

Abstract
Polycystic ovary syndrome (PCOS) is a very common endocrine disorder characterized by chronic anovulation, clinical and/or biochemical hyperandrogenism, and/or polycystic ovaries. But most experts consider that hyperandrogenism is the main characteristic of PCOS. Several theories propose different mechanisms to explain PCOS manifestations: (1) a primary enzymatic default in the ovarian and/or adrenal steroidogenesis; (2) an impairment in gonadotropin releasing hormone (GnRH) secretion that promotes luteal hormone (LH) secretion; or (3) alterations in insulin actions that lead to insulin resistance with compensatory hyperinsulinemia. However, in the past 20 years there has been growing evidence supporting that defects in insulin actions or in the insulin signalling pathways are central in the pathogenesis of the syndrome. Indeed, most women with PCOS are metabolically insulin resistant, in part due to genetic predisposition and in part secondary to obesity. But some women with typical PCOS do not display insulin resistance, which supports the hypothesis of a genetic predisposition specific to PCOS that would be revealed by the development of insulin resistance and compensatory hyperinsulinemia in most, but not all, women with PCOS. However, these hypotheses are not yet appropriately confirmed, and more research is still needed to unravel the true pathogenesis underlying this syndrome. The present review thus aims at discussing new concepts and findings regarding insulin actions in PCOS women and how it is related to hyperandrogenemia.
Keywords: Polycystic ovary syndrome, Hyperandrogenism, Insulin, Insulin signalling pathways, Insulin resistance, Free fatty acids
1. Introduction
The polycystic ovary syndrome (PCOS) affects 6–10% of women of childbearing age and is one of the commonest endocrine disorders [1,2]. Defining this syndrome is a difficult task because of its multiform symptoms. During a National Institute of Health meeting in 1990, many experts in the field decided on the criteria that must be retained to established a diagnostic of PCOS, both for clinical and research purposes [3]. They conclude that PCOS is a diagnostic of exclusion that associates hyperandrogenism and ovulatory dysfunction. Hyperandrogenism is defined by the state characterized or caused by excessive production and/or secretion of androgens, which is usually manifested by acne, hirsutism or frontal alopecia. Hyperandrogenemia refers to increased blood levels of androgens. During the Rotterdam consensus workshop group meeting in 2003 [4], the experts revised PCOS diagnostic criteria and concluded that it is necessary to have 2 of the 3 subsequent criteria: [i] oligo and/or anovulation, [ii] clinical and/or biochemical signs of hyperandrogenism, and [iii] polycystic ovaries. These criteria also recognize that other androgen excess or related disorders should be excluded before making the diagnosis of PCOS. In 2006, the Androgen Excess-PCOS (AE-PCOS) Society Task Force resolved that PCOS should be first considered a disorder of androgen excess or hyper-androgenism [5]. It was considered that there may be forms of PCOS without overt evidence of hyperandrogenism, but that more data were required before validating this supposition. Therefore, the AE-PCOS Society Task Force proposed in 2009 a novel definition of PCOS based on available data. They declare that PCOS should be defined by the presence of hyperandrogenism (clinical and/or biochemical), ovarian dysfunction (oligo-anovulation and/or polycystic ovaries), and the exclusion of related disorders [1].
PCOS definition is an evolving and difficult task because a combination of environmental and genetic factors influences PCOS pathophysiology and manifestations [6], but also because many aspects of this syndrome remain to be discovered. Indeed, PCOS is now recognized to be associated with important concomitant and future metabolic consequences. In fact, most women with PCOS display impaired glucose tolerance and are at higher risk for developing type 2 diabetes mellitus (T2DM) [2,7]. Recent clinical data showed the positive impacts of insulin-sensitizing drugs, such as metformin, to improve metabolic, ovarian and androgenic status in PCOS women (for review, see Nestler [8]). As hyperandrogenism remains the main feature of PCOS, because up to 70–80% of PCOS women exhibit clinical manifestations of hyperandrogenism [9], it is thus becoming of great importance to understand the mechanisms by which insulin resistance or insulin actions may produce hyperandrogenemia in PCOS women. The aim of this review is to discuss new concepts and findings regarding insulin actions in PCOS women and how it is related to hyperandrogenemia. For a list of the abbreviations used in this review, please refer to Table 1.
Table 1.
List of abbreviations.
| ACTH | Adrenocorticotropin hormone |
| AE-PCOS | Androgen Excess-PCOS society |
| BMI | Body mass index |
| CHO | Carbohydrates |
| DHEA | Dehydroepiandrosterone |
| FFA | Free fatty acid |
| GLUT4 | Glucose transporter 4 |
| GnRH | Gonadotropines releasing hormone |
| Grb2 | Growth factor receptor-bound protein 2 |
| GSK-3 | Glycogen synthase kinase-3 |
| hCG | Human chorionic gonadotrophin |
| HMGCoA reductase | 3-Hydroxy-3-methyl-glutaryl-CoA reductase |
| HSL | Hormone-sensitive lipase |
| IRS-1 | Insulin receptor substrate 1 |
| LH | luteal hormone |
| LDL | Low-density lipoprotein |
| NADPH | Nicotinamide adenine dinucleotide phosphate-oxidase |
| OGTT | Oral glucose tolerance test |
| MAPK | Mitogen-activated protein kinase |
| MUFA | Monounsaturated fatty acids |
| PCOS | Polycystic ovary syndrome |
| PI-3K | Phosphoinositide 3-kinase |
| PKB/C | Protein kinase B/C |
| PPAR | Peroxisome proliferator-activated receptor |
| PUFA | Polyunsaturated fatty acids |
| P450c17 | Cytochrome P450 α-hydroxylase |
| SHBG | Sex-hormone binding globulin |
| SHC | src homologous and collagen protein |
| SREBP | Steroid regulatory element-binding protein |
| TNF-α | Tumor necrosis factor-α |
| TZD | Thiazolidinedione |
| T2DM | Type 2 diabetes mellitus |
| WHR | Waist-to-hip ratio |
| 3βHSD2 | 3β-Hydroxysteroid dehydrogenase type 2 |
2. Hyperandrogenism
2.1. Steroidogenesis
Since androgen excess is the main feature of PCOS, it is of great importance to clearly define how these androgens are normally produced. Androgens are part of the steroid hormone family. For the matter of this paper, we will review only steroidogenesis occurring within the ovary and the adrenal gland. In both human tissues, cholesterol is the precursor for pregnenolone being then converted to steroid hormones following a series of enzymatic processes (Fig. 1). Cholesterol can be delivered either by circulating lipoproteins (mostly low-density lipoproteins [LDL] in human) or by de novo biosynthesis via the rate-limiting enzyme 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGCoA reductase) [10,11]. In the ovary, the first steps of androgen formation are performed in LH-stimulated thecal cells, as these cells express the cytochrome P450c17 gene (see below), with the synthesis of DHEA (dehydroepiandrosterone) and androstenedione [11–13]. Most of these precursors will be converted to estrogens by granulosa cells, which express the enzyme P450aromatase [14]. But ovaries will also directly secrete androgens in the circulation, mainly as androstenedione and testosterone. Interestingly, ovarian androgens will not significantly feedback on LH production, such that an excess in free testosterone or androstenedione will not reduce ovarian production of these androgens in women, as opposed to men.
Fig. 1.
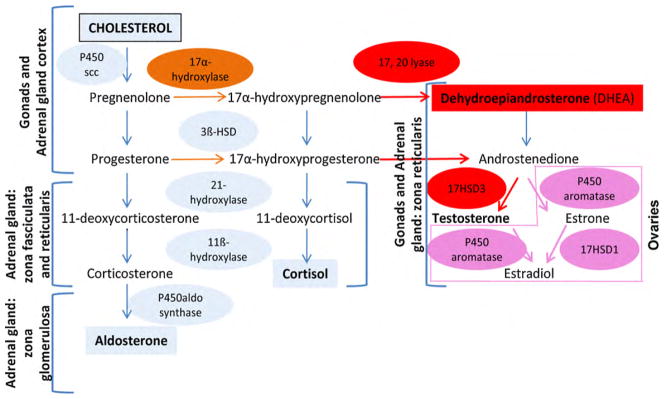
Steroidogenesis occurring both in gonads and adrenal gland of human origin. As cholesterol is the precursor for all steroids, each zone of the adrenal gland or cell types of the ovary expresses specific enzymes necessary for appropriate steroid production. The ovaries, more particularly the thecal cells, possesses the P450c17 enzyme having both the 17α-hydroxylase/17,20-lyase activities needed for androgens secretions, DHEA/testosterone. Granulosa cells expresses the P450aromatase enzyme necessary for estrogens production. The adrenal gland has the capacity to secrete mineralocorticoid (aldosterone) due to the presence of the P450aldo synthase enzyme in the zona glomerulosa. The adrenal gland zona fasciculata, and to a much lesser extent the zona reticularis, produces glucocorticoid (such as cortisol in human) because they express the 17α-hydroxylase activity of the P450c17 enzyme. The zona reticularis expresses to a larger extent than the zona fasciculata both 17α-hydroxylase and 17,20-lyase activities of the P450c17 that are necessary to produce androgens, adapted from Ref. [13].
Adrenal production of androgens is frequently observed in mammals, such as in primates, dogs, bovine, pigs, etc... However, rodents’ adrenals do not produce androgens [15]. In women, adrenal gland contribution to androgen production is very important. Indeed, ovarian and adrenal glands contribute approximately half and half to circulating testosterone in women of reproductive age [16]: they each contribute directly to approximately 25% of total testosterone production and to 25% of total androstenedione secretion, which is in turn converted peripherally to testosterone [17]. Adrenal glands are, however, the major source of circulating testosterone in postmenopausal women because at this stage, ovaries progressively diminish their androgen production [18]. The cortex of the adrenal gland is composed of three layers and each has distinct enzymatic cascades resulting in three different types of steroids. The outer part of the adrenal gland (zona glomerulosa) has the capacity to secrete mineralocorticoids, such as aldosterone. In humans, the inner parts of the adrenal cortex (zona fasciculata and zona reticularis) produce androgen such as DHEA and androstenedione. The zona fasciculata is relatively less efficient in producing androgen and thus secretes mainly glucocorticoids, namely cortisol [19,20]. The most potent stimulus of adrenocortical cells is unquestionably the adrenocorticotropin hormone (ACTH), which induces a substantial increase in all steroids, both in vivo and in vitro conditions [21]. In both men and women, adrenal androgens do not significantly feedback on ACTH production, which is mainly under the control of cortisol. Accordingly, both adrenal and ovarian androgen production is not significantly regulated by circulating androgen levels in women.
2.1.1. The key enzyme for androgen biosynthesis: P450c17
P450c17 is a very important enzyme for steroid production and most importantly, for androgen biosynthesis. To further emphasize on its importance, P450c17 was also termed the “qualitative regulator of steroidogenesis” as it determines which class of steroid will be produced [22]. Class of steroid is either dependent upon P450c17 absence or upon expression of its two different enzymatic activities. In fact, P450c17 is an enzyme coded by one single gene, having both 17α-hydroxylase and 17,20-lyase activities, and showing some species-related differences [19,23,24]. Thus, human adrenal gland zona fasciculata mostly expresses the 17α-hydroxylase activity, thus favoring cortisol production [25]. The 17,20-lyase specific activity of P450c17 is weak in zona fasciculata, but strong in zona reticularis in order to produce DHEA and androstenedione [26]. In the ovary, the general consensus is that only thecal cells express both the 17α-hydroxylase and 17,20-lyase activities of the P450c17 enzyme [27], although one report showed the presence of P450c17 within human cultured granulosa cells [28]. Thus, androgen formation is dependent upon the 17,20-lyase/17α-hydroxylase activities ratio. This ratio is regulated at the post-translational level and lyase activity is favored by: (1) a high molar ratios of P450 oxidoreductase (flavoprotein carrying electron from NADPH) to P450c17, (2) the serine/threonine phosphorylation of P450c17, and (3) by the presence of cytochrome b5. These last two factors promote the interaction of P450oxidoreductase with P450c17 [29].
2.2. Origins of hyperandrogenemia in PCOS
In PCOS, the ovaries produce up to 60% of androgens, while the adrenals contribute the remaining 40% [30]. It is established that androgens incoming from both the ovary and the adrenal are the underlying sources of hyperandrogenemia in PCOS women. When ovarian androgen synthesis is suppressed with GnRH agonists, PCOS women were found to have higher androgen levels in comparison to normal women, thus suggesting adrenal overproduction of androgens [31–35]. Similarly, when adrenal androgen synthesis is suppressed with dexamethasone, PCOS women again display higher androgen levels in comparison to normal women, indicating exaggerated ovarian production [36,37]. Low levels of sex-hormone binding globulin (SHBG) also contribute to high free testosterone levels in women with PCOS, by reducing testosterone binding. SHBG levels are negatively correlated with the circulating levels of insulin or with the degree of insulin resistance in women with or without PCOS, as shown in many studies. Moreover, a study found that reducing insulin levels in obese PCOS women with diazoxide, a drug that only decreases insulin secretion without modifying insulin sensitivity, caused an increase in SHBG levels [38]. This suggests that insulin can directly suppress SHBG secretion by the liver and that compensatory hyperinsulinemia, rather than insulin resistance, explains low SHBG levels in obese women with PCOS.
A dysfunction of the hypothalamic-pituitary-ovarian or adrenal axis was proposed to be the cause of hyperandrogenemia in women with PCOS. But it was demonstrated that chronic suppression of LH or ACTH did not alter the exaggerated 17α-hydroxyprogesterone response to LH/human chorionic gonadotrophin (hCG) [34] or ACTH [39] stimulation in PCOS patients, as compared to control women. These results suggest that chronic LH stimulation is not implicated in ovarian androgenic hyper-secretion typical of PCOS. On the other hand, several studies have demonstrated that treatments aimed at improving insulin resistance in lean and obese PCOS women (e.g. weight loss, metformin, D-chiro-inositol and peroxisome proliferator-activated receptor gamma (PPARγ) agonists) reduce androgen levels [2] and improve the exaggerated androgenic response to LH [40–42] or ACTH [43–46] stimulation tests. Taken together, these studies suggest that the androgenic hyper-responsiveness that characterizes women with PCOS is probably due to factors controlled by insulin sensitization rather than LH, ACTH or ovarian steroids per se.
We conducted a randomized-controlled trial using two insulin-sensitizing drugs (metformin and rosiglitazone, a PPARγ agonist), alone or in combination, in 100 non-obese women with PCOS having normal insulin levels, both during fasting and following an oral glucose tolerance test (OGTT) [47]. Metformin has some direct effects on insulin sensitization, but its main mechanism of action is the reduction of hepatic glucose production, which reduces the need for insulin stimulation. PPARγ agonists are insulin sensitizers that have been shown to increase insulin-stimulated glucose metabolism in adipose, muscle and hepatic tissues, while decreasing compensatory hyperinsulinemia [48], but insulin levels are unchanged in subjects with normal insulin sensitivity. In our trial, testosterone levels were decreased (Fig. 2a) in actively treated groups comparatively to placebo. Despite normoinsulinemia at baseline, metformin reduced insulin levels, but not rosiglitazone (Fig. 2b). Therefore, metformin may have improved hyperandrogenemia in these women mainly by reducing insulin levels, which decreased below normal baseline levels. These results suggest that even in non-obese normoinsulinemic PCOS women, hyperandrogenemia is related to insulin action and might result from increased insulin action on androgen biosynthesis [49]. On the other hand, in our study population, rosiglitazone reversed hyperandrogenemia without decreasing insulin levels, suggesting that PPARγ agonists might directly improve this androgenic hyper-responsiveness to insulin [49].
Fig. 2.

Serum free testosterone, fasting insulin levels and insulin sensitivity (HOMA IS), in women with PCOS having normal insulin levels, before and after the administration of insulin-sensitizing drugs or placebo for 6 months. (a) Testosterone concentrations and (b) fasting insulin levels and HOMA IS are shown as data represented by mean and 95% confidence interval. *P < 0.05 for comparison with the group given placebo and †P < 0.05 for comparison with the group given rosiglitazone, using Tukey HSD tests after ANCOVA analysis, adapted from Baillargeon et al. [47], with permission.
3. Insulin action in PCOS women
3.1. Insulin molecular signalling pathways
Insulin’s actions are mediated via its receptor through two major pathways: the phosphatidylinositol 3-kinase (PI-3K)/Akt pathway implicated in the metabolic effects of insulin and the mitogen-activated protein kinase (MAPK) pathway responsible for the proliferative effects of insulin. Insulin receptor is part of the tyrosine-kinase family. It is a tetrameric protein consisting of two α- and two β-subunits. Activation of the receptor, following insulin binding, leads to conformational changes thus increasing kinase activity necessary for substrates phosphorylation, such as insulin receptor substrate (IRS) family [50]. These phosphorylated proteins are recognized by effector molecules, such as PI3K that further activates Akt. Akt is then the major effector for signal transduction of glucose regulation and metabolism [51]. For example, Akt activation potentiates glucose transporter 4 (GLUT4) translocation from intracellular compartments to the plasma membrane, thus increasing glucose uptake by the cells. In addition of Akt, recent data showed that glycogen synthase kinase-3 (GSK-3) and membrane-associated non-classical protein kinase C (PKC) effectors are involved in the insulin transduction pathway leading to glucose metabolism [52]. Insulin is involved in many other metabolic pathways of glucose, such as the inhibition of gluconeogenesis and glycogenolysis [53,54]. Insulin is also implicated in lipid metabolism, such as lipid synthesis and inhibition of their catabolism. These modulations require an increase in the transcription factor steroid regulatory element-binding protein (SREBP)-1c [55]. In adipocytes, insulin also inhibits lipolysis through inhibition of the enzyme hormone-sensitive lipase (HSL) [56]. Recent data showed that protein kinase B (PKB) is required for the regulation of lipogenesis and for the antilipolysis effect of insulin [57].
Growth signal induced by insulin receptor activation can be obtained either via IRS and growth factor receptor-bound protein 2 (Grb2) association or following recruitment of the adaptor protein, SHC (src homologous and collagen protein) that also recognizes Grb2. Grb2 is able to activate the SOS factor, itself activating Ras. These cascades of phosphorylations lead to MAPK activation, such as JNK, Erk1/2 and p38. These molecules are involved in cell proliferation and apoptosis. Recent data reported that the growth and survival effects mediated by glucose on β-cells of mice were mediated through the activation of insulin receptor and IRS-2, thus providing a dominant role for insulin in the regeneration and function of the pancreas [58]. Furthermore, in rat ovarian thecal-interstitial cells, it was demonstrated that insulin increases proliferation by acting, in part, through increased phosphorylation of the MAPK3/1 and the PI3K pathways [59].
3.2. Defective insulin actions in PCOS
PCOS is a common and well-defined clinical model of insulin resistance and pre-diabetic state. Insulin sensitivity is decreased by an average of 35% to 40% in women with PCOS, as compared to matched controls, similar to what is seen in women with non-insulin-dependent diabetes mellitus [60–62]. Thus, most women with PCOS are insulin resistant and develop compensatory hyperinsulinemia [1], which seems to play a critical role in the syndrome’s pathogenesis [1,2]. However, those features are not essential to develop PCOS since a subgroup of women with typical PCOS are neither insulin resistant nor hyperinsulinemic. Nevertheless, in those lean, normoinsulinemic and normally insulin-sensitive PCOS women, increased free testosterone and androstenedione were significantly reduced following diazoxide-induced lowering of insulin levels [63]. Notably, suppression of insulin secretion with diazoxide did not alter testosterone levels in healthy, non-obese women [64]. These results suggest that hyperandrogenemia is related to insulin action even in lean PCOS women with normal insulin sensitivity and levels [49].
3.2.1. Cellular mechanisms of metabolic insulin resistance
Most of the recent studies in PCOS women done with tissues like muscle, adipocytes and ovaries have shown that causative defects of insulin resistance probably involve insulin post-binding signalling pathways [62,65–67]. It has been demonstrated that early steps in insulin signalling (maximal rate of glucose uptake, abundance of GLUT4, inhibition of lipolysis stimulated by insulin) were all decreased in PCOS women compared to controls, even if the number and affinity of insulin receptors are not obviously decreased in adipocytes from obese PCOS women [68]. These findings were also observed in PCOS women presenting no obesity, glucose intolerance, or increased waist-to-hip ratio [69], suggesting that they may be intrinsic to the syndrome. Moreover, muscles biopsied from PCOS obese women during insulin-glucose clamp protocols presented impaired insulin-stimulated association of IRS-1 with PI-3K, concomitant with a decrease in glucose transport in vivo [62]. Therefore, defects in the metabolic actions of insulin in PCOS women appear to implicate an early step in insulin signalling and were independent of obesity and T2DM.
In cultured myotubes obtained after biopsies from obese PCOS women, the activity of PI3-kinase was significantly decreased, when normalized for total IRS-1 abundance, as well as insulin signalling via IRS-2-associated PI-3K [70]. This study also found that the phosphorylation of IRS-1 at a key regulatory site on Ser312 was constitutively increased in PCOS, which may have contributed to its signalling defect. This study highlights one very important point: decreased insulin-induced glucose uptake observed in PCOS women in vivo seems to be secondary to impaired association between PI3K and IRS1, suggesting that in vivo factors are involved in this defect. Another study, in skeletal muscle of obese PCOS women, demonstrated that decreased insulin action on peripheral glucose metabolism is associated with impaired insulin signalling at the level of Akt and AS160. Akt is an important mediator of insulin-stimulated GLUT4 translocation and glucose transport [71], and seems to be dependent on the phosphorylation of AS160 at several sites by Akt [72–74]. In obese PCOS women, a decrease in insulin-mediated phosphorylation of AS160 and of Akt (at Thr308 and Ser473) has been described [75]. This may be due to dephosphorylation of Akt by protein phosphatases, including protein phosphatase 2A (PP2A), that are activated by ceramide metabolites of palmitate [76].
In perpetuated cultured skin fibroblasts, it was observed that in approximately 50% of PCOS subjects, autophosphorylation of insulin receptors following insulin stimulation was decreased [66]. It has been also reported that the number or affinity of insulin receptors were not affected compared to normal subjects [66,77]. Furthermore, PCOS insulin receptors displayed a constitutive increase in the phosphorylation of their serine residues and a decrease in insulin-stimulated phosphorylation of their tyrosine residues. Insulin receptors were also less efficient to phosphorylate IRSs, suggesting that the activity of insulin receptors were impaired by this exaggerated serine phosphorylation [66]. This insulin receptor’s state seems to be independent of the presence of obesity and non-insulin-dependent diabetes mellitus, suggesting that this defect might be specific to PCOS. Moreover, this defect was probably due to an extrinsic factor that increases the serine phosphorylation state of the cells, because normal insulin signalling was restored by inhibitors of serine kinase activity [67].
Interestingly, FFA metabolites (e.g. diacylglycerols and ceramides) that accumulate in cells have been postulated to activate intracellular serine/threonine kinases [78] and protein phosphatases, such as PP2A [76]. Most women with PCOS present increased circulating levels of free fatty acids (FFAs), which have been shown to cause insulin resistance in vivo [79,80]. In fact, several in vivo studies demonstrated that elevating circulating FFAs leads to peripheral tissue insulin resistance [81–83]. Accumulation of plasma fatty acids in muscle and liver tissues induce mitochondrial dysfunction, oxidative stress, inflammation and immune disorders [84]. On the other hand, high circulating FFA levels have been shown in vivo to increase the production of all androgens in normal women [85]. Another study found that male rats fed on a high monounsaturated fatty acid (MUFA) diet for 6 weeks display increased testosterone levels (vs. low MUFA diet), and that free fatty acid with different degree of saturation increased testosterone levels to significantly different extent [86]. In addition, serine phosphorylation of P450c17 have been shown to increase its 17,20-lyase activity [87,88]. Therefore, FFAs may be the serine phosphorylation key factor that induces both an increase in P450c17 activity and a defect in the insulin signalling pathways causing insulin resistance (Fig. 3). Moreover, the saturated fatty acid (SFA) palmitate was found to decrease MAPK activity in vitro in rat fibroblasts [89], and evidence suggests that such defect in the MAPK component of the insulin signalling pathway may contribute to increase androgen biosynthesis both in adrenal and ovarian tissues, as discussed in the next section.
Fig. 3.

Proposed cellular mechanisms involved in insulin-stimulated androgen biosynthesis, PCOS-associated defects, free fatty acids-induced insulin resistance and increased androgen production, and PPARγ actions. Insulin binds to its receptor resulting in tyrosine phosphorylation of the receptor and insulin receptor substrates (IRSs) such as the IRS-1. IRS-1 activates phosphoinositide-3-kinase (PI-3K) and Akt, which mediate insulin-stimulated glucose metabolism. Serine phosphorylation of IRS-1 prevents its binding with PI-3K and inhibits insulin signalling. Furthermore, serine phosphorylation of P450c17 increases its 17,20-lyase activity and thus androgen biosynthesis. Interestingly, serine phosphorylation of IRS-1 is constitutively increased in PCOS women and increased by fatty acids (FAs) accumulation, and PPARγ agonists increase tyrosine phosphorylation of IRS-1. On the other hand, insulin-stimulated androgen production has been shown to be reduced by specific inhibition of PI-3K and increased by specific inhibition of MEK. MEK/ERK activity was found to be constitutively reduced in PCOS women, activated by PPARγ agonists and inhibited by FFAs. It was also suggested that P450c17 activity may be stimulated by other players of the MAPK pathway, such as MKK3/6-p38 and MKK4/7-JNK, which are at least normally functional in women with PCOS, adapted from Baillargeon [92].
3.2.2. Cellular mechanisms of insulin-related hyperandrogenism
As previously discussed, PCOS is also characterized by insulin-related hyperandrogenemia, which implies an important role of insulin in the regulation of ovarian androgen biosynthesis [41,90,91]. In fact, multiple studies have shown that insulin stimulates androgenesis in normal ovarian in vitro models [92]. Indeed, PCOS thecal cells in culture show increased androgen responsiveness to insulin and LH when compared to normal thecal cells [93,94]. Physiological doses of insulin are able to activate androgen production in PCOS thecal cells, while higher concentrations of insulin are necessary in normal thecal cells. In both type of the-cal cells, the combination of physiological doses of LH and insulin synergistically increases androgen biosynthesis [93,95]. Androgenesis is also increased in long-term cultures of PCOS thecal cells, as compared with normal ones, suggesting that this hyperandrogenism may be an intrinsic property of PCOS thecal cells [96,97]. Taken together, these observations support that PCOS thecal cells present an androgenic hyper-responsiveness that involves a crosstalk between LH and insulin pathways. Similarly, insulin increases basal and ACTH-stimulated production of androgens or expression of P450c17 in normal cultured human [98,99] and bovine [100] adrenal cells.
Wu et al. [101] found that PCOS luteinized granulosa cells have a selective increase in insulin activation of its mitogenic pathway, via the MAP kinase pathway, concomitant to resistance in the metabolic pathway of insulin action. The same group also found, in cultured porcine thecal cells, that dexamethasone induces resistance to insulin-mediated glucose transport with increased testosterone production and expression of P450c17 [102]. These studies also showed that a PPAR-γ agonist, an insulin sensitizer, can reverse both the increased insulin-stimulated mitogenic pathway and hyperandrogenism, on one side, and the insulin resistance of the metabolic pathway, in the other side. These observations support the possibility that increased insulin action on androgen production may co-exist with normal or reduced metabolic activity of insulin in PCOS.
The cellular mechanisms by which insulin regulates androgenesis are not well understood, but potential pathways are proposed and illustrated in Fig. 3. Insulin acts through its own receptor in the-cal [94,103] or fasciculata [104] cells. Specific blockade of PI-3K in normal human thecal cells markedly inhibits the combined insulin and LH stimulation of P450c17 activity [105]. It was also suggested that insulin may stimulate P450c17 activity through some players of the MAPK pathway such as MKK3/p38 and MKK4/JNK (see for review: [106] and Fig. 3). On the other hand, specific inhibition of MEK/ERK, another component of the MAPK insulin pathway, increases P450c17 activity [105]. Increased expression of P450c17 after inhibition of MEK/ERK was also found in human adrenal cells [107]. The attenuated MEK/ERK signal could stimulate androgen production via a reduction in c-fos expression, because c-fos was shown to inhibit P450c17 expression in a thecal cell tumor model [108]. Since insulin stimulates MEK/ERK activity, such a defect would not contribute to insulin-stimulated androgen production, but it could promote baseline androgenesis or its responsiveness to stimulation with insulin (via PI-3K, MKK3/p38 and MKK4/JNK pathways) and LH/ACTH.
3.2.3. Obesity and PCOS
At diagnosis, the prevalence of overweight and obesity in the PCOS population is above 50% in the United States [109] and between 30% and 50% in Europe [110]. Obesity by itself is associated with insulin resistance and compensatory hyperinsulinemia, which is worst following intra-abdominal accumulation of fat. Indeed, the visceral fat depot metabolism is more active than the subcutaneous one [84]. Intra-abdominal fat tissue is more sensitive to lipolysis and releases more FFAs in the circulation, and produces several cytokines (i.e. tumor necrosis factor-α [TNF-α], IL-6, leptin, resistin) involved in insulin resistance [84]. As previously mentioned, circulating FFAs can accumulate in non-adipose tissues, causing lipotoxicity and insulin resistance (for a review, please refer to [84]). During obesity development, insulin resistance is also related to TNF-α that enhances serine phosphorylation of IRS-1 and inhibits insulin receptor signalling [111]. Furthermore, insulin resistance associated to obesity induces leptin resistance and reduced adiponectin levels, two factors that may reduce fatty acid oxidation and promote lipotoxicity [84,112].
Obesity is not an essential feature of PCOS, but by aggravating the degree of insulin resistance and hyperinsulinemia, obesity will precipitate the clinical manifestations of the syndrome in predisposed women or will aggravate them in those already affected [49,113]. It is probably because of its pathophysiologic role in the syndrome, in association with genetic or other primary predisposition, that women with PCOS are on average more obese or abdominally overweight than normal women. Of note, overweight in PCOS is characterized by a central distribution, with increased visceral rather than subcutaneous fat, which is more closely associated with insulin resistance [110,114–116], as previously discussed. Even in lean women matched for body mass index (BMI, defined by weight in kg divided by height in m2), PCOS women have a higher percentage of body fat, a larger waist-to-hip ratio (WHR) and increased accumulation of intra-abdominal peritoneal and visceral fat than their matched controls [110,114–116].
As compared to non-obese PCOS women, obese women with PCOS have more menstrual irregularities and uterine dysfunctional bleeding, as well as an increased prevalence of infertility, which were also associated with an abdominal distribution of fat [113,117,118]. Obese women with or without PCOS display increased risk of miscarriage, gestational diabetes and pre-eclampsia [119]. Moreover, PCOS women who are obese tend to have higher hirsutism and acne scores than their lean counterparts. Indeed, SHBG levels are reduced in obese PCOS women, especially if they present with abdominal obesity [113,120]. Lower SHBG levels increase the bioavailability of testosterone and thus further increase hyperandrogenemia. Obese PCOS women also have a higher risk of developing glucose intolerance or diabetes than lean PCOS women [120,121]. Therefore, since obesity is an important environmental factor exacerbating the clinical symptoms and metabolic risks of the syndrome, it is essential in the management of PCOS to start by lifestyle modifications and to put emphasis on weight loss in all obese and overweight women with PCOS.
4. Management of insulin-related hyperandrogenism and insulin resistance in PCOS women
4.1. Weight loss and exercise
Lifestyle modification, such as diet re-calibration and increased physical activity, is considered as the first-line treatment for PCOS women [122,123], particularly when their BMI exceeds 25 kg/m2. In order to improve fertility, 343 overweight infertile women with PCOS were randomized to either clomiphene citrate alone, the insulin sensitizer metformin alone, the combination of both, or a lifestyle modification program (low-calorie diet and risk-free exercise for 30 min/day) [124]. Lifestyle group women did better than the medicated groups with regard to waist circumference, LDL and insulin levels, while SHBG was improved equally in lifestyle and metformin groups. More importantly, pregnancy rate was higher in the lifestyle group (20%) than in the combination group (14.8%), although this difference did not reach statistical significance. A recent clinical trial randomized 30 obese, insulin-resistant PCOS women to lifestyle modification with the addition of metformin or placebo for 4 months [125]. The authors found that a small decrease in body weight trough lifestyle changes was enough to improve menstrual cycles in these PCOS women and that metformin offered additive effects regarding insulin resistance and hyperandrogenism. Thus, a modest weight loss in obese PCOS women of only 5% of initial body weight can result in pregnancy [126], while a weight loss of 5–10% can reduce hyperandrogenism and insulin levels [127].
There are no conclusive data regarding the optimal composition of the diet in order to improve clinical consequences of PCOS. Twenty-eight overweight PCOS women were randomized to a low-or high-protein diet for 12 weeks [128]. Both diet decreased weight (7.5%) and abdominal fat (12.5%), and improved pregnancy rates, menstrual cyclicity, lipid profile, and insulin resistance, but without significant difference based on diet composition. Similarly, a randomized-controlled trial comparing high-protein and high-carbohydrate diets did not find significant differences in weight loss and clinical or biochemical improvements between diets [129]. However, based on the hypothesis that accumulation of fatty acids in androgen-secreting cells may play an important role in PCOS pathophysiology, fat composition of the diet might prove to be more important than other macronutrients. For example, in male rats saturated fatty acids were more prone to accumulate in cells and to increase androgen levels than polyunsaturated fatty acid (PUFA), and to a lesser extent than MUFA [86]. Accordingly, a prospective study found that after a 3-month habitual diet, partly replacing fat by PUFAs for another 3 months improved glucose homeostasis, plasma lipids and sex steroids in women with PCOS [130]. A cross-over study comparing eucaloric diets either enriched with MUFA or low in carbohydrates (CHO), evidenced that both interventions lowered fasting insulin levels and circulating triglycerides, but the acute insulin response to glucose was lower following the low CHO diet relative to the MUFA diet [131]. However, diets were tested for only 16 days, which was probably too short for fat modulation to impact on insulin sensitivity and testosterone levels. Since very few studies assessed the role of dietary fat content modulation in women with PCOS, we propose that further investigations should be done in order to better characterize and understand the effects of dietary fat in PCOS management.
4.2. Insulin-sensitizing drugs
Following failure of non-pharmacological methods, medical treatments for the management of insulin-related hyperandrogenism and insulin resistance can be suggested to women with PCOS. Indeed, all insulin-sensitizing or insulin-lowering agents used for treatment of type 2 diabetes, namely metformin, thiazolidinediones (TZDs, PPARγ agonists), D-chiro- or myo-inositols, and acarbose, have been shown to improve hyperandrogenemia [2,122,132], both in lean and obese women with PCOS. Metformin is a biguanide who mainly acts by reducing hepatic glucose production, but also improves insulin sensitivity to some extent. This drug also reduces appetite in many PCOS women and is thus often [133], but not always [134], associated with more weight loss. Metformin has been shown to be effective in all women with PCOS, even those without insulin resistance and hyperinsulinemia [47,135–137], but tends to be more effective in lean as compared to obese PCOS women [138]. The effects of metformin in PCOS are probably mainly mediated through a reduction in insulin levels, which is observed both in insulin-sensitive and insulin-resistant PCOS women because of the reduction in hepatic glucose production. Metformin also seems to reduce androgen production by a direct action on the ovaries [139,140], which could be related to improvement in intracellular accumulation of FFA. But this hypothesis needs to be verified in vitro.
TZDs are other insulin-sensitizing agents that can be used for the treatment of PCOS manifestations. TZDs binding to gamma peroxisome proliferator activator receptors (PPARγ receptors), induce gene transcription and activate genes that encode for insulin action and normal FFA metabolism in adipocytes and androgen-secreting cells. TZDs, unlike metformin, are true sensitizers, such that insulin levels will be maintained stable in individuals with normal insulin sensitivity. To date, three molecules were released: troglitazone, rosiglitazone and pioglitazone, but troglitazone was withdrawn from the market because of idiosyncratic hepatotoxicity. Several studies have demonstrated the therapeutic benefits of one or another TZD on insulin resistance, ovulatory dysfunction and hyperandrogenism in PCOS women [141–144]. Similar to metformin, TZDs were found to improve hyperandrogenism and ovulation rates even in lean women with PCOS [47,135] and with normal insulin levels [47]. TZDs seem at least as effective as metformin for clinical improvement of PCOS [2,122]. For example, in obese PCOS patients treated over a 12 weeks period with metformin, orlistat (weight loss inducer) or pioglitazone, features of hyperandrogenemia were equally reduced with the three drugs [145].
PPARγ receptors were found in adrenal fasciculata and ovarian thecal cells, and ligands of these receptors decreased P450c17 and 3βHSD2 activity in human adrenal cells, and LH- and/or insulin-stimulated testosterone production in porcine thecal [146,147] and human ovarian cells [148]. PPARγ agonists have also been shown to reverse the enhanced expression of P450c17 induced by specific inhibition of MEK/ERK in human adrenal cells [107] (Fig. 3). Thus, PPARγ seems directly implicated in androgen production and its activation may improve some of the insulin signalling protein defects associated with PCOS hyperandrogenemia described in previous sections (Fig. 3). Furthermore, since all insulin-sensitizing therapies decrease circulating FFA levels by improving adipocyte insulin sensitivity; this might be a common mechanism by which insulin sensitization improves hyperandrogenemia (see previous discussion).
5. Conclusion
In summary, PCOS is a very common endocrine disorder that affects the quality of life of women suffering from its multiform symptoms. Moreover, those women are at greater risk to develop metabolic syndrome and T2DM. The main feature of PCOS is hyper-androgenism and evidence suggest that insulin resistance or insulin action play critical roles in its pathophysiology. The aim of this review was to discuss on new insights and findings regarding insulin actions in PCOS women and how it is related to hyper-androgenemia. It remains difficult to understand the mechanisms involved because many defects are observed and they might be caused either by genetic predisposition, environmental impact or both. New studies are necessary to elucidate the pathophysiology of PCOS in order to develop better treatments with beneficial short-term and long-term effects.
References
- 1.Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, Witchel SF. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril. 2009;91(2):456–488. doi: 10.1016/j.fertnstert.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 2.Baillargeon JP. Use of insulin sensitizers in polycystic ovarian syndrome. Curr Opin Investig Drugs. 2005;6(10):1012–1022. [PubMed] [Google Scholar]
- 3.Zawadzki JK, Dunaif A. Current Issues in Endocrinology and Metabolism: Polycystic Ovary Syndrome. Blackwell Scientific Publications; Cambridge, MA: 1992. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach; pp. 377–384. [Google Scholar]
- 4.Anonymous. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81(1):19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, Witchel SF. Positions statement: criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: an Androgen Excess Society guideline. J Clin Endocrinol Metab. 2006;91(11):4237–4245. doi: 10.1210/jc.2006-0178. [DOI] [PubMed] [Google Scholar]
- 6.Deligeoroglou E, Kouskouti C, Christopoulos P. The role of genes in the polycystic ovary syndrome: predisposition and mechanisms. Gynecol Endocrinol. 2009 doi: 10.1080/09513590903015619. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 7.Gagnon C, Baillargeon JP. Suitability of recommended limits for fasting glucose tests in women with polycystic ovary syndrome. CMAJ. 2007;176(7):933–938. doi: 10.1503/cmaj.060607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nestler JE. Metformin for the treatment of the polycystic ovary syndrome. N Engl J Med. 2008;358(1):47–54. doi: 10.1056/NEJMct0707092. [DOI] [PubMed] [Google Scholar]
- 9.Diamanti-Kandarakis E, Christakou CD, Kandaraki E, Alexandraki KI. Early onset adiposity: a pathway to polycystic ovary syndrome in adolescents? Hormones (Athens) 2007;6(3):210–217. [PubMed] [Google Scholar]
- 10.Kraemer FB. Adrenal cholesterol utilization. Mol Cell Endocrinol. 2007;265–266:42–45. doi: 10.1016/j.mce.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Wood JR, Strauss JF., III Multiple signal transduction pathways regulate ovarian steroidogenesis. Rev Endocr Metab Disord. 2002;3(1):33–46. doi: 10.1023/a:1012748718150. [DOI] [PubMed] [Google Scholar]
- 12.Jamnongjit M, Hammes SR. Ovarian steroids: the good, the bad, and the signals that raise them. Cell Cycle. 2006;5(11):1178–1183. doi: 10.4161/cc.5.11.2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Payne AH, Hales DB. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr Rev. 2004;25(6):947–970. doi: 10.1210/er.2003-0030. [DOI] [PubMed] [Google Scholar]
- 14.Mendelson CR, Kamat A. Mechanisms in the regulation of aromatase in developing ovary and placenta. J Steroid Biochem Mol Biol. 2007;106(1–5):62–70. doi: 10.1016/j.jsbmb.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vinson GP. Adrenocortical zonation and ACTH. Microsc Res Tech. 2003;61(3):227–239. doi: 10.1002/jemt.10331. [DOI] [PubMed] [Google Scholar]
- 16.Piltonen T, Koivunen R, Morin-Papunen L, Ruokonen A, Huhtaniemi IT, Tapanainen JS. Ovarian and adrenal steroid production: regulatory role of LH/HCG. Hum Reprod. 2002;17(3):620–624. doi: 10.1093/humrep/17.3.620. [DOI] [PubMed] [Google Scholar]
- 17.Parker CR., Jr . Androgen Excess Disorders in Women. 2. Humana Press; Totowa, NJ: 2006. Androgens throughout the life of women; pp. 35–48. [Google Scholar]
- 18.Couzinet B, Meduri G, Lecce MG, Young J, Brailly S, Loosfelt H, Milgrom E, Schaison G. The postmenopausal ovary is not a major androgen-producing gland. J Clin Endocrinol Metab. 2001;86(10):5060–5066. doi: 10.1210/jcem.86.10.7900. [DOI] [PubMed] [Google Scholar]
- 19.Endoh A, Kristiansen SB, Casson PR, Buster JE, Hornsby PJ. The zona reticularis is the site of biosynthesis of dehydroepiandrosterone and dehydroepiandrosterone sulfate in the adult human adrenal cortex resulting from its low expression of 3 beta-hydroxysteroid dehydrogenase. J Clin Endocrinol Metab. 1996;81(10):3558–3565. doi: 10.1210/jcem.81.10.8855801. [DOI] [PubMed] [Google Scholar]
- 20.Miller WL. Steroidogenic enzymes. Endocr Dev. 2008;13:1–18. doi: 10.1159/000134751. [DOI] [PubMed] [Google Scholar]
- 21.Sewer MB, Waterman MR. ACTH modulation of transcription factors responsible for steroid hydroxylase gene expression in the adrenal cortex. Microsc Res Tech. 2003;61(3):300–307. doi: 10.1002/jemt.10339. [DOI] [PubMed] [Google Scholar]
- 22.Miller WL. Androgen biosynthesis from cholesterol to DHEA. Mol Cell Endocrinol. 2002;198(1–2):7–14. doi: 10.1016/s0303-7207(02)00363-5. [DOI] [PubMed] [Google Scholar]
- 23.Brock BJ, Waterman MR. Biochemical differences between rat and human cytochrome P450c17 support the different steroidogenic needs of these two species. Biochemistry. 1999;38(5):1598–1606. doi: 10.1021/bi9821059. [DOI] [PubMed] [Google Scholar]
- 24.Hyatt PJ, Bhatt K, Tait JF. Steroid biosynthesis by zona fasciculata and zona reticularis cells purified from the mammalian adrenal cortex. J Steroid Biochem. 1983;19(1C):953–959. doi: 10.1016/0022-4731(83)90039-0. [DOI] [PubMed] [Google Scholar]
- 25.Ishimura K, Fujita H. Light and electron microscopic immunohistochemistry of the localization of adrenal steroidogenic enzymes. Microsc Res Tech. 1997;36(6):445–453. doi: 10.1002/(SICI)1097-0029(19970315)36:6<445::AID-JEMT2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 26.Tee MK, Dong Q, Miller WL. Pathways leading to phosphorylation of p450c17 and to the posttranslational regulation of androgen biosynthesis. Endocrinology. 2008;149(5):2667–2677. doi: 10.1210/en.2007-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasano H, Okamoto M, Mason JI, Simpson ER, Mendelson CR, Sasano N, Silverberg SG. Immunolocalization of aromatase, 17 alpha-hydroxylase and side-chain-cleavage cytochromes P-450 in the human ovary. J Reprod Fertil. 1989;85(1):163–169. doi: 10.1530/jrf.0.0850163. [DOI] [PubMed] [Google Scholar]
- 28.Moran FM, VandeVoort CA, Overstreet JW, Lasley BL, Conley AJ. Molecular target of endocrine disruption in human luteinizing granulosa cells by 2,3,7,8-tetrachlorodibenzo-p-dioxin: inhibition of estradiol secretion due to decreased 17alpha-hydroxylase/17,20-lyase cytochrome P450 expression. Endocrinology. 2003;144(2):467–473. doi: 10.1210/en.2002-220813. [DOI] [PubMed] [Google Scholar]
- 29.Miller WL, Auchus RJ, Geller DH. The regulation of 17,20 lyase activity. Steroids. 1997;62(1):133–142. doi: 10.1016/s0039-128x(96)00172-9. [DOI] [PubMed] [Google Scholar]
- 30.Cedars MI, Steingold KA, de Ziegler D, Lapolt PS, Chang RJ, Judd HL. Long-term administration of gonadotropin-releasing hormone agonist and dexamethasone: assessment of the adrenal role in ovarian dysfunction. Fertil Steril. 1992;57(3):495–500. doi: 10.1016/s0015-0282(16)54890-0. [DOI] [PubMed] [Google Scholar]
- 31.Barnes RB, Rosenfield RL, Burstein S, Ehrmann DA. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N Engl J Med. 1989;320(9):559–565. doi: 10.1056/NEJM198903023200904. [DOI] [PubMed] [Google Scholar]
- 32.Carmina E, Koyama T, Chang L, Stanczyk FZ, Lobo RA. Does ethnicity influence the prevalence of adrenal hyperandrogenism and insulin resistance in polycystic ovary syndrome? Am J Obstet Gynecol. 1992;167(6):1807–1812. doi: 10.1016/0002-9378(92)91779-a. [DOI] [PubMed] [Google Scholar]
- 33.Ehrmann DA, Rosenfield RL, Barnes RB, Brigell DF, Sheikh Z. Detection of functional ovarian hyperandrogenism in women with androgen excess. N Engl J Med. 1992;327(3):157–162. doi: 10.1056/NEJM199207163270304. [DOI] [PubMed] [Google Scholar]
- 34.Gilling-Smith C, Story H, Rogers V, Franks S. Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome. Clin Endocrinol (Oxf) 1997;47(1):93–99. doi: 10.1046/j.1365-2265.1997.2321049.x. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman DI, Klove K, Lobo RA. The prevalence and significance of elevated dehydroepiandrosterone sulfate levels in anovulatory women. Fertil Steril. 1984;42(1):76–81. doi: 10.1016/s0015-0282(16)47961-6. [DOI] [PubMed] [Google Scholar]
- 36.Lachelin GC, Judd HL, Swanson SC, Hauck ME, Parker DC, Yen SS. Long term effects of nightly dexamethasone administration in patients with polycystic ovarian disease. J Clin Endocrinol Metab. 1982;55(4):768–773. doi: 10.1210/jcem-55-4-768. [DOI] [PubMed] [Google Scholar]
- 37.Rittmaster RS, Thompson DL. Effect of leuprolide and dexamethasone on hair growth and hormone levels in hirsute women: the relative importance of the ovary and the adrenal in the pathogenesis of hirsutism. J Clin Endocrinol Metab. 1990;70(4):1096–1102. doi: 10.1210/jcem-70-4-1096. [DOI] [PubMed] [Google Scholar]
- 38.Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, Clore JN, Blackard WG. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72(1):83–89. doi: 10.1210/jcem-72-1-83. [DOI] [PubMed] [Google Scholar]
- 39.Devesa J, Perez-Fernandez R, Lima L, Cabezas-Cerrato J. Adrenal cortex and type II polycystic ovary syndrome. Gynecol Endocrinol. 1987;1(3):269–277. doi: 10.3109/09513598709023614. [DOI] [PubMed] [Google Scholar]
- 40.Jakubowicz DJ, Nestler JE. 17 alpha-Hydroxyprogesterone responses to leuprolide and serum androgens in obese women with and without polycystic ovary syndrome offer dietary weight loss. J Clin Endocrinol Metab. 1997;82(2):556–560. doi: 10.1210/jcem.82.2.3753. [DOI] [PubMed] [Google Scholar]
- 41.Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17 alpha activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N Engl J Med. 1996;335(9):617–623. doi: 10.1056/NEJM199608293350902. [DOI] [PubMed] [Google Scholar]
- 42.Nestler JE, Jakubowicz DJ. Lean women with polycystic ovary syndrome respond to insulin reduction with decreases in ovarian P450c17 alpha activity and serum androgens. J Clin Endocrinol Metab. 1997;82(12):4075–4079. doi: 10.1210/jcem.82.12.4431. [DOI] [PubMed] [Google Scholar]
- 43.Arslanian SA, Lewy V, Danadian K, Saad R. Metformin therapy in obese adolescents with polycystic ovary syndrome and impaired glucose tolerance: amelioration of exaggerated adrenal response to adrenocorticotropin with reduction of insulinemia/insulin resistance. J Clin Endocrinol Metab. 2002;87(4):1555–1559. doi: 10.1210/jcem.87.4.8398. [DOI] [PubMed] [Google Scholar]
- 44.Guido M, Romualdi D, Suriano R, Giuliani M, Costantini B, Apa R, Lanzone A. Effect of pioglitazone treatment on the adrenal androgen response to corticotrophin in obese patients with polycystic ovary syndrome. Hum Reprod. 2004;19(3):534–539. doi: 10.1093/humrep/deh145. [DOI] [PubMed] [Google Scholar]
- 45.la Marca A, Morgante G, Paglia T, Ciotta L, Cianci A, De Leo V. Effects of metformin on adrenal steroidogenesis in women with polycystic ovary syndrome. Fertil Steril. 1999;72(6):985–989. doi: 10.1016/s0015-0282(99)00407-0. [DOI] [PubMed] [Google Scholar]
- 46.Romualdi D, Giuliani M, Draisci G, Costantini B, Cristello F, Lanzone A, Guido M. Pioglitazone reduces the adrenal androgen response to corticotropin-releasing factor without changes in ACTH release in hyperinsulinemic women with polycystic ovary syndrome. Fertil Steril. 2007;88(1):131–138. doi: 10.1016/j.fertnstert.2006.11.076. [DOI] [PubMed] [Google Scholar]
- 47.Baillargeon JP, Jakubowicz DJ, Iuorno MJ, Jakubowicz S, Nestler JE. Effects of metformin and rosiglitazone, alone and in combination, in nonobese women with polycystic ovary syndrome and normal indices of insulin sensitivity. Fertil Steril. 2004;82(4):893–902. doi: 10.1016/j.fertnstert.2004.02.127. [DOI] [PubMed] [Google Scholar]
- 48.Gardner OS, Dewar BJ, Graves LM. Activation of mitogen-activated protein kinases by peroxisome proliferator-activated receptor ligands: an example of nongenomic signaling. Mol Pharmacol. 2005;68(4):933–941. doi: 10.1124/mol.105.012260. [DOI] [PubMed] [Google Scholar]
- 49.Baillargeon JP, Nestler JE. Commentary: polycystic ovary syndrome: a syndrome of ovarian hypersensitivity to insulin? J Clin Endocrinol Metab. 2006;91(1):22–24. doi: 10.1210/jc.2005-1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patti ME, Kahn CR. The insulin receptor—a critical link in glucose homeostasis and insulin action. J Basic Clin Physiol Pharmacol. 1998;9(2–4):89–109. doi: 10.1515/jbcpp.1998.9.2-4.89. [DOI] [PubMed] [Google Scholar]
- 51.Czech MP, Corvera S. Signaling mechanisms that regulate glucose transport. J Biol Chem. 1999;274(4):1865–1868. doi: 10.1074/jbc.274.4.1865. [DOI] [PubMed] [Google Scholar]
- 52.Kim do M, Jang HJ, Han SJ, Ha ES, Kim YK, Park JW, Song KE, Jung SH, Ahn SM, Choi SE, Kim HJ, Kim DJ, Lee HC, Lee KW. Classical PKC is not associated with defective insulin signaling in patients with impaired glucose tolerance. Diabetes Res Clin Pract. 2009;83(3):334–340. doi: 10.1016/j.diabres.2008.11.035. [DOI] [PubMed] [Google Scholar]
- 53.Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab. 2000;11(9):351–356. doi: 10.1016/s1043-2760(00)00323-4. [DOI] [PubMed] [Google Scholar]
- 54.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6(1):87–97. [PubMed] [Google Scholar]
- 55.Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Gold-stein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci USA. 1999;96(24):13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anthonsen MW, Ronnstrand L, Wernstedt C, Degerman E, Holm C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem. 1998;273(1):215–221. doi: 10.1074/jbc.273.1.215. [DOI] [PubMed] [Google Scholar]
- 57.Berggreen C, Gormand A, Omar B, Degerman E, Goransson O. Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am J Physiol Endocrinol Metab. 2009;296(4):E635–E646. doi: 10.1152/ajpendo.90596.2008. [DOI] [PubMed] [Google Scholar]
- 58.Assmann A, Ueki K, Winnay JN, Kadowaki T, Kulkarni RN. Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol Cell Biol. 2009;29(11):3219–3228. doi: 10.1128/MCB.01489-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kwintkiewicz J, Spaczynski RZ, Foyouzi N, Pehlivan T, Duleba AJ. Insulin and oxidative stress modulate proliferation of rat ovarian theca-interstitial cells through diverse signal transduction pathways. Biol Reprod. 2006;74(6):1034–1040. doi: 10.1095/biolreprod.105.049908. [DOI] [PubMed] [Google Scholar]
- 60.Book CB, Dunaif A. Selective insulin resistance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1999;84(9):3110–3116. doi: 10.1210/jcem.84.9.6010. [DOI] [PubMed] [Google Scholar]
- 61.Ciaraldi TP, el Roeiy A, Madar Z, Reichart D, Olefsky JM, Yen SS. Cellular mechanisms of insulin resistance in polycystic ovarian syndrome. J Clin Endocrinol Metab. 1992;75(2):577–583. doi: 10.1210/jcem.75.2.1322430. [DOI] [PubMed] [Google Scholar]
- 62.Dunaif A, Wu X, Lee A, Diamanti-Kandarakis E. Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS) Am J Physiol Endocrinol Metab. 2001;281(2):E392–E399. doi: 10.1152/ajpendo.2001.281.2.E392. [DOI] [PubMed] [Google Scholar]
- 63.Baillargeon JP, Carpentier A. Role of insulin in the hyperandrogenemia of lean women with polycystic ovary syndrome and normal insulin sensitivity. Fertil Steril. 2007;88(4):886–893. doi: 10.1016/j.fertnstert.2006.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nestler JE, Singh R, Matt DW, Clore JN, Blackard WG. Suppression of serum insulin level by diazoxide does not alter serum testosterone or sex hormone-binding globulin levels in healthy, nonobese women. Am J Obstet Gynecol. 1990;163(4 Pt 1):1243–1246. doi: 10.1016/0002-9378(90)90698-7. [DOI] [PubMed] [Google Scholar]
- 65.Diamanti-Kandarakis E, Papavassiliou AG. Molecular mechanisms of insulin resistance in polycystic ovary syndrome. Trends Mol Med. 2006;12(7):324–332. doi: 10.1016/j.molmed.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 66.Dunaif A, Xia J, Book CB, Schenker E, Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest. 1995;96(2):801–810. doi: 10.1172/JCI118126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li M, Youngren JF, Dunaif A, Goldfine ID, Maddux BA, Zhang BB, Evans JL. Decreased insulin receptor (IR) autophosphorylation in fibroblasts from patients with PCOS: effects of serine kinase inhibitors and IR activators. J Clin Endocrinol Metab. 2002;87(9):4088–4093. doi: 10.1210/jc.2002-020363. [DOI] [PubMed] [Google Scholar]
- 68.Ciaraldi TP, Morales AJ, Hickman MG, Odom-Ford R, Olefsky JM, Yen SS. Cellular insulin resistance in adipocytes from obese polycystic ovary syndrome subjects involves adenosine modulation of insulin sensitivity. J Clin Endocrinol Metab. 1997;82(5):1421–1425. doi: 10.1210/jcem.82.5.3961. [DOI] [PubMed] [Google Scholar]
- 69.Ek I, Arner P, Bergqvist A, Carlstrom K, Wahrenberg H. Impaired adipocyte lipolysis in nonobese women with the polycystic ovary syndrome: a possible link to insulin resistance? J Clin Endocrinol Metab. 1997;82(4):1147–1153. doi: 10.1210/jcem.82.4.3899. [DOI] [PubMed] [Google Scholar]
- 70.Corbould A, Kim YB, Youngren JF, Pender C, Kahn BB, Lee A, Dunaif A. Insulin resistance in the skeletal muscle of women with PCOS involves intrinsic and acquired defects in insulin signaling. Am J Physiol Endocrinol Metab. 2005;288(5):E1047–E1054. doi: 10.1152/ajpendo.00361.2004. [DOI] [PubMed] [Google Scholar]
- 71.Hojlund K, Wojtaszewski JF, Birk J, Hansen BF, Vestergaard H, Beck-Nielsen H. Partial rescue of in vivo insulin signalling in skeletal muscle by impaired insulin clearance in heterozygous carriers of a mutation in the insulin receptor gene. Diabetologia. 2006;49(8):1827–1837. doi: 10.1007/s00125-006-0312-6. [DOI] [PubMed] [Google Scholar]
- 72.Larance M, Ramm G, Stockli J, van Dam EM, Winata S, Wasinger V, Simpson F, Graham M, Junutula JR, Guilhaus M, James DE. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280(45):37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 73.Sano H, Hsu DK, Apgar JR, Yu L, Sharma BB, Kuwabara I, Izui S, Liu FT. Critical role of galectin-3 in phagocytosis by macrophages. J Clin Invest. 2003;112(3):389–397. doi: 10.1172/JCI17592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exo-cytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mpl Biol Cell. 2004;15(10):4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hojlund K, Glintborg D, Andersen NR, Birk JB, Treebak JT, Frosig C, Beck-Nielsen H, Wojtaszewski JF. Impaired insulin-stimulated phosphorylation of Akt and AS160 in skeletal muscle of women with polycystic ovary syndrome is reversed by pioglitazone treatment. Diabetes. 2008;57(2):357–366. doi: 10.2337/db07-0706. [DOI] [PubMed] [Google Scholar]
- 76.Cazzolli R, Carpenter L, Biden TJ, Schmitz-Peiffer C. A role for protein phosphatase 2A-like activity, but not atypical protein kinase Czeta, in the inhibition of protein kinase B/Akt and glycogen synthesis by palmitate. Diabetes. 2001;50(10):2210–2218. doi: 10.2337/diabetes.50.10.2210. [DOI] [PubMed] [Google Scholar]
- 77.Ciaraldi TP, Morales AJ, Hickman MG, Odom-Ford R, Yen SS, Olefsky JM. Lack of insulin resistance in fibroblasts from subjects with polycystic ovary syndrome. Metabolism. 1998;47(8):940–946. doi: 10.1016/s0026-0495(98)90348-1. [DOI] [PubMed] [Google Scholar]
- 78.Tanti JF, Gual P, Gremeaux T, Gonzalez T, Barres R, Marchand-Brustel Y. Alteration in insulin action: role of IRS-1 serine phosphorylation in the retroregulation of insulin signalling. Ann Endocrinol (Paris) 2004;65(1):43–48. doi: 10.1016/s0003-4266(04)95629-6. [DOI] [PubMed] [Google Scholar]
- 79.Morin-Papunen LC, Vauhkonen I, Koivunen RM, Ruokonen A, Tapanainen JS. Insulin sensitivity, insulin secretion, and metabolic and hormonal parameters in healthy women and women with polycystic ovarian syndrome. Hum Reprod. 2000;15(6):1266–1274. doi: 10.1093/humrep/15.6.1266. [DOI] [PubMed] [Google Scholar]
- 80.Holte J, Bergh T, Berne C, Wide L, Lithell H. Restored insulin sensitivity but persistently increased early insulin secretion after weight loss in obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1995;80(9):2586–2593. doi: 10.1210/jcem.80.9.7673399. [DOI] [PubMed] [Google Scholar]
- 81.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23(2):201–229. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 82.Lam TK, van de WG, Giacca A. Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab. 2003;284(2):E281–E290. doi: 10.1152/ajpendo.00332.2002. [DOI] [PubMed] [Google Scholar]
- 83.Bachmann OP, Dahl DB, Brechtel K, Machann J, Haap M, Maier T, Loviscach M, Stumvoll M, Claussen CD, Schick F, Haring HU, Jacob S. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes. 2001;50(11):2579–2584. doi: 10.2337/diabetes.50.11.2579. [DOI] [PubMed] [Google Scholar]
- 84.Carpentier AC. Postprandial fatty acid metabolism in the development of lipotoxicity and type 2 diabetes. Diabetes Metab. 2008;34(2):97–107. doi: 10.1016/j.diabet.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 85.Mai K, Bobbert T, Reinecke F, Andres J, Maser-Gluth C, Wudy SA, Mohlig M, Weickert MO, Hartmann MF, Schulte HM, Diederich S, Pfeiffer AF, Spranger J. Intravenous lipid and heparin infusion-induced elevation in free fatty acids and triglycerides modifies circulating androgen levels in women: a randomized, controlled trial. J Clin Endocrinol Metab. 2008;93(10):3900–3906. doi: 10.1210/jc.2008-0714. [DOI] [PubMed] [Google Scholar]
- 86.Gromadzka-Ostrowska J. Effects of dietary fat on androgen secretion and metabolism. Reprod Biol. 2006;6(Suppl 2):13–20. [PubMed] [Google Scholar]
- 87.Pandey AV, Miller WL. Regulation of 17,20 lyase activity by cytochrome b5 and by serine phosphorylation of P450c17. J Biol Chem. 2005;280(14):13265–13271. doi: 10.1074/jbc.M414673200. [DOI] [PubMed] [Google Scholar]
- 88.Zhang LH, Rodriguez H, Ohno S, Miller WL. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: implications for adrenarche and the polycystic ovary syndrome. Proc Natl Acad Sci USA. 1995;92(23):10619–10623. doi: 10.1073/pnas.92.23.10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Usui I, Takata Y, Imamura T, Morioka H, Sasaoka T, Sawa T, Ishihara H, Ishiki M, Kobayashi M. Fatty acid induced insulin resistance in rat-1 fibroblasts overexpressing human insulin receptors: impaired insulin-stimulated mitogen-activated protein kinase activity. Diabetologia. 1997;40(8):894–901. doi: 10.1007/s001250050765. [DOI] [PubMed] [Google Scholar]
- 90.Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18(6):774–800. doi: 10.1210/edrv.18.6.0318. [DOI] [PubMed] [Google Scholar]
- 91.Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev. 1995;16(3):322–353. doi: 10.1210/edrv-16-3-322. [DOI] [PubMed] [Google Scholar]
- 92.Baillargeon JP. The Polycystic Ovary Syndrome—Current Concepts on Pathogenesis and Clinical Care. Springer; New York: 2007. Insulin action in polycystic ovary syndrome: in vivo and in vitro; pp. 43–68. [Google Scholar]
- 93.Nelson-DeGrave VL, Wickenheisser JK, Hendricks KL, Asano T, Fujishiro M, Legro RS, Kimball SR, Strauss JF, III, McAllister JM. Alterations in mitogen-activated protein kinase kinase and extracellular regulated kinase signaling in theca cells contribute to excessive androgen production in polycystic ovary syndrome. Mol Endocrinol. 2005;19(2):379–390. doi: 10.1210/me.2004-0178. [DOI] [PubMed] [Google Scholar]
- 94.Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83(6):2001–2005. doi: 10.1210/jcem.83.6.4886. [DOI] [PubMed] [Google Scholar]
- 95.Willis D, Mason H, Gilling-Smith C, Franks S. Modulation by insulin of follicle-stimulating hormone and luteinizing hormone actions in human granulosa cells of normal and polycystic ovaries. J Clin Endocrinol Metab. 1996;81(1):302–309. doi: 10.1210/jcem.81.1.8550768. [DOI] [PubMed] [Google Scholar]
- 96.Qin KN, Rosenfield RL. Role of cytochrome P450c17 in polycystic ovary syndrome. Mol Cell Endocrinol. 1998;145(1–2):111–121. doi: 10.1016/s0303-7207(98)00177-4. [DOI] [PubMed] [Google Scholar]
- 97.Wood JR, Nelson VL, Ho C, Jansen E, Wang CY, Urbanek M, McAllister JM, Mosselman S, Strauss JF., III The molecular phenotype of polycystic ovary syndrome (PCOS) theca cells and new candidate PCOS genes defined by microarray analysis. J Biol Chem. 2003;278(29):26380–26390. doi: 10.1074/jbc.M300688200. [DOI] [PubMed] [Google Scholar]
- 98.Kristiansen SB, Endoh A, Casson PR, Buster JE, Hornsby PJ. Induction of steroidogenic enzyme genes by insulin and IGF-I in cultured adult human adrenocortical cells. Steroids. 1997;62(2):258–265. doi: 10.1016/s0039-128x(96)00223-1. [DOI] [PubMed] [Google Scholar]
- 99.l’Allemand D, Penhoat A, Lebrethon MC, Ardevol R, Baehr V, Oelkers W, Saez JM. Insulin-like growth factors enhance steroidogenic enzyme and corticotropin receptor messenger ribonucleic acid levels and corticotropin steroidogenic responsiveness in cultured human adrenocortical cells. J Clin Endocrinol Metab. 1996;81(11):3892–3897. doi: 10.1210/jcem.81.11.8923834. [DOI] [PubMed] [Google Scholar]
- 100.Kramer RE, Buster JE, Andersen RN. Differential modulation of ACTH-stimulated cortisol and androstenedione secretion by insulin. J Steroid Biochem. 1990;36(1–2):33–42. doi: 10.1016/0022-4731(90)90111-5. [DOI] [PubMed] [Google Scholar]
- 101.Wu XK, Zhou SY, Liu JX, Pollanen P, Sallinen K, Makinen M, Erkkola R. Selective ovary resistance to insulin signaling in women with polycystic ovary syndrome. Fertil Steril. 2003;80(4):954–965. doi: 10.1016/s0015-0282(03)01007-0. [DOI] [PubMed] [Google Scholar]
- 102.Qu J, Wang Y, Wu X, Gao L, Hou L, Erkkola R. Insulin resistance directly contributes to androgenic potential within ovarian theca cells. Fertil Steril. 2009;91(5 Suppl):1990–1997. doi: 10.1016/j.fertnstert.2008.02.167. [DOI] [PubMed] [Google Scholar]
- 103.Hernandez ER, Resnick CE, Holtzclaw WD, Payne DW, Adashi EY. Insulin as a regulator of androgen biosynthesis by cultured rat ovarian cells: cellular mechanism(s) underlying physiological and pharmacological hormonal actions. Endocrinology. 1988;122(5):2034–2043. doi: 10.1210/endo-122-5-2034. [DOI] [PubMed] [Google Scholar]
- 104.Penhoat A, Chatelain PG, Jaillard C, Saez JM. Characterization of insulin-like growth factor I and insulin receptors on cultured bovine adrenal fasciculata cells. Role of these peptides on adrenal cell function. Endocrinology. 1988;122(6):2518–2526. doi: 10.1210/endo-122-6-2518. [DOI] [PubMed] [Google Scholar]
- 105.Munir I, Yen HW, Geller DH, Torbati D, Bierden RM, Weitsman SR, Agarwal SK, Magoffin DA. Insulin augmentation of 17alpha-hydroxylase activity is mediated by phosphatidyl inositol 3-kinase but not extracellular signal-regulated kinase-1/2 in human ovarian theca cells. Endocrinology. 2004;145(1):175–183. doi: 10.1210/en.2003-0329. [DOI] [PubMed] [Google Scholar]
- 106.Wickenheisser JK, Nelson-DeGrave VL, McAllister JM. Human ovarian theca cells in culture. Trends Endocrinol Metab. 2006;17(2):65–71. doi: 10.1016/j.tem.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 107.Kempna P, Hofer G, Mullis PE, Fluck CE. Pioglitazone inhibits androgen production in NCI-H295R cells by regulating gene expression of CYP17 and HSD3B2. Mol Pharmacol. 2007;71(3):787–798. doi: 10.1124/mol.106.028902. [DOI] [PubMed] [Google Scholar]
- 108.Beshay VE, Havelock JC, Sirianni R, Ye P, Suzuki T, Rainey WE, Carr BR. The mechanism for protein kinase C inhibition of androgen production and 17alpha-hydroxylase expression in a theca cell tumor model. J Clin Endocrinol Metab. 2007;92(12):4802–4809. doi: 10.1210/jc.2007-1394. [DOI] [PubMed] [Google Scholar]
- 109.Yildiz BO, Knochenhauer ES, Azziz R. Impact of obesity on the risk for polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93(1):162–168. doi: 10.1210/jc.2007-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Svendsen PF, Nilas L, Norgaard K, Jensen JE, Madsbad S. Obesity, body composition and metabolic disturbances in polycystic ovary syndrome. Hum Reprod. 2008;23(9):2113–2121. doi: 10.1093/humrep/den211. [DOI] [PubMed] [Google Scholar]
- 111.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 112.Poretsky L, Cataldo NA, Rosenwaks Z, Giudice LC. The insulin-related ovarian regulatory system in health and disease. Endocr Rev. 1999;20(4):535–582. doi: 10.1210/edrv.20.4.0374. [DOI] [PubMed] [Google Scholar]
- 113.Gambineri A, Pelusi C, Vicennati V, Pagotto U, Pasquali R. Obesity and the polycystic ovary syndrome. Int J Obes Relat Metab Disord. 2002;26(7):883–896. doi: 10.1038/sj.ijo.0801994. [DOI] [PubMed] [Google Scholar]
- 114.Yildirim B, Sabir N, Kaleli B. Relation of intra-abdominal fat distribution to metabolic disorders in nonobese patients with polycystic ovary syndrome. Fertil Steril. 2003;79(6):1358–1364. doi: 10.1016/s0015-0282(03)00265-6. [DOI] [PubMed] [Google Scholar]
- 115.Michelmore K, Ong K, Mason S, Bennett S, Perry L, Vessey M, Balen A, Dunger D. Clinical features in women with polycystic ovaries: relationships to insulin sensitivity, insulin gene VNTR and birth weight. Clin Endocrinol (Oxf) 2001;55(4):439–446. doi: 10.1046/j.1365-2265.2001.01375.x. [DOI] [PubMed] [Google Scholar]
- 116.Rebuffe-Scrive M, Cullberg G, Lundberg PA, Lindstedt G, Bjorntorp P. Anthropometric variables and metabolism in polycystic ovarian disease. Horm Metab Res. 1989;21(7):391–397. doi: 10.1055/s-2007-1009245. [DOI] [PubMed] [Google Scholar]
- 117.Pasquali R, Gambineri A, Pagotto U. The impact of obesity on reproduction in women with polycystic ovary syndrome. Bjog-An Int J Obstet Gynaecol. 2006;113(10):1148–1159. doi: 10.1111/j.1471-0528.2006.00990.x. [DOI] [PubMed] [Google Scholar]
- 118.Hirschberg AL. Polycystic ovary syndrome, obesity and reproductive implications. Womens Health (Lond Engl) 2009;5(5):529–540. doi: 10.2217/whe.09.39. [DOI] [PubMed] [Google Scholar]
- 119.Linne Y. Effects of obesity on women’s reproduction and complications during pregnancy. Obes Rev. 2004;5(3):137–143. doi: 10.1111/j.1467-789X.2004.00147.x. [DOI] [PubMed] [Google Scholar]
- 120.Plymate SR, Fariss BL, Bassett ML, Matej L. Obesity and its role in polycystic ovary syndrome. J Clin Endocrinol Metab. 1981;52(6):1246–1248. doi: 10.1210/jcem-52-6-1246. [DOI] [PubMed] [Google Scholar]
- 121.Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84(1):165–169. doi: 10.1210/jcem.84.1.5393. [DOI] [PubMed] [Google Scholar]
- 122.Baillargeon JP, Farid NR. Diagnosis and Management of Polycystic Ovarian Disease. Springer Science + Business Media Inc; New York: 2009. Medical treatment; pp. 209–232. [Google Scholar]
- 123.Anonymous. Consensus on infertility treatment related to polycystic ovary syndrome. Fertil Steril. 2008;89(3):505–522. doi: 10.1016/j.fertnstert.2007.09.041. [DOI] [PubMed] [Google Scholar]
- 124.Karimzadeh MA, Javedani M. An assessment of lifestyle modification versus medical treatment with clomiphene citrate, metformin, and clomiphene citrate-metformin in patients with polycystic ovary syndrome. Fertil Steril. 2009 doi: 10.1016/j.fertnstert.2009.02.078. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 125.Fux OC, Wior M, Iraci GS, Kaplan R, Torres D, Gaido MI, Wyse EP. Clinical, metabolic, and endocrine parameters in response to metformin and lifestyle intervention in women with polycystic ovary syndrome: a randomized, double-blind, and placebo control trial. Gynecol Endocrinol. 2009 doi: 10.3109/09513590903215581. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 126.Sastre ME, Prat MO, Checa MA, Carreras RC. Current trends in the treatment of polycystic ovary syndrome with desire for children. Ther Clin Risk Manag. 2009;5(2):353–360. doi: 10.2147/tcrm.s3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Moran L, Norman RJ. Understanding and managing disturbances in insulin metabolism and body weight in women with polycystic ovary syndrome. Best Pract Res Clin Obstet Gynaecol. 2004;18(5):719–736. doi: 10.1016/j.bpobgyn.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 128.Moran LJ, Noakes M, Clifton PM, Tomlinson L, Galletly C, Norman RJ. Dietary composition in restoring reproductive and metabolic physiology in overweight women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88(2):812–819. doi: 10.1210/jc.2002-020815. [DOI] [PubMed] [Google Scholar]
- 129.Stamets K, Taylor DS, Kunselman A, Demers LM, Pelkman CL, Legro RS. A randomized trial of the effects of two types of short-term hypocaloric diets on weight loss in women with polycystic ovary syndrome. Fertil Steril. 2004;81(3):630–637. doi: 10.1016/j.fertnstert.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 130.Kasim-Karakas SE, Almario RU, Gregory L, Wong R, Todd H, Lasley BL. Metabolic and endocrine effects of a polyunsaturated fatty acid-rich diet in polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(2):615–620. doi: 10.1210/jc.2003-030666. [DOI] [PubMed] [Google Scholar]
- 131.Douglas CC, Gower BA, Darnell BE, Ovalle F, Oster RA, Azziz R. Role of diet in the treatment of polycystic ovary syndrome. Fertil Steril. 2006;85(3):679–688. doi: 10.1016/j.fertnstert.2005.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bloomgarden ZT, Futterweit W, Poretsky L. Use of insulin-sensitizing agents in patients with polycystic ovary syndrome. Endocr Pract. 2001;7(4):279–286. doi: 10.4158/EP.7.4.279. [DOI] [PubMed] [Google Scholar]
- 133.Glueck CJ, Aregawi D, Agloria M, Winiarska M, Sieve L, Wang P. Sustainability of 8% weight loss, reduction of insulin resistance, and amelioration of atherogenic-metabolic risk factors over 4 years by metformin-diet in women with polycystic ovary syndrome. Metabolism. 2006;55(12):1582–1589. doi: 10.1016/j.metabol.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 134.Tang T, Glanville J, Orsi N, Barth JH, Balen AH. The use of metformin for women with PCOS undergoing IVF treatment. Hum Reprod. 2006;21(6):1416–1425. doi: 10.1093/humrep/del025. [DOI] [PubMed] [Google Scholar]
- 135.Yilmaz M, Biri A, Karakoc A, Toruner F, Bingol B, Cakir N, Tiras B, Ayvaz G, Arslan M. The effects of rosiglitazone and metformin on insulin resistance and serum androgen levels in obese and lean patients with polycystic ovary syndrome. J Endocrinol Invest. 2005;28(11):1003–1008. doi: 10.1007/BF03345339. [DOI] [PubMed] [Google Scholar]
- 136.Nawrocka J, Starczewski A. Effects of metformin treatment in women with polycystic ovary syndrome depends on insulin resistance. Gynecol Endocrinol. 2007;23(4):231–237. doi: 10.1080/09513590701260193. [DOI] [PubMed] [Google Scholar]
- 137.Tan S, Hahn S, Benson S, Dietz T, Lahner H, Moeller LC, Schmidt M, Elsenbruch S, Kimmig R, Mann K, Janssen OE. Metformin improves polycystic ovary syndrome symptoms irrespective of pre-treatment insulin resistance. Eur J Endocrinol. 2007;157(5):669–676. doi: 10.1530/EJE-07-0294. [DOI] [PubMed] [Google Scholar]
- 138.Maciel GA, Soares JM, Alves da Motta EL, Abi HM, de Lima GR, Baracat EC. Nonobese women with polycystic ovary syndrome respond better than obese women to treatment with metformin. Fertil Steril. 2004;81(2):355–360. doi: 10.1016/j.fertnstert.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 139.Mansfield R, Galea R, Brincat M, Hole D, Mason H. Metformin has direct effects on human ovarian steroidogenesis. Fertil Steril. 2003;79(4):956–962. doi: 10.1016/s0015-0282(02)04925-7. [DOI] [PubMed] [Google Scholar]
- 140.Rice S, Pellatt L, Ramanathan K, Whitehead SA, Mason HD. Metformin inhibits aromatase via an ERK (extracellular signal-regulated kinase)—mediated pathway. Endocrinology. 2009;150(10):4794–4801. doi: 10.1210/en.2009-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Azziz R, Ehrmann D, Legro RS, Whitcomb RW, Hanley R, Fereshetian AG, O’Keefe M, Ghazzi MN. Troglitazone improves ovulation and hirsutism in the polycystic ovary syndrome: a multicenter, double blind, placebo-controlled trial. J Clin Endocrinol Metab. 2001;86(4):1626–1632. doi: 10.1210/jcem.86.4.7375. [DOI] [PubMed] [Google Scholar]
- 142.Brettenthaler N, De Geyter C, Huber PR, Keller U. Effect of the insulin sensitizer pioglitazone on insulin resistance, hyperandrogenism, and ovulatory dysfunction in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89(8):3835–3840. doi: 10.1210/jc.2003-031737. [DOI] [PubMed] [Google Scholar]
- 143.Aroda VR, Ciaraldi TP, Burke P, Mudaliar S, Clopton P, Phillips S, Chang RJ, Henry RR. Metabolic and hormonal changes induced by pioglitazone in polycystic ovary syndrome: a randomized, placebo-controlled clinical trial. J Clin Endocrinol Metab. 2009;94(2):469–476. doi: 10.1210/jc.2008-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sepilian V, Nagamani M. Effects of rosiglitazone in obese women with polycystic ovary syndrome and severe insulin resistance. J Clin Endocrinol Metab. 2005;90(1):60–65. doi: 10.1210/jc.2004-1376. [DOI] [PubMed] [Google Scholar]
- 145.Cho LW, Jayagopal V, Kilpatrick ES, Atkin SL. The mean and the biological variation of insulin resistance does not differ between polycystic ovary syndrome and type 2 diabetes. Ann Clin Biochem. 2009;46(Pt 3):218–221. doi: 10.1258/acb.2008.008146. [DOI] [PubMed] [Google Scholar]
- 146.Veldhuis JD, Zhang G, Garmey JC. Troglitazone, an insulin-sensitizing thiazolidinedione, represses combined stimulation by LH and insulin of de novo androgen biosynthesis by thecal cells in vitro. J Clin Endocrinol Metab. 2002;87(3):1129–1133. doi: 10.1210/jcem.87.3.8308. [DOI] [PubMed] [Google Scholar]
- 147.Schoppee PD, Garmey JC, Veldhuis JD. Putative activation of the peroxisome proliferator-activated receptor gamma impairs androgen and enhances progesterone biosynthesis in primary cultures of porcine theca cells. Biol Reprod. 2002;66(1):190–198. doi: 10.1095/biolreprod66.1.190. [DOI] [PubMed] [Google Scholar]
- 148.Seto-Young D, Paliou M, Schlosser J, Avtanski D, Park A, Patel P, Holcomb K, Chang P, Poretsky L. Direct thiazolidinedione action in the human ovary: insulin-independent and insulin-sensitizing effects on steroidogenesis and insulin-like growth factor binding protein-1 production. J Clin Endocrinol Metab. 2005;90(11):6099–6105. doi: 10.1210/jc.2005-0469. [DOI] [PubMed] [Google Scholar]