model formulated for a classic receptor-ligand interaction.
Anti-carboxylate antibodies (mAbGB3.1)-enriched sRAGE from bovine lungs showed a higher bioactivity to block NF-κB activation than that of sRAGESf9 [25], and sRAGE purified from bovine lungs potently blocks monocyte migration [13]. These results are consistent with our finding that sRAGE from mammalian origins has higher bioactivities versus that of non-mammalian origin. While the N-glycoforms of sRAGE from lung tissues are unknown, the majority of sRAGECHO belongs to sialylated complex type N-glycoforms (Fig. 2 and Table S1). Srikrishna and colleagues also showed that only a small portion of RAGE expressed in lung tissues are carboxylated [25], whereas CHO cells with their high capacity for glycosylation have often been used as the host cell by the pharmaceutical industry for the production of therapeutic glycoproteins [22, 37, 38]. Despite the observed high efficacy in our acute injury model, the exact N-glycan structure in sRAGECHO that is critical for the bioactivity remains unknown. Future studies to elucidate the specific N-glycan structure that impacts RAGE signaling and ligand interactions should help to design an even more potent sRAGE through glyco-bioengineering for clinic applications.
Therapeutic window also contributes to sRAGECHO efficacy to restrict neointimal growth
Our strategy to administer sRAGECHO immediately after the arterial injury may also contribute to the observed high efficacy. Our studies showed that administered sRAGECHO is rapidly and specifically recruited to the locus where the trauma occurred (Fig. 7). Since RAGE alarmin ligands HMGB1 and s100B are released from injured and necrotic cells [39], scavenging these ligands at early acute injury stage may circumvent the positive feed of RAGE signaling [9], and thus offset the inflammatory circuit and impede the ensuing tissue remodeling in the vessel. Studies of erythropoietin (EPO) in a rat model have also shown that a single dose administered immediately after coronary ligation effectively protects cardiac tissues from ischemic damage and apoptosis, whereas multiple administrations of EPO after the surgery not only do not add additional therapeutic benefits, but also results in adverse side effects, including an increased hematocrit and thrombosis [40, 41]. Similarly, a delayed administration of sRAGESf9 was also ineffective to restrict neointimal growth in a murine model [6]. Our results showing that a single dose administered immediately after the acute injury is sufficient to significantly reduce neointimal remodeling (Fig. 5) reinforce the concept that using a small, but effective dose of sRAGE immediately after the injury is likely to be beneficial at least for an acute injury.
We did not observe any adverse effects on rats that were administered sRAGECHO in the present study (Table S3). It remains to be determined whether a high dose of sRAGECHO will trigger monocyte reaction [20]. If the observed monocyte inflammatory response is due to the use of a high dose of sRAGE, our finding that the effective dose of sRAGECHO is significantly lower than that of sRAGESf9 may provide an alternative avenue to prevent this side-effect, while maintaining therapeutic efficacy. Because our model system deals with acute injury only, it is unclear whether multiple administrations of low dose of sRAGECHO in chronic models will impact the physiology of the animal, or elicit inflammatory responses. Future studies using different models may address this question more adequately and aid further development of sRAGE as a therapeutic protein applicable in multiple pathological conditions.
Supplementary Material
Highlights.
The specific N-glycoform modification is the key underlying sRAGE bioactivity;
Markedly reduced sRAGE dose to attenuate neointimal hyperplasia and inflammation;
Provide a molecular target for glycobioengineering of sRAGE as a therapeutic protein;
Blocking RAGE alarmin ligands during acute injury phase offsets neointimal growth.
Acknowledgments
We thank Robert Monticone for rat VSMCs and advice on cell migration assays. We also thank reviewers of this manuscript for their inputs that improve our work. The work was supported by the intramural research program of the NIH, National Institute on Aging (LL, MIT, RPX and EGL), and the Korea Research Foundation grant KRF-2009-013-E00008 (SP). WW was supported in part by the Oak Ridge Institute for Science and Education’s Research Associates Program at NIH; JP was a recipient of the 2011 Johns Hopkins “Excellent in Medical Student Research Award”; RAR and DB were supported by intramural research training awards from NIH.
Footnotes
Conflicts of interest
NIH has filed a patent based on this work. The authors (S.P., W. W., R.-P. X. M.I.T, E.G.L., or L. L) have not received any loyalties from this patent.
References
- 1.Inoue T, Node K. Molecular basis of restenosis and novel issues of drug-eluting stents. Circ J. 2009;73:615–621. doi: 10.1253/circj.cj-09-0059. [DOI] [PubMed] [Google Scholar]
- 2.Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009;78:539–552. doi: 10.1016/j.bcp.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu CB, Sun Y, Edvinsson L. Cardiovascular risk factors regulate the expression of vascular endothelin receptors. Pharmacol Ther. 2010;127:148–155. doi: 10.1016/j.pharmthera.2010.04.013. [DOI] [PubMed] [Google Scholar]
- 4.Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 6.Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, Andrassy M, Marso SP, Duda S, Arnold B, Liliensiek B, Nawroth PP, Stern DM, Schmidt AM, Naka Y. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. 2003;111:959–972. doi: 10.1172/JCI17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Z, Wang K, Penn MS, Marso SP, Lauer MA, Forudi F, Zhou X, Qu W, Lu Y, Stern DM, Schmidt AM, Lincoff AM, Topol EJ. Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation. 2003;107:2238–2243. doi: 10.1161/01.CIR.0000063577.32819.23. [DOI] [PubMed] [Google Scholar]
- 8.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 9.Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, Pinsky D, Stern D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem. 1994;269:9889–9897. [PubMed] [Google Scholar]
- 10.Lin L, Park S, Lakatta EG. RAGE signaling in inflammation and arterial aging. Front Biosci. 2009;14:1403–1413. doi: 10.2741/3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–1206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy MA, Li SL, Sahar S, Kim YS, Xu ZG, Lanting L, Natarajan R. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J Biol Chem. 2006;281:13685–13693. doi: 10.1074/jbc.M511425200. [DOI] [PubMed] [Google Scholar]
- 13.Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–1182. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 14.Bierhaus A, Illmer T, Kasper M, Luther T, Quehenberger P, Tritschler H, Wahl P, Ziegler R, Muller M, Nawroth PP. Advanced glycation end product (AGE)-mediated induction of tissue factor in cultured endothelial cells is dependent on RAGE. Circulation. 1997;96:2262–2271. doi: 10.1161/01.cir.96.7.2262. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem. 1997;272:16498–16506. doi: 10.1074/jbc.272.26.16498. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem. 2000;275:25781–25790. doi: 10.1074/jbc.M001235200. [DOI] [PubMed] [Google Scholar]
- 17.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo J. 2007;26:1129–1139. doi: 10.1038/sj.emboj.7601552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, Okamoto H, Watanabe T, Yamamoto H. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003;370:1097–1109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Bukulin M, Kojro E, Roth A, Metz VV, Fahrenholz F, Nawroth PP, Bierhaus A, Postina R. Receptor for advanced glycation end products is subjected to protein ectodomain shedding by metalloproteinases. J Biol Chem. 2008;283:35507–35516. doi: 10.1074/jbc.M806948200. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Wang H, Piper MG, McMaken S, Mo X, Opalek J, Schmidt AM, Marsh CB. sRAGE induces human monocyte survival and differentiation. J Immunol. 2010;185:1822–1835. doi: 10.4049/jimmunol.0903398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varki A, Cummings RD, Esco JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. Essentials of glycobiology. 2. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2009. [PubMed] [Google Scholar]
- 22.Ghaderi D, Zhang M, Hurtado-Ziola N, Varki A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol Genet Eng Rev. 2012;28:147–175. doi: 10.5661/bger-28-147. [DOI] [PubMed] [Google Scholar]
- 23.Park SJ, Kleffmann T, Hessian PA. The G82S polymorphism promotes glycosylation of the receptor for advanced glycation end products (RAGE) at asparagine 81: comparison of wild-type rage with the G82S polymorphic variant. J Biol Chem. 2011;286:21384–21392. doi: 10.1074/jbc.M111.241281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srikrishna G, Huttunen HJ, Johansson L, Weigle B, Yamaguchi Y, Rauvala H, Freeze HH. N-Glycans on the receptor for advanced glycation end products influence amphoterin binding and neurite outgrowth. J Neurochem. 2002;80:998–1008. doi: 10.1046/j.0022-3042.2002.00796.x. [DOI] [PubMed] [Google Scholar]
- 25.Srikrishna G, Nayak J, Weigle B, Temme A, Foell D, Hazelwood L, Olsson A, Volkmann N, Hanein D, Freeze HH. Carboxylated N-glycans on RAGE promote S100A12 binding and signaling. J Cell Biochem. 2010;110:645–659. doi: 10.1002/jcb.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, Varki N, Kronenberg M, Freeze HH, Srikrishna G. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis. 2008;29:2035–2043. doi: 10.1093/carcin/bgn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pang J, Zeng X, Xiao RP, Lakatta EG, Lin L. Design, generation, and testing of mammalian expression modules that tag membrane proteins. Protein Sci. 2009;18:1261–1271. doi: 10.1002/pro.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomiya N, Kurono M, Ishihara H, Tejima S, Endo S, Arata Y, Takahashi N. Structural analysis of N-linked oligosaccharides by a combination of glycopeptidase, exoglycosidases, and high-performance liquid chromatography. Anal Biochem. 1987;163:489–499. doi: 10.1016/0003-2697(87)90253-3. [DOI] [PubMed] [Google Scholar]
- 29.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 30.Renard C, Chappey O, Wautier MP, Nagashima M, Lundh E, Morser J, Zhao L, Schmidt AM, Scherrmann JM, Wautier JL. Recombinant advanced glycation end product receptor pharmacokinetics in normal and diabetic rats. Mol Pharmacol. 1997;52:54–62. doi: 10.1124/mol.52.1.54. [DOI] [PubMed] [Google Scholar]
- 31.Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A. S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem. 2007;282:31317–31331. doi: 10.1074/jbc.M703951200. [DOI] [PubMed] [Google Scholar]
- 32.Reeves RH, Yao J, Crowley MR, Buck S, Zhang X, Yarowsky P, Gearhart JD, Hilt DC. Astrocytosis and axonal proliferation in the hippocampus of S100b transgenic mice. Proc Natl Acad Sci U S A. 1994;91:5359–5363. doi: 10.1073/pnas.91.12.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei W, Lampe L, Park S, Vangara BS, Waldo GS, Cabantous S, Subaran SS, Yang D, Lakatta EG, Lin L. Disulfide Bonds within the C2 Domain of RAGE Play Key Roles in Its Dimerization and Biogenesis. PLoS One. 2012;7:e50736. doi: 10.1371/journal.pone.0050736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee YC. Biochemistry of carbohydrate-protein interaction. Faseb J. 1992;6:3193–3200. doi: 10.1096/fasebj.6.13.1397841. [DOI] [PubMed] [Google Scholar]
- 35.Dam TK, Brewer CF. Maintenance of cell surface glycan density by lectin-glycan interactions: a homeostatic and innate immune regulatory mechanism. Glycobiology. 2010;20:1061–1064. doi: 10.1093/glycob/cwq084. [DOI] [PubMed] [Google Scholar]
- 36.Dam TK, Brewer CF. Multivalent lectin-carbohydrate interactions energetics and mechanisms of binding. Adv Carbohydr Chem Biochem. 2010;63:139–164. doi: 10.1016/S0065-2318(10)63005-3. [DOI] [PubMed] [Google Scholar]
- 37.Durocher Y, Butler M. Expression systems for therapeutic glycoprotein production. Curr Opin Biotechnol. 2009;20:700–707. doi: 10.1016/j.copbio.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 38.Werner RG, Kopp K, Schlueter M. Glycosylation of therapeutic proteins in different production systems. Acta Paediatr Suppl. 2007;96:17–22. doi: 10.1111/j.1651-2227.2007.00199.x. [DOI] [PubMed] [Google Scholar]
- 39.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 40.Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, Talan MI. Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc Natl Acad Sci U S A. 2003;100:11612–11617. doi: 10.1073/pnas.1930406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moon C, Krawczyk M, Paik D, Lakatta EG, Talan MI. Cardioprotection by recombinant human erythropoietin following acute experimental myocardial infarction: dose response and therapeutic window. Cardiovasc Drugs Ther. 2005;19:243–250. doi: 10.1007/s10557-005-3189-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.