

Abstract
Western blot analysis demonstrated that PC-12 cells express monomeric and dimeric forms of serine racemase (m-SR, d-SR) and that 1321N1 cells express m-SR. Quantitative RT-PCR and functional studies demonstrated that PC-12 cells express homomeric and heteromeric forms of nicotinic acetylcholine receptors (nAChR) while 1321N1 cell primarily express the α7-nAChR subtype. The effect of nAChR agonists and antagonists on SR activity and expression was examined by following concentration-dependent changes in intracellular D-Ser levels and SR protein expression. Incubation with (S)-nicotine increased D-Ser levels, which was attenuated by the α7-nAChR antagonist methyllycaconitine (MLA). Treatment of PC-12 cells with mecamylamine (MEC) produced a bimodal reduction of D-Ser reflecting MEC inhibition of homomeric and heteromeric nAChRs, while a unimodal curve was observed with 1321N1 cells, reflecting predominant expression of α7-nAChR. The nAChR subtype selectivity was probed using α7-nAChR selective inhibitors MLA and (R,S)-dehydronorketamine and α3β4-nAChR specific inhibitor AT-1001. The compounds reduced D-Ser in PC-12 cells, but only MLA and (R,S)-dehydronorketamine were effective in 1321N1 cells. Incubation of PC-12 and 1321N1 cells with (S)-nicotine, MEC and AT-1001 did not affect m-SR or d-SR expression, while MLA and (R,S)-dehydronorketamine increased m-SR expression but not SR mRNA levels. Treatment with cycloheximide indicated that increased m-SR was due to de novo protein synthesis associated with phospho-active forms of ERK1/2, MARCKS, Akt and rapamycin-sensitive mTOR. This effect was attenuated by treatment with the pharmacological inhibitors U0126, LY294002 and rapamycin, which selectively block the activation of ERK1/2, Akt and mTOR, respectively, and siRNAs directed against ERK1/2, Akt and mTOR. We propose that nAChR-associated changes in Ca2+ flux affect SR activity, but not expression, and that MLA and (R,S)-dehydronorketamine bind to allosteric sites on the α7-nAChR and promote multiple signaling cascades that converge at mTOR to increase m-SR levels.
Keywords: Serine racemase, D-Serine, nicotinic acetylcholine receptors, mTOR
1. Introduction
D-Serine (D-Ser) is an N-methyl D-aspartate receptor (NMDAR) co-agonist that plays a key role in neurotransmission [1]. Recent studies have demonstrated that synaptic NMDARs have a preferential affinity for D-Ser relative to the NMDAR co-agonist glycine and that these receptors play a key role in long-term potentiation (LTP) and NMDA-induced neurotoxicity [2]. A potential connection between endogenous D-Ser levels and CNS diseases and pathological states has been established with the observation that increased CNS levels of D-Ser are associated with amyotrophic lateral sclerosis and Alzheimer’s disease [1] while decreased CNS concentrations have been associated with schizophrenia [3, 4]. In addition, a reduction of endogenous D-Ser concentration in the rostral anterior cortex of the rat attenuated pain-related negative emotions, and led to the suggestion that reducing D-Ser concentrations and, thereby, NMDAR activity, may be a new strategy for reducing chronic pain-induced emotional distress [5].
Endogenous D-Ser is produced by the serine racemase (SR)-mediated enantio-conversion of L-Ser, and the inhibition and augmentation in the activity and expression of SR are currently areas of pharmacological and clinical interest [3, 4]. Since SR is a pyridoxal-5′-phosphate-dependent enzyme whose activation is dependent upon posttranslational binding of divalent cations such as Mg2+ and Ca2+ [4], a potential approach to the modulation of SR activity is the alteration of intracellular Ca2+ concentrations. The approach is supported by previous studies which demonstrated that 1) the addition of a calcium chelator to the incubation media decreased D-Ser release from rat neuronal cultures [6]; 2) paclitaxel- and vincristine-induced neuropathic pain (static mechanical allodynia and mechanic hyperalgesia) in the rat is significantly ameliorated by drugs that decrease extracellular and intracellular Ca+2 availability [7], and 3) NMDA-associated LTP of synaptic transmission in the CA1 region of the rat hippocampus can be regulated by Ca+2-dependent release of D-Ser from astrocytes [2]. In addition, we have recently demonstrated that incubation of PC-12 cells with gabapentin and (S)-pregabalin, inhibitors of the α2-δ subunit of voltage-gated Ca+2 channels, produced concentration-dependent decreases in intracellular D-Ser [8], suggesting that gabapentin-associated attenuation of Ca2+ influx may contribute to its clinical efficacy in a wide-range of neuropathic pain conditions [9,10]. This hypothesis is supported by the observation that gabapentin-related reductions in hyperalgesia and allodynia were reversed by D-Ser [9].
The current study explores the relationship between attenuation of the activity of neuronal nicotinic acetylcholine receptors (nAChRs) and the expression and activity of SR. The nAChR was chosen for study as this ligand-gated ion channel affects Ca2+ flux [11,12] and intracellular free Ca2+ concentrations via release from intracellular Ca2+ stores [13]. In order to explore this possibility, we have studied the effect of nAChR modulators on basal SR activity in PC-12 and 1321N1 cells. PC-12 cells were chosen as Western blot analysis has demonstrated that this cell line expresses monomeric and dimeric forms of SR, m-SR and d-SR [14], and Western blot analysis, ligand binding and functional studies have indicated that this cell line expresses heteromeric and homomeric forms of the nAChR [11, 12, 15, 16, 17]. The same experimental approaches have shown that 1321N1 cells express m-SR [14] and the α7-nAChR subtype [13, 18] with some expression of an unidentified heteromeric nAChR subtype(s) [18]. The cells were treated with sub-type specific nAChR antagonists, and the effects on SR activity assessed by measuring relative changes in intracellular D-Ser concentrations using a validated enantioselective capillary electrophoresis – laser-induced fluorescence assay [14]. Changes in intracellular D-Ser concentrations produced by the chelating agent ethylene glycol tetraacetic acid (EGTA) and the nAChR agonist (S)-nicotine were also examined. The effect of the test compounds on the expression of SR and phosphorylation of relevant intracellular signal transduction proteins were determined using Western blot technique, while the effect on SR gene expression was measured using quantitative RT- PCR. The results demonstrate for the first time that incubation with nAChR antagonists decreases intracellular D-Ser concentrations and that specific inhibition of α7-nAChR activity induces de novo SR protein expression via multiple signaling cascades that converge at mTOR. The results may afford a novel therapeutic strategy for the treatment of pain and neurological disorders associated with altered levels of endogenous D-Ser.
2. Materials and Methods
2.1. Materials
D-Serine (D-Ser), D-arginine (D-Arg), methyllycaconitine (MLA), 2-hydroxypropyl-β-cyclodextrin (HP-β-CD), acetonitrile, cycloheximide, fluorescein isothiocyanate (FITC), ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) and (S)-nicotine were obtained from Sigma-Aldrich (St. Louis, MO). (R,S)-dehydronorketamine (DHNK) was purchased from Cerillant (Round Rock, TX). Dihydro-β-erythroidine hydrobromide (DHβE) was purchased from Tocris (Minneapolis, MN). AT-1001 was kindly provided by Dr. N. Zaveri (Astraea Therapeutics, Mountain View, CA). Mecamylamine (MEC) was obtained from Ascent Scientific (Princeton, NJ), rapamycin was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and U0126 and LY294002 were from Calbiochem (La Jolla, CA). De-ionized water was obtained from a Milli-Q system (Millipore, Billerica, MA). All other chemicals used were of analytical grade.
2.2. Maintenance and treatment of cell lines
The PC-12 pheochromocytoma cell line derived from rat adrenal medulla was obtained from American Type Culture Collection (Manassas, VA). The human-derived 1321N1 astrocytoma cell line was obtained from European Collection of Cell Cultures (Sigma-Aldrich). Dulbecco’s modified eagle medium with glutamine, RPMI-1640, trypsin solution, phosphate-buffered saline, fetal bovine serum (FBS), sodium pyruvate (0.1 M), L-glutamine (0.2 M) and penicillin/streptomycin solution (containing 10,000 units/ml penicillin and 10,000 μg/ml streptomycin) were obtained from Quality Biological (Gaithersburg, MD), horse serum (heat inactivated) was purchased from Biosource (Rockville, MD) and HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer [1 M, pH 7.4] was obtained from Mediatech Inc. (Manassas, VA). The PC-12 cells were maintained in RPMI-1640 supplemented with 1 mM HEPES buffer, 10% horse serum, 5% FBS, 1% sodium pyruvate, 5 % L-glutamine and 1% penicillin/streptomycin, and the 1321N1 cells were maintained in Dulbecco’s modified eagle medium with L-glutamine supplemented with 10% FBS and 1% penicillin/streptomycin.
2.3. RNA extraction, cDNA synthesis and quantitative RT-PCR
The expression of the nicotinic acetylcholine receptors nAChR (CHRN) subunits was analyzed in PC-12 and 1321N1 cell lines. Cells were seeded on 100 × 20 mm tissue culture plates and maintained at 37 °C under humidified 5% CO2 in air until they reached >70% confluence and then collected for analysis. Total RNA was isolated by using the RNeasy mini kit (Qiagen, Valencia, CA). RNA concentration and quality was measured using the NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). To obtain cDNA, 1 μg total RNA was reverse-transcribed using the Promega reverse transcription kit (Promega Corporation, Madison, WI). Quantitative RT-PCR reactions were performed to determine the expression of the different subunits of CHRN mRNA using the PrimeTime qPCR Assays and Primers (IDT DNA Technologies, Coralville, IA) following the manufacturer’s instructions. Normalization was carried out using 18S and GAPDH. The genes and the catalog numbers used in this study are listed in the Supplementary Material (Table S1).
2.4. Determination of intracellular D-Ser concentrations
Intracellular D-Ser concentrations were measured using a previously described and validated capillary electrophoresis-laser induced fluorescence (CE-LIF) method performed using a P/ACE MDQ system equipped with a laser-induced fluorescence detector (Beckman Instruments, Fullerton, CA) [14]. In brief, at the completion of the incubations, the cells were collected, sedimented by centrifugation (1000 rpm, 5 min), and the supernatant discarded. The cell pellet was resuspended in 1.00 ml of water, and 0.05 ml of D-Arg [100 μM in water] was added as internal standard, followed by 4.0 ml of acetonitrile. The resulting suspension was sonicated for 20 min, centrifuged for 15 min at 2500 × g at 4 °C and the supernatant collected and stream dried under nitrogen. The residue was dissolved in 0.9 ml of borate buffer [80mM, pH 9.3] followed by 0.1 ml of FITC solution (3 mg/ml in acetone) and the resulting solution was placed in darkness for 12 h at room temperature. The samples were analyzed using an uncoated fused-silica capillary (50 μm I.D., effective length 50 cm), a running buffer composed of 500 μM HP-β-CD solution prepared in borate buffer [80 mM, pH 9.3] and detection at λ = 488 nm (excitation) and λ = 520 nm (emission). Quantification was accomplished using area ratios calculated for FITC-D-Ser with FITC-D-Arg as the internal standard.
2.5. Measurement of m-SR and d-SR expression by Western blotting
The expression level of m-SR and d-SR proteins was determined using polyacrylamide gel electrophoresis under denaturing conditions following established procedures. Cells were lysed in radioimmunoprecipitation buffer containing EGTA and EDTA (Boston BioProducts, Ashland, MA). The lysis buffer contained a protease inhibitor cocktail composed of 4-(2-aminoethyl)benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin, and aprotinin (Sigma-Aldrich). Protein concentrations were determined using the bicinchoninic acid reagent (Thermo Fisher Scientific, Waltham, MA). Proteins (20 μg/well) were separated on 4 to 12% precast gels (Invitrogen, Carlsbad, CA) using SDS-polyacrylamide gel electrophoresis under reducing conditions and then electrophoretically transferred onto polyvinylidene fluoride membrane (Invitrogen). Western blots were performed according to standard methods [19], which involved blocking in 5% non-fat milk and incubated with the primary antibody of interest, followed by incubation with a secondary antibody conjugated with the enzyme horseradish peroxidase. The detection of immunoreactive bands was performed by using the ECL Plus Western Blotting Detection System (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). The quantification of bands was done by volume densitometry using ImageJ software (National Institutes of Health, Bethesda, MD) and normalization to β-actin. The primary antibody for d-SR was obtained from Santa Cruz Biotechnology (cat. # sc-48741; Santa Cruz, CA), and the antibody that recognizes both m-SR and d-SR was purchased from Abcam, Inc. (cat. # ab45434; Cambridge, MA). The primary antibody for β-actin was from Abcam. The antibodies were used at a dilution recommended by the manufacturer.
2.6. Effects of MEC and MLA on intracellular D-Ser concentration and m-SR and d-SR expression
Cells were seeded on 100 × 20 mm tissue culture plates and maintained at 37 °C under humidified 5% CO2 in air until they reached >70% confluence. The original media was replaced with media containing the test compounds and the plates were incubated for an additional 36 h, unless otherwise indicated. The medium was removed, and the cells collected for analysis. When MEC was studied the concentrations used were: 0 – 250 μM; the study of the effects of MLA was performed using the following concentrations: 0 – 0.01 μM. The effect of the co-addition of MEC (10 μM) and MLA (0.050 μM) was also determined. The cells were assessed for intracellular D-Ser concentration and m-SR and d-SR expression as described above. The intracellular D-Ser concentrations were determined in this experiment and all of the following experiments using three replicate dishes each day for three independent days. Determination of SR protein expression for all studies was carried out on one set of dishes each day for three separate days.
2.7. Effect of MLA on SRR mRNA expression in 1321N1 cells
1321N1 cells were treated with MLA (0.05 μM) and cells were collected at 0, 1, 2, 4, 8 and 10 h post treatment. RNA isolation and cDNA synthesis were carried out as described above. Quantitative real-time PCR reactions were performed using the TaqMan gene expression assays (Applied Biosystems Inc., Foster City, CA) following the manufacturer’s instructions in order to determine the expression of SRR mRNA (Hs00222592_m1) using 18S RNA as the internal control. The SRR mRNA expression was determined using three replicate dishes each day for three independent days.
2.8. Effects of dihydro-β-erythroidine hydrobromide (DHβE), AT-1001, (R,S)-DHNK and (S)-nicotine on intracellular D-Ser concentrations and expression of m-SR and d-SR
PC-12 and 1321N1 cells were incubated for 36 h with serial concentrations of DhβE, 0 – 2 μM; AT-1001, 0 – 0.5 μM; (R,S)-DHNK, 0 – 0.45 μM; and (S)-nicotine, 0.01 – 100 μM. The intracellular D-Ser concentrations and expression of m-SR and d-SR were determined as described above.
2.9. Effects of EGTA, (S)-nicotine and MLA on m-SR and d-SR expression and activity
PC-12 and 1321N1 cells were incubated for 12 h with EGTA (1000 μM), MLA (0.05 μM), (S)-nicotine (10 μM), or the combinations of EGTA + MLA or (S)-nicotine + MLA. Cells were treated for 1 h with MLA prior to the addition of the second agent. The intracellular D-Ser concentrations and expression of m-SR and d-SR were determined as described above.
2.10. Effects of cycloheximide, U0126, LY294004 and rapamycin on m-SR expression and activity
1321N1 cells were incubated with cycloheximide (10 μg/ml) or MLA (0.05 μM), or the combination of cycloheximide and MLA for 3 to 36 h. In a second series of experiments, the cells were incubated for 12 h in the presence of U0126 (10 μM), LY294004 (10 μM), rapamycin (0.02 μM), MLA (0.050 μM), or the combinations of U0126 + MLA, LY294004 + MLA, or rapamycin + MLA in which MLA was added 1 h after the initiation of the incubation. Similar experiments were conducted to study the effect of a 12-h incubation with MEC (10 μM) alone or in combination with cycloheximide, U0126, LY294004 and rapamycin. The intracellular D-Ser concentrations and expression of m-SR levels were determined as described above.
2.11. Effects of MLA on pErk1/2, pMARCKS, pAKT and pmTOR levels
1321N1 cells were incubated with MLA (0.05 μM) for 15 to 360 min, after which the levels of phosphorylated and total forms of ERK1/2 (pErk1/2, Thr202/Tyr204), MARCKS (pMARCKS, Ser152/156), Akt (pAKT, Ser473) and mTOR (pmTOR, Ser2448) were determined by Western blotting technique. The primary antibodies for pERK1/2, pMARCKS, pAkt, pmTOR, Erk1/2 (total), Akt (total) and mTOR (total) were obtained from Cell Signaling Technology (Beverly, MA). The expression of total and phosphorylated forms of the signaling proteins was determined using one set of dishes each day for three separate days.
2.12. RNA Interference
1321N1 cells were transfected with siRNA oligos (0.75 μg) against ERK1/2, Akt, mTOR or a non-silencing control siRNA using 6 μl of siRNA Transfection Reagent (cat. # sc-29528, Santa Cruz Biotechnology, Santa Cruz, CA) following the manufacturer’s protocol. The siRNA used for this study was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). mTOR siRNA (cat. # sc-35409) is a pool of 3 target-specific 19–25 nt siRNAs designed to knock down mTOR gene expression. ERK 1 (cat. # sc-29307), ERK 2 (cat. # sc-35335), Akt 1 (cat. # sc-29195) and Akt 2 (cat. # sc-29197) siRNAs are target-specific 19–25 nt siRNA designed to knock down corresponding gene expressions. For transfection, ERK 1 and ERK 2 oligos were mixed together to knockdown ERK1/2 and same for Akt knockdown where Akt 1 and Akt 2 were added together. A non-silencing siRNA (cat. # sc-37007) was used as control. These siRNAs have been validated to perform efficient knockdown with minimal off-target effects [42–44]. Following 48 h of siRNA treatment, cells were washed with PBS, and maintained in serum-free medium for 3 h before the MLA (50 nM) treatment for 1 h, after which the levels of phosphorylated and total forms of ERK1/2 (pERK1/2, Thr202/Tyr204), Akt (pAKT, Ser473) and mTOR (pmTOR, Ser2448) were determined as described above. In a second series of experiments, the cells after 24 h of transfection were incubated 50 nM of MLA for 36 h. The intracellular D-Ser concentrations and expression of m-SR levels were determined as described above.
2.13. Statistical Analysis
The effect of the test compounds on intracellular D-Ser concentration (“response”) is reported as ‘average percent change ± standard deviation’. The”response” versus drug concentration sigmoidal dose-response curves (IC50 curves) were determined for each of the repeated sets using the ‘nonlinear regression (curve fit)’ model contained within the Prism 4 software package (GraphPad Software, Inc., La Jolla, CA) running on a personal computer. The statistical significance of the concentration-dependent effects on intracellular D-Ser concentrations for MEC, MLA and the combination of MEC and MLA was determined using ANOVA for repeated measures with a 2×6 model. A p<0.05 was set for statistical significance and the analyses were performed using Systat version 10.2 software (SYSTAT Software, Inc., www.systat.com). All other statistical comparisons were performed using a Student’s t-test program contained within the Prism 4 software package.
3. Results
3.1. Expression of nicotinic acetylcholine receptors nAChR (CHRN) subunits in PC-12 and 1321N1 cells
The data from the quantitative RT-PCR studies indicated that the PC-12 cell line expressed the α3, α5, α7, β2, and β4 subunits of the nAChR, which is consistent with previous studies that indicated expression of multiple heteromeric nAChR subtypes in PC-12 cells as well as the homomeric α7-nAChR (Table 1). The nAChR subunits expressed in 1321N1 cells were α5, α7, β2, and β4 (Table 1). Previous studies had established that the α7-nAChR subtype is the primary nAChR expressed in this cell line and that a κ-bungarotoxin-sensitive heteromeric αxβy-nAChR subtype(s) is also expressed. The data suggest that this subtype(s) is composed of the α5 subunit in combination with β2 and/or β4 subunits. However, the expression and functional activity of the potential subtype(s) had not been determined. The results from the current study indicate that subtypes containing the α6 subunit and the α4β2-nAChR and α9-nAChR subtypes were not expressed in either cell line.
Table 1.
Expression of the different nAChR subunit genes in 1321N1 and PC-12 cells determined in this study and previously reported expressed subtypes; where:
3.2. Effect of MEC on intracellular D-Ser concentrations in PC-12 and 1321N1 cells
The effects on intracellular D-Ser concentration of MEC, as well as the other agents used in this study, are presented as percentage change relative to the initial intracellular D-Ser concentration for each experiment. The results are presented in this manner because wide variations in initial intracellular D-Ser concentrations made it impossible to extract meaningful data based upon absolute D-Ser concentrations obtained after treatment with the test agents.
The 36-h incubation of PC-12 cells with MEC (0 – 250 μM) produced a concentration-dependent reduction in intracellular D-Ser levels with a maximum decrease of 44 ± 14% (***P < 0.001) observed at a MEC concentration of 250 μM (Fig. 1A). The plot of the average percentage decrease in D-Ser levels versus MEC concentration produced a biphasic curve (Fig. 1A). The data was used to calculate two IC50 values, 3.1 ± 1.2 μM and 99.5 ± 24.5 μM, associated with the MEC-induced decrease in intracellular D-Ser. MEC is a non-competitive inhibitor of both homomeric and heteromeric nAChRs with an inhibitory potency (IC50 value) ranging from 0.64 μM (α3β4-nAChR) to 6.9 μM (α7-nAChR), respectively [20]. The biphasic response of MEC in PC-12 cells is consistent with the inhibition of the basal activity of heteromeric and homomeric nAChR subtypes.
Fig. 1.

Effects of MEC and MLA on the intracellular D-Ser levels in PC-12 and 1321N1 cells. Cells were incubated with increasing concentrations of MEC (0 – 250 μM) (A) or MLA (0 – 0.1 μM) (B) for 36h followed by the determination of intracellular D-Ser content. IC50 values for MEC in PC-12 cells were 3.1 ± 1.2 μM (curve 1) and 99.5 ± 24.5 μM (curve 2); IC50 value for MEC in 1321N1 cells was 53.8 ± 8.1 μM; IC50 values for MLA were 0.05 ± 0.01 μM (PC-12) and 0.05 ± 0.01 μM (1321N1). C, Cells were incubated either with MLA (0.05 μM), MEC (10 μM) or the combination MLA+MEC for 36h. D, Cells were incubated with MLA (0.05 μM), EGTA (1 mM) or the combination MLA+EGTA for 12 h, where the experiments were done in triplicate on 3 independent days. Results are shown as average ± SD. All of the observed decreases in intracellular D-Ser were significant at p < 0.05.
A similar concentration-dependent reduction in relative D-Ser intracellular levels was observed when 1321N1 cells were treated with MEC, with a maximum reduction of 34 ± 11% (***P < 0.001) produced by 250 μM MEC (Fig. 1A). The plot of the average percentage decrease in D-Ser levels versus MEC concentration produced a monophasic curve (Fig. 1A), with a calculated IC50 value of 54 ± 8 μM, which is consistent with the effect of the antagonist at the α7-nAChR.
3.3. Effect of MLA on intracellular D-Ser concentrations in PC-12 and 1321N1 cells
Previous studies have determined that MLA is a selective competitive antagonist of the α7-nAChR at concentrations ≤0.1 μM [21] with some activity at the α9-nAChR [22]. In the present study, PC-12 and 1321N1 cells were incubated with MLA in concentrations ranging from 0.01 to 0.1 μM in order to selectively affect α7-nAChR activity. In PC-12 cells, incubation with increasing concentrations of MLA produced significant reductions in the relative intracellular D-Ser levels, with a maximum reduction of 22 ± 8% (***P < 0.001) at 0.1 μM MLA (Fig. 1B). The plot of the average percentage decrease in relative D-Ser levels versus MLA concentrations produced a monophasic curve (Fig. 1B) with a calculated IC50 value of 0.05 ± 0.01 μM. The treatment of 1321N1 cells with MLA also produced a concentration-dependent decrease in relative intracellular D-Ser levels, with a maximum decrease of 56 ± 5% (***P < 0.001) produced by 0.1 μM of MLA (Fig. 1B). The greater percentage reduction of D-Ser in 1321N1 cells relative to PC-12 cells probably reflects a higher relative expression of α7-nAChR in the 1321N1 cells. The plot of the average percentage decrease in D-Ser levels versus MLA concentrations produced a monophasic curve (Fig. 1B), with a calculated IC50 value of 0.05 ± 0.01 μM. The data suggest that the MLA-mediated decreases in the relative intracellular levels of D-Ser reflect the compound’s inhibitory effects on α7-nAChR activity and not on α9-nAChR activity since this subtype is not expressed in these cell lines.
3.4. Effect of simultaneous treatment of MEC and MLA on intracellular D-Ser concentrations in PC-12 and 1321N1 cells
In order to determine whether the effects of MEC and MLA on intracellular D-Ser concentrations were independent of each other, the cells were treated with MEC (10 μM) and MLA (0.05 μM) alone and in combination. MEC significantly reduced intracellular D-Ser concentrations in PC-12 and 1321N1 cells by 29 ± 5% and 12 ± 2%, respectively (Fig. 1C, ***P < 0.001), and treatment with MLA (0.05 μM) resulted in 19 ± 8% and 18 ± 5% reductions (***P < 0.001). Simultaneous treatment with MEC and MLA augmented the reduction of intracellular D-Ser concentrations in PC-12 and 1321N1 cells by 37 ± 4% and 22 ± 4%, respectively (***P < 0.001). The ANOVA analysis of the data suggests that the effect on the intracellular D-Ser concentrations due to co-treatment with MEC and MLA was additive (data not shown), suggesting a concerted inhibition of α7-nAChR and the heteromeric nAChRs expressed in these cell lines.
3.5. Effect of MEC and MLA on the expression of m-SR and d-SR in PC-12 and 1321N1 cells
Previous studies in our laboratory using Western blot techniques have demonstrated that PC-12 cells express m-SR and d-SR while only m-SR is expressed in the 1321N1 cell line [14]. Here, increasing concentrations of MEC (0 – 250 μM) produced no significant change in the expression of m-SR or d-SR in either cell line (data not shown).
The treatment of PC-12 cells with MLA (0 – 0.1 μM) had little effect on d-SR levels, but produced a concentration-dependent stimulation of m-SR protein expression, with a >2.5-fold increase at 0.1 μM of MLA (Fig. 2A). The treatment of 1321N1 cells with the same range of MLA concentrations also produced a significant ~4-fold increase in m-SR expression and this effect appeared to plateau at 0.06 μM (Fig. 2B). The higher level of α7-nAChR expression in 1321N1 cells may explain their greater sensitivity to MLA as compared to PC-12 cells. SR gene expression was studied in 1321N1 cells at 0, 1, 2, 4, 8 and 10 h post treatment with 0.05 μM MLA and no significant change was detected at any time point (data not shown).
Fig. 2. Modulation of SR protein levels by MLA and the calcium chelator EGTA.

A, PC-12 cells were treated with different concentrations of MLA for 36 h and then analyzed for SR expression by Western blot analysis using primary antibodies that detected both the m-SR and d-SR. B, SR expression was determined after a 36-h MLA treatment in 1321N1 cells. C, Bars represent relative levels of m-SR in both cell types after quantification and normalization with β-actin, which was used as loading control. Effect of EGTA on m-SR protein expression was assessed after a 12-h treatment either with MLA, EGTA, or MLA plus EGTA in PC-12 cells (D) and 1321N1 cells (E). F, Relative levels of m-SR in both PC-12 and 1321N1 cells after quantification and normalization with β-actin are shown. Data represents the average ± SD of three independent experiments. *, ** and ***, P< 0.05, 0.01 and 0.001 as compared with the control cells. Molecular mass numbers (kDa) are shown on the left of the immunoblots.
3.6. Effect of EGTA and MLA on intracellular D-Ser concentrations and m-SR and d-SR expression in PC-12 and 1231N1 cells
Addition of EGTA to the incubation medium for 12 h reduced the intracellular D-Ser concentrations in PC-12 and 1321N1 cells by 17 ± 3% and 12 ± 2%, respectively (Fig. 1D, *P < 0.05), consistent with previous reports showing that chelation of extracellular Ca2+ reduces SR activity in astrocytes as determined by measurement of D-Ser levels in the extracellular media [6, 2]. Incubation with 0.05 μM MLA for 12 h decreased intracellular D-Ser levels by 11 ± 1% and 7.3 ± 0.7% in PC-12 and 1321N1 cells, respectively, while treatment with MLA (1 h) followed by EGTA reduced the intracellular D-Ser concentration in an additive manner to 28 ± 5% and 19 ± 1% (Fig. 1D, *P < 0.05).
Incubation of 1321N1 cells with EGTA had no effect on the expression of m-SR (Fig. 2D) and similar treatment did not affect m-SR or d-SR protein levels in PC-12 cells (Fig. 2C, Fig. S1A). The combination of EGTA + MLA resulted in higher m-SR expression in 1321N1 and PC-12 cells (Fig. 2C), but did not affect the expression of d-SR in PC-12 cells (Fig. S1A). The results suggest that the increased expression of m-SR is a Ca2+–independent process that is initiated by MLA binding to the α7-nAChR.
3.7. Effect of (S)-nicotine on intracellular D-Ser concentrations and m-SR and d-SR expression in PC-12 and 1231N1 cells
nAChR agonists have been shown to increase intracellular Ca2+ levels [23]. Thus, we investigated the effect of (S)-nicotine on intracellular D-Ser concentrations and expression of m-SR and d-SR. The addition of (S)-nicotine, 0.01 – 10.0 μM, to the incubation media enhanced intracellular D-Ser levels in PC-12 and 1321N1 cells in a concentration-dependent manner, EC50 = ~2 μM (Fig. 3A), with 10 μM (S)-nicotine producing 31.7 ± 0.8 % and 35.2 ± 0.3% increases, respectively. The (S)-nicotine response was attenuated by pretreatment with 0.05 μM MLA, indicating the involvement of α7-nAChRs in the observed increase in intracellular D-Ser levels (Fig. 3B). Incubation of 1321N1 and PC-12 cells with (S)-nicotine had no effect on the expression of m-SR or d-SR protein levels (data not shown). The results support the hypothesis that antagonist binding to the α7-nAChR initiates the increased expression of m-SR.
Fig. 3.

Effects of (S)-nicotine and (R,S)-DHNK on the intracellular D-Ser levels. PC-12 and 1321N1 cells were incubated with increasing concentrations of S-nicotine (0 – 10 μM) (A) or (R,S)-DHNK (0.05 – 0.45 μM) (C) for 36h followed by the determination of intracellular D-Ser content. EC50 values for (S)-nicotine in PC-12 and 1321N1 cells were 1.9 ± 0.5 μM and 1.8 ± 0.2 μM, respectively; IC50 values for (R,S)-DHNK in PC-12 and 1321N1 cells were 0.115 and 0.035 μM, respectively. B, Cells were incubated with either MLA (0.05 μM), (S)-nicotine (10 μM) or the combination of MLA + (S)-nicotine for 12h. Results are shown as average ± SD, where the experiments were done in triplicate on 3 independent days. All of the observed decreases in intracellular D-Ser were significant at p < 0.05.
3.8. Effect of subtype selective nAChR antagonists on intracellular D-Ser concentrations and m-SR and d-SR expression in PC-12 and 1231N1 cells
In order to determine the nAChR subtypes associated with the effect of MEC and MLA on intracellular D-Ser concentrations and SR expression, PC-12 and 1321N1 cells were treated with DhβE, a selective antagonist for α4β2-nAChR and α4β4-nAChR [23], AT-1001, which is a selective α3β4-nAChR antagonist [23], and the selective α7-nAChR antagonist (R,S)-DHNK [24]. Cells treated with DHβE produced no significant change in intracellular D-Ser levels (data not shown), which is consistent with the lack of expression of α4β2-nAChR and α4β4-nAChR in these cell lines. Treatment of PC-12 cells with AT-1001 (0.01 – 0.5 μM) decreased intracellular D-Ser content in a concentration-dependent manner, IC50 = 0.087 μM, while no effect was observed in 1321N1 cells, which is consistent with the expression of α3β4-nAChR only in PC-12 cells. The incubation of PC-12 and 1321N1 cells with (R,S)-DHNK (0.05 – 0.45 μM) lowered intracellular D-Ser levels in a concentration-dependent manner, IC50 = 0.115 μM and IC50 = 0.035 μM, respectively (Fig. 3C), which is consistent with the expression of α7-nAChR in both cell lines. Under these conditions, the m-SR or d-SR protein levels were unaffected by DHβE or AT-1001 in either cell line (data not shown). However, incubation of PC-12 and 1321N1 cells with (R,S)-DHNK significantly increased expression of m-SR protein (Fig. 4A–C) without change in d-SR levels in PC-12 cells (Fig. S1B).
Fig. 4.
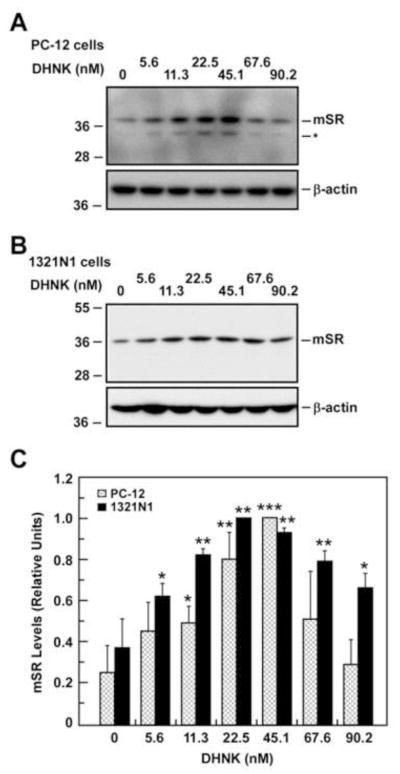
Expression of m-SR protein after treatment with different concentrations of (R,S)-dehydronorketamine (DHNK) for 36 h. Western blot analysis in PC-12 cells (A) and 1321N1 cells (B) with anti-SR antibody shows the expression of m-SR. C, Relative levels of m-SR in both PC-12 and 1321N1 cells after quantification and normalization with β-actin are shown in bars. Data represents the average ± SD of three independent experiments. *, ** and ***, P< 0.05, 0.01 and 0.001 as compared with the control cells.
3.9. Effect of cycloheximide on the expression and activity of m-SR and d-SR in PC-12 and 1231N1 cells
In order to determine if the MLA-induced increase in m-SR was a result of de novo protein synthesis, PC-12 and 1321N1 cells were treated with the protein synthesis inhibitor, cycloheximide. Cells were incubated either with cycloheximide (10 μg/ml), MLA (0.05 μM), or the combination of cycloheximide + MLA, and the effects on m-SR and d-SR expression were assessed at 12, 24 and 36 h. No significant changes in d-SR expression were observed in PC-12 cells at any point in these studies (data not shown). Cell treatment with cycloheximide produced no significant changes in m-SR expression up to the 24-h time-point while a ~50% reduction was observed at 36 h in both cell lines (Fig. 5C and 5D).
Fig. 5.

Effect of cycloheximide (CX) on m-SR protein expression after 12, 24, and 36 h treatment with CX, MLA or CX + MLA. Western blot analysis in PC-12 cells (A) and 1321N1 cells (B) using anti-SR antibody for the detection of m-SR. C, Graphs represent relative m-SR expression after quantification and normalization with β-actin in PC-12 cells (C) and 1321N1 cells (D). Data represents the average ± SD of three independent experiments. *, ** and ***, P< 0.05, 0.01 and 0.001 as compared with the control cells.
When PC-12 and 1321N1 cells were treated with 0.05 μM MLA, there was a ~1.5 to 2-fold increase in m-SR expression at 12-h, which was maintained through the 36-h time point (Fig. 5A and 5B). The co-incubation with cycloheximide attenuated the effect of MLA on m-SR at 12-h while blocking the MLA-associated response at the 24-h and 36-h time points (Fig. 5A and 5B). The effect on SR activity of MLA, cycloheximide and the combination of the agents was measured 12-h post treatment. Treatment of PC-12 cells with MLA resulted in no significant reduction, whereas cycloheximide and the combination of cycloheximide plus MLA produced a 41 ± 4% and 42 ± 3% reduction in intracellular D-Ser levels, respectively. The same treatments in 1321N1 cells resulted in reductions of 15 ± 4%, 23 ± 10% and 28 ± 8%, respectively. The data indicate that the newly synthesized m-SR protein generated in response to MLA treatment does not appear to be enzymatically active.
3.10. Effect of MLA on phospho-active forms of ERK1/2, MARCKS, Akt and mTOR in 1321N1 cells
To elucidate whether MLA-mediated inhibition of basal α7-nAChR activity was associated with activation of classical intracellular signal transduction molecules, 1321N1 cells were treated for 1 h with 0.05 μM MLA and the levels of phospho-active and total forms of ERK1/2, MARCKS, Akt and mTOR were then determined. The data demonstrate that there were significant increases in the ratio of phosphorylated/total form of these signaling proteins (Fig. 6A and 6B). Since the potent pharmacological inhibitors U0126, LY294002 and rapamycin selectively block the activation of MEK1/ERK1/2, phosphoinositide 3-kinase/Akt, and mTOR, respectively [25], we examined the effect of these inhibitors on the constitutive and MLA-induced increases in m-SR expression. While having no effect on basal m-SR protein levels, treatment of the cells for 1 h with U0126 or LY294002 followed by 12-h incubation with 0.05 μM MLA markedly attenuated the MLA-associated increase in m-SR expression (Fig. 6C and 6D). The same effect was observed after mTOR inhibition with rapamycin (Fig. 6C and 6D). As expected, when 10 μM MEC was used in place of MLA, no significant effect on m-SR expression was observed with any of the treatments (data not shown).
Fig. 6. Effects of pharmacological inhibitors on SR expression in basal and MLA-treated 1321N1 cells.

A, 1321N1 cells were treated with MLA for 1 h and processed for Western blot analysis. Signals associated with phosphorylated active and total forms of mTOR, MARCKS, AKT and Erk1/2 are shown. B, Bars represent the ratio of phosphorylated versus total forms after quantification. C, m-SR protein levels were determined after a 12-h treatment either with U0126 (UO), LY294002 (LY) or rapamycin (rapa) with or without addition of MLA. β-actin was used as a loading control. D, Relative m-SR levels after quantification and normalization with β-actin are shown in bars. Data represents the average ± SD of three independent experiments. * and ***, P< 0.05 and 0.001 as compared with the control cells.
The treatment of 1321N1 cells with U0126, LY294002 or rapamycin had no effect on the intracellular concentrations of D-Ser nor did co-incubation with MLA or MEC affect the reduction in intracellular D-Ser produced by the individual nAChR antagonists (data not shown).
3.11. Effect of MLA on phospho-active forms of ERK1/2, Akt, mTOR, and m-SR in transfected 1321N1 cells
In order to further investigate the possible association between the phospho-active forms of ERK1/2, Akt and mTOR, and the MLA-induced increase in m-SR expression, 1321N1 cells were transfected with siRNA against ERK1/2, Akt and mTOR, which resulted in reductions in the expression of the respective proteins (Fig. 7A). Transfection with the control siRNA significantly increased the basal level of pmTOR relative to the native 1321N1 cells and had no effect on the basal pAkt and pERK1/2 levels (Fig. 7B–D). Transfection with the control siRNA did not affect the significant increase in pmTOR, pAkt and pERK1/2 levels produced by incubation with MLA (Fig. 7B–D). Transfection with siRNAs directed against mTOR, Akt and ERK1/2 had little effect on the basal levels of pmTOR, pAkt and pERK1/2 relative to the native 1321N1 cells, but the MLA-associated increases were blocked in the transfected cells (Fig. 7B–D). Further, we observed that siRNA-associated reductions in the expression of ERK1/2, Akt and mTOR attenuated the MLA-induced increase in m-SR expression (Fig. 7E), but did not affect the intracellular concentrations of D-Ser (data not shown).
Fig. 7.

Effect of MLA on phospho-active forms of ERK1/2, Akt, mTOR, and m-SR in transfected 1321N1 cells. 1321N1 Cells were transfected with siRNA against ERK1/2, Akt and mTOR, treated with MLA and processed for Western blot analysis. A. Signals associated with phosphorylated active and total forms of mTOR, AKT and ERK1/2. B. Bars represent pmTOR after quantification in control and transfected 1321N1 cells before and after treatment with MLA. C. Bars represent pAkt after quantification in control and transfected 1321N1 cells before and after treatment with MLA. D. Bars represent the pERK1/2 after quantification in control and transfected 1321N1 cells before and after treatment with MLA. E. Relative m-SR levels after quantification and normalization with β-actin are shown in bars. Data represents the average ± SD of three independent experiments. * and ***, P< 0.05 and 0.001 as compared with the control cells.
4. Discussion
We have recently demonstrated that the voltage-gated calcium channel inhibitors gabapentin and (S)-pregabalin significantly reduce intracellular D-Ser concentrations in PC-12 cells [8]. This observation led us to examine the effect of changes in the activity of nAChRs on the relative intracellular concentrations of D-Ser, as these ion-gated channels contribute to intracellular Ca2+ flux [15, 26]. The relationship between nAChR activity and intracellular concentrations of D-Ser was initially investigated using the nAChR agonist (S)-nicotine, which has been shown to increase intracellular Ca2+ [24, 27]. Incubation of PC-12 and 1321N1 cells with (S)-nicotine increased intracellular D-Ser levels in a concentration-dependent manner, which was attenuated by MLA (Fig. 3A, 3B), indicating that (S)-nicotine-associated activation of the α7-nAChR activity in PC-12 and 1321N1 cells contributed to the observed increase in intracellular D-Ser concentrations.
Since PC-12 cells express heteromeric and homomeric nAChRs, we investigated the effect on intracellular D-Ser concentrations of MEC, a non-competitive, open-channel antagonist of heteromeric and homomeric nAChRs [16, 20] as MEC has been shown to reduce intracellular Ca2+ concentrations [24]. In this study, MEC produced a significant reduction in the relative intracellular D-Ser levels in a biphasic concentration-dependent manner (Fig. 1A), most likely reflecting the effect of MEC at multiple nAChRs.
The multi-receptor nature of the MEC inhibition was probed using the α7-nAChR selective inhibitors MLA [21] and (R,S)-dehydronorketamine [24], the α3β4-nAChR specific inhibitor AT-1001 [23] and the α4β2/α4β4-nAChR inhibitor DHβE [28]. Incubation of PC-12 cells with MLA and (R,S)-DHNK produced monophasic concentration-dependent reductions in relative D-Ser concentrations (Fig. 3C). The addition of AT-1001 to the incubation media also produced a monophasic concentration-dependent reduction in intracellular D-Ser levels while DhβE had no effect. The latter finding was expected because the PC-12 cell line does not express the α4 subunit of the nAChR (Table 1). The results indicate the inhibition of the α7-nAChR and α3β4-nAChR contributes to the observed reduction in intracellular D-Ser concentration in PC-12 cells and that the biphasic nature of the effect is most likely due to the relative potency of MEC at multiple heteromeric and homomeric nAChRs and the relative expression of these receptors.
Incubation of 1321N1 cells with MEC, MLA and (R,S)-DHNK reduced the relative intracellular D-Ser concentrations in monophasic concentration-dependent manners. AT-1001 and DHβE had no effect on the intracellular D-Ser levels in this cell line. The latter finding was expected as the 1321N1 cell line does not express the α2, α3, α4, or α9 subunits of the nAChR.
The data indicate that the inhibition of basal activity of homomeric and heteromeric nAChR lowers intracellular Ca2+, which, in turn, reduces the concentration of Ca2+–activated SR and basal intracellular D-Ser concentrations. This process is depicted in Fig. 8. The results obtained using combinations of MEC and MLA (Fig. 1C) further suggests that the observed effects are the result of independent interactions at individual nAChR subtypes.
Fig. 8.
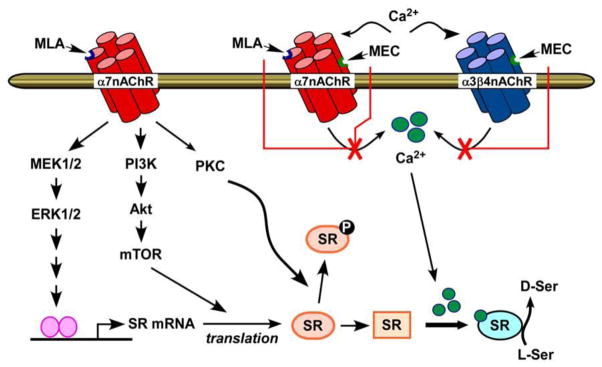
Schematic representation of modulation of SR expression and activity by the nAChR antagonists, MEC and MLA.
In our initial studies on the effect on SR activity, we observed that changes in intracellular D-Ser concentrations produced by L-Ser, glycine, gabapentin and (S)-pregabalin were not associated with alterations in m-SR or d-SR expression [8, 14]. This led us to examine the effect of (S)-nicotine and select nAChR antagonists on the expression of m-SR and d-SR in PC-12 cells and m-SR in 1321N1 cells.
While the intracellular Ca2+ concentrations in PC-12 and 1321N1 cells were increased by treatment with (S)-nicotine there was no change in m-SR or d-SR protein levels in either cell lines and the same result was observed when the intracellular Ca2+ concentrations were reduced using EGTA, MEC and AT-1001. These results suggest that alterations of intracellular Ca2+ concentrations have no direct effect on SR expression. Unexpectedly, treatment of PC-12 and 1321N1 cells with the α7-nAChR inhibitors MLA and (R,S)-DHNK produced a significant concentration-dependent increase in m-SR protein levels in both cell lines, but did not affect d-SR expression in PC-12 cells. The data suggest that antagonism of the α7-nAChR plays a role in the increased m-SR expression and the data obtained with AT-1001 supports the notion that inhibition of the α3β4-nAChR has no significant effect. The fact that MEC also inhibits α7-nAChR activity suggests that differences in the mode of inhibition could account for the observed properties of MEC vs MLA and (R,S)-DHNK. MEC is a non-competitive inhibitor of the α7-nAChR that binds within the central lumen of the receptor [20]. MLA is a selective competitive inhibitor at extracellular “orthosteric” binding sites located at the interface between two adjacent subunits and may also act as a non-competitive inhibitor of nAChR-positive allosteric modulators within an intrasubunit transmembrane cavity [29]. It has been suggested that the latter effect is due to binding to orthosteric binding sites, which results in the α7-nAChR adopting a conformation state that is unfavorable for the binding of nAChR-positive allosteric modulators and/or blocking allosteric binding sites [29]. (R,S)-DHNK is a negative allosteric modifier of the α7-nAChR, which does not act as an open-channel blocker or competitively inhibit at the “orthosteric” binding sites [24]. Thus, the source of the differential effects on m-SR expression of MLA and (R,S)-DHNK relative to MEC may lie in the binding of MLA and (R,S)-DHNK binding to “allosteric” site(s) on the α7-nAChR, which triggers the intracellular events leading to the increased production of SR protein.
The increase in m-SR expression by MLA and (R,S)-DHNK was unexpected since previous studies have demonstrated that the nAChR-associated induction of gene expression is an agonist-mediated process [11, 30, 31, 32]. For example, it has been demonstrated that in PC-12 cells and in the presence of the positive allosteric modulator PNU-120596, α7-nAChR agonists activated extracellular signal-regulated kinase-1 and -2 (ERK1/2) leading to CREB phosphorylation and transactivation of target genes [32]. Although MLA and chelation of extracellular Ca2+ attenuated these effects, the action of MLA alone on basal ERK1/2 activity was not examined [32]. We observed no increase in SR gene expression in response to MLA, indicating that a CREB-independent pathway may be responsible for the induction of de novo m-SR protein synthesis both in PC-12 and 1321N1 cells. Indeed, treatment of 1321N1 cells with MLA alone increased the phospho-active forms of important signaling intermediates, including the Akt/mTOR kinases, which are critical regulators of protein translation. This increase was attenuated by specific inhibitors and siRNAs of ERK1/2, Akt and mTOR. Thus, our data suggest that MLA promotes the convergence of multiple signaling cascades at mTOR to increase m-SR levels, but the newly synthesized protein is inactive.
One potential source of the production of an “inactive” form of m-SR is the observation that SR is the target of a number of posttranslational modifications. It has been shown that protein kinase C (PKC)-mediated phosphorylation of SR on serine residues decreases D-Ser levels in astrocytes and neuronal cultures [33]. Phosphorylation of SR on Thr227, which correlates with its translocation from the cytosol to the plasma membrane, also results in lower synthesis of D-Ser [34, 35]. In our study, MLA treatment of 1321N1 cells elicited PKC activation as evidenced by the increase in MARCKS phosphorylation (Fig. 6A). From these data, we speculate that MLA-mediated increase in PKC activity promotes SR phosphorylation, thus resulting in an “inactive” form of m-SR (Fig. 8).
5. Conclusions
The data from this study are consistent with the results from our previous study utilizing gabapentin and (S)-pregabalin [8] and indicate that the reduction of intracellular Ca+2 and the resulting attenuation of SR activity may be a useful therapeutic approach. In particular, the preferential affinity of synaptic NMDARs for D-Ser [2, 35] and the presynaptic expression of nAChRs, particularly α7-nAChRs [37, 38, 39], suggest that nAChR inhibitors, especially selective α7-nAChR antagonists, may be useful in the treatment of CNS pathologies associated with NMDA-induced neurotoxicity. The potential therapeutic utility of competitive nAChR antagonists has been previously suggested [40] and the data from this study suggest that non-competitive inhibitors may also be useful.
Supplementary Material
Highlights.
Nicotinic acetylcholine receptor antagonists inhibit serine racemase activity
Nicotinic acetylcholine receptor antagonists reduce intracellular D-serine
α7-Nicotinic acetylcholine receptor antagonists increase p-mTOR expression
Serine racemase expression is increased via mTOR pathway
Acknowledgments
This work was supported by funding from the Intramural Research Program of the National Institute on Aging/NIH.
List of nonstandard abbreviations
- nAChR
nicotinic acetylcholine receptor
- MLA
methyllycaconitine
- MEC
mecamylamine
- D-Ser
D-serine
- m-SR
monomeric serine racemase
- d-SR
dimeric serine racemase
- DHNK
dehydronorketamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wolosker H, Dumin E, Balan L, Foltyn V. D-Amino acids in the brain: D-Serine in neurotransmission and neurodegeneration. The FEBS Journal. 2008;275:3514–3526. doi: 10.1111/j.1742-4658.2008.06515.x. [DOI] [PubMed] [Google Scholar]
- 2.Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sethuraman R, Lee T, Tachibana S. D-Serine regulation: A possible therapeutic approach for central nervous diseases and chronic pain. Mini-Reviews in Medicinal Chemistry. 2009;9:813–819. doi: 10.2174/138955709788452630. [DOI] [PubMed] [Google Scholar]
- 4.Jirásková-Vanícková J, Ettrich R, Vorlová B, Hoffman H, Lepšík M, Jansa P, Konvalinka J. Inhibition of human serine racemase, an emerging target for medicinal chemistry. Current Drug Targets. 2011;12:1037–1055. doi: 10.2174/138945011795677755. [DOI] [PubMed] [Google Scholar]
- 5.Ren WH, Guo JD, Cao H, Wang H, Wang PF, Sha H, Rong R, Zhao ZQ, Zhang YQ. Is endogenous D-Serine in the rostral anterior cingulate cortex necessary for pain-related negative affect? Journal of Neurochemistry. 2006;96:1636–1647. doi: 10.1111/j.1471-4159.2006.03677.x. [DOI] [PubMed] [Google Scholar]
- 6.Kartvelishvily E, Shleper M, Balan L, Dumin E, Wolosker H. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. The Journal of Biological Chemistry. 2006;281:14151–14162. doi: 10.1074/jbc.M512927200. [DOI] [PubMed] [Google Scholar]
- 7.Siau C, Bennett GJ. Dysregulation of cellular calcium homeostasis in chemotherapy evoked painful peripheral neuropathy. Anesthesia and Analgesia. 2006;102:1485–1490. doi: 10.1213/01.ane.0000204318.35194.ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh NS, Paul RK, Torjman MC, Wainer IW. Gabapentin and (S)-pregabalin decrease intracellular d-serine concentrations in PC-12 cells. Neuroscience Letters. 2012;535:90–94. doi: 10.1016/j.neulet.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor CP. Mechanisms of analgesia by gabapentin and pregabalin – Calcium channel α2-δ [Cavα2-δ] ligands. Pain. 2009;142:13–16. doi: 10.1016/j.pain.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Turan A, Kaya G, Karamanlioglu B, Pamukcu Z, Apfel CC. Effect of oral gabapentin on postoperative epidural analgesia. British Journal of Anaesthesia. 2006;96:242–246. doi: 10.1093/bja/aei294. [DOI] [PubMed] [Google Scholar]
- 11.Nakayama H, Shimoke K, Isosaki M, Satoh H, Yoshizumi M, Ikeuchi T. Subtypes of neuronal nicotinic acetylcholine receptors involved in nicotine-induced phosphorylation of extracellular signal-regulated protein kinase in PC-12h cells. Neuroscience Letters. 2006;392:101–104. doi: 10.1016/j.neulet.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 12.Kouhen REL, Hu M, Anderson DJ, Li J, Gopalakrishnan M. Pharmacology of α7 nicotinic acetylcholine receptor mediated extracellular signal-regulated kinase signalling in PC-12 cells. British Journal Pharmacology. 2009;156:638–648. doi: 10.1111/j.1476-5381.2008.00069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma G, Vijayaraghavan S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proceedings of the National Academy of Science of the United States of America. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh NS, Paul RK, Sichler M, Moaddel R, Bernier M, Wainer IW. Capillary electrophoresis-laser-induced fluorescence (CE-LIF) assay for measurement of intracellular D-serine and serine racemase activity. Analytical Biochemistry. 2012;421:460–466. doi: 10.1016/j.ab.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickinson JA, Hanrott KE, Mok MH, Kew JN, Wonnacott S. Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC-12 cells. Journal of Neurochemistry. 2007;100:1089–1096. doi: 10.1111/j.1471-4159.2006.04273.x. [DOI] [PubMed] [Google Scholar]
- 16.Rogers SW, Mandelzys A, Deneris ES, Cooper E, Heinemann S. The expression of nicotinic acetylcholine receptors by PC-12 cells treated with NGF. Journal of Neuroscience. 1992;12:4611–4623. doi: 10.1523/JNEUROSCI.12-12-04611.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virginio C, Giacometti A, Aldegheri L, Rimland JM, Terstappen GC. Pharmacological properties of rat alpha 7 nicotinic receptors expressed in native and recombinant cell systems. European Journal of Pharmacology. 2002;445:153–161. doi: 10.1016/s0014-2999(02)01750-8. [DOI] [PubMed] [Google Scholar]
- 18.Kitabatake T, Moaddel R, Cole R, Gandhari M, Frazier C, Hartenstein J, Rosenberg A, Bernier M, Wainer IW. Characterization of a multiple ligand-gated ion channel cellular membrane affinity chromatography column and identification of endogenously expressed receptors in astrocytoma cell lines. Analytical Chemistry. 2008;80:8673–8680. doi: 10.1021/ac8016407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Tubuly AA. SDS-PAGE and Western Blotting. Methods in Molecular Medicine. 2000;40:391–405. doi: 10.1385/1-59259-076-4:391. [DOI] [PubMed] [Google Scholar]
- 20.Papke RL, Sanberg PR, Shytle RD. Analysis of mecamylamine stereoisomers on human nicotinic receptor subtypes. Journal of Pharmacology and Experimental Therapeutics. 2001;297:646–656. [PubMed] [Google Scholar]
- 21.Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. Methyllycaconitine is a potent antagonist of α-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. Journal of Pharmacology and Experimental Therapeutics. 2002;302:197–204. doi: 10.1124/jpet.302.1.197. [DOI] [PubMed] [Google Scholar]
- 22.Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB. Mixed nicotinic–muscarinic properties of the α9 nicotinic cholinergic receptor. Neuropharmacology. 2000;39:2515–2524. doi: 10.1016/s0028-3908(00)00124-6. [DOI] [PubMed] [Google Scholar]
- 23.Toll L, Zaveri NT, Polgar WE, Jiang F, Khroyan TV, Zhou W, Xie XS, Stauber GB, Costello MR, Leslie FM. AT-1001: A High Affinity and Selective α3β4 Nicotinic Acetylcholine Receptor Antagonist Blocks Nicotine Self-Administration in Rats. Neuropsychopharmacology. 2012;37:1367–1376. doi: 10.1038/npp.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, Jimenez L, Rosenberg A, Tran T, Xiao Y, Zarate CA, Wainer IW. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. European Journal of Pharmacology. 2013;698:228–234. doi: 10.1016/j.ejphar.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. The Biochemical Journal. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen JX, Yakel JL. Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacologica Sinica. 2009;30:673–680. doi: 10.1038/aps.2009.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carlisle DL, Hopkins TM, Gaither-Davis A, Silhanek MJ, Luketich JD, Christie NA, Siegfried JM. Nicotine signals through muscle-type and neuronal nicotinic acetylcholine receptors in both human bronchial epithelial cells and airway fibroblasts. Respiratory Research. 2004;5:27. doi: 10.1186/1465-9921-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alimohammadi H, Silver WL. Evidence for Nicotinic Acetylcholine Receptors on Nasal Trigeminal Nerve Endings of the Rat. Chemical Senses. 2000;25:61–66. doi: 10.1093/chemse/25.1.61. [DOI] [PubMed] [Google Scholar]
- 29.Gill JK, Savolainen M, Young GT, Zwart R, Sher E, Millar NS. Agonist activation of α7 nicotinic acetylcholine receptors via an allosteric transmembrane site. Proceedings of the National Academy of Science of the United States of America. 2011;108:5867–5872. doi: 10.1073/pnas.1017975108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gueorguiev VD, Zeman RJ, Hiremagalur B, Menezes A, Sabban EL. Differing temporal roles of Ca2+ and cAMP in nicotine-elicited elevation of tyrosine hydroxylase mRNA. The American Journal of Physiology. 1999;276:C54–C65. doi: 10.1152/ajpcell.1999.276.1.C54. [DOI] [PubMed] [Google Scholar]
- 31.Nakayama H, Numakawa T, Ikeuchi T, Hatanaka H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC-12h cells. Journal of Neurochemistry. 2001;79:489–498. doi: 10.1046/j.1471-4159.2001.00602.x. [DOI] [PubMed] [Google Scholar]
- 32.Gubbins EJ, Gopalakrishnan M, Li J. Alpha7 nAChR-mediated activation of MAP kinase pathways in PC-12 cells. Brain Research. 2010;1328:1–11. doi: 10.1016/j.brainres.2010.02.083. [DOI] [PubMed] [Google Scholar]
- 33.Vargas-Lopes C, Madeira C, Kahn SA, Albino do Couto I, Bado P, Houzel JC, De Miranda J, de Freitas MS, Ferreira ST, Panizzutti R. Protein kinase C activity regulates D-serine availability in the brain. Journal of Neurochemistry. 2011;116:281–290. doi: 10.1111/j.1471-4159.2010.07102.x. [DOI] [PubMed] [Google Scholar]
- 34.Balan L, Foltyn VN, Zehl M, Dumin E, Dikopoltsev E, Knoh D, Ohno Y, Kihara A, Jensen ON, Radzishevsky IS, Wolosker H. Feedback inactivation of D-serine synthesis by NMDA receptor-elicited translocation of serine racemase to the membrane. Proceedings of the National Academy of Science of the United States of America. 2009;106:7589–7594. doi: 10.1073/pnas.0809442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foltyn VN, Zehl M, Dikopoltsev E, Jensen ON, Wolosker H. Phosphorylation of mouse serine racemase regulates D-serine synthesis. FEBS Letters. 2010;584:2937–2941. doi: 10.1016/j.febslet.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 36.Papouin T, Ladépéche L, Ruel J, Sacchi S, Labasque M, Hanini M, Groc L, Pollegioni L, Mothet J-P, Oliet SHR. Cell. 2012;150:633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 37.Marchi M, Grilli M. Presynaptic nicotinic receptors modulating neurotransmitter release in the central nervous system: functional interactions with other coexisting receptors. Progress in Neurobiology. 2010;92:105–111. doi: 10.1016/j.pneurobio.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 38.Ge S, Dani JA. Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. The Journal of Neuroscience. 2005;25:6084–6091. doi: 10.1523/JNEUROSCI.0542-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones IW, Wonnacott S. Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. The Journal of Neuroscience. 2004;24:11244–11252. doi: 10.1523/JNEUROSCI.3009-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dwoskin LP, Crooks PA. Competitive neuronal nicotinic receptor antagonists: a new direction for drug discovery. The Journal of Pharmacology and Experimental Therapeutics. 2001;298:395–402. [PubMed] [Google Scholar]
- 41.Scholze P, Koth G, Orr-Urtreger A, Huck S. Subunit composition of α5-containing nicotinic receptors in the rodent habenula. Journal of Neurochemistry. 2012;121:551–560. doi: 10.1111/j.1471-4159.2012.07714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kong D, Banerjee S, Huang W, Li Y, Wang Z, Kim H-RC, Sarkar FH. Mammalian Target of Rapamycin Repression by 3,3′-Diindolylmethane Inhibits Invasion and Angiogenesis in Platelet-Derived Growth Factor-D–Overexpressing PC3 Cells. Cancer Research. 2008;68:1927–1934. doi: 10.1158/0008-5472.CAN-07-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ning L, Chen H, Kunnimalaiyaan M. Focal Adhesion Kinase, a Downstream Mediator of Raf-1 Signaling, Suppresses Cellular Adhesion, Migration, and Neuroendocrine Markers in BON Carcinoid Cells. Molecular Cancer Research. 2010;8:775–782. doi: 10.1158/1541-7786.MCR-09-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xin M, Deng X. Nicotine Inactivation of the Proapoptotic Function of Bax through Phosphorylation. The Journal of Biological Chemistry. 2005;280:10781–10789. doi: 10.1074/jbc.M500084200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.