

Abstract
The development and implementation of arenavirus reverse genetics represents a significant breakthrough in the arenavirus field 4. The use of cell-based arenavirus minigenome systems together with the ability to generate recombinant infectious arenaviruses with predetermined mutations in their genomes has facilitated the investigation of the contribution of viral determinants to the different steps of the arenavirus life cycle, as well as virus-host interactions and mechanisms of arenavirus pathogenesis 1, 3, 11 . In addition, the development of trisegmented arenaviruses has permitted the use of the arenavirus genome to express additional foreign genes of interest, thus opening the possibility of arenavirus-based vaccine vector applications 5 . Likewise, the development of single-cycle infectious arenaviruses capable of expressing reporter genes provides a new experimental tool to improve the safety of research involving highly pathogenic human arenaviruses 16 . The generation of recombinant arenaviruses using plasmid-based reverse genetics techniques has so far relied on the use of rodent cell lines 7,19 , which poses some barriers for the development of Food and Drug Administration (FDA)-licensed vaccine or vaccine vectors. To overcome this obstacle, we describe here the efficient generation of recombinant arenaviruses in FDA-approved Vero cells.
Keywords: Virology, Issue 78, Infection, Infectious Diseases, Microbiology, Molecular Biology, Cellular Biology, Medicine, Biomedical Engineering, Viruses, arenaviruses, plasmid transfection, recombinant virus, reverse genetics techniques, vaccine/vaccine vector seed development, clinical applications
Introduction
Arenaviruses are enveloped viruses with a bisegmented, negative-stranded RNA genome 3 that belong to the Arenaviridae family. The arenavirus genome encodes four proteins in an ambisense fashion from two separate viral segments 3. The large (L) segment encodes the RNA-dependent RNA polymerase (L) and the small RING (Really Interesting New Gene) finger protein (Z) that serves as the main driving force of virus budding. The small (S) segment encodes the viral nucleoprotein (NP) and the surface glycoprotein (GP) (Figure 1).
Several members of the Arenaviridae family are responsible for lethal hemorrhagic fever (HF) in humans 3. Of principle concerns are Lassa virus (LASV) and Junín virus (JUNV), which are known to cause high mortality in hospitalized patients 8, 9 . Although these viruses are endemic to West Africa and rural Argentina, respectively, there are increasing concerns of importation of LASV and JUNV to non-endemic areas due to increased travel 10. Additionally, although not normally associated with severe disease in humans, the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) is considered a neglected pathogen as there have been cases of lethal infection in immunocompromised patients 6, 15 and is responsible for congenital birth defects and spontaneous abortions in pregnant women 2, 13 . Currently there are no US FDA-approved vaccines against arenaviruses and treatment is limited to the nucleoside analog ribavirin, which is only partially effective and often associated with significant side effects.
The introduction of plasmid-based reverse genetics 4 and generation of recombinant arenaviruses 7, 19 have greatly advanced the field of arenavirus research. Currently, rodent cells (such as BHK-21) are used for the generation of recombinant arenaviruses due to the species-specific murine RNA polymerase I (pol-I) promoter directing the initial transcription of the S and L segments. However virus rescue in BHK-21 cells are not an approved method for the generation of recombinant arenaviruses as potential vaccine seed candidates. Here we document the use of the human pol-I promoter for efficient rescue of the prototypic Old World (OW) LCMV and the New World (NW) JUNV Candid#1 strain in Vero cells. Using a similar methodology, we generated recombinant trisegmented LCMV (r3LCMV) and Candid#1 (r3Candid#1) arenaviruses that contain two additional foreign genes encoded in two different S RNA segments 5 . This new system not only follows a highly reproducible and simple protocol but can be immediately implemented in the generation of recombinant arenaviruses as potential vaccine or vaccine vector seeds.
Protocol
1. Arenavirus Rescue Transfection
Our rescue system was based on the use of both polymerase II protein expression plasmids encoding the nucleoprotein (NP) and RNA-dependent-RNA-polymerase (L), the viral trans-acting factors required for RNA replication and gene expression of the arenavirus genome 7, 19 , and plasmids capable to direct intracellular synthesis, via the cellular RNA polymerase I (pol-I), of the S and L antigenome RNA species 7 . In our studies we use the pCAGGs protein expression plasmid, which uses the chicken β-actin promoter and polyadenylanation (pA) signal sequences, and the human pol-I plasmid, which uses the human polymerase I promoter and murine terminator sequences (Figure 2). The S and L RNA segments are cloned into the hpol-I plasmid in an antigenomic orientation to allow for the generation of genomic RNA segments upon transcription by hpol-I. For the rescue of wild-type recombinant LCMV (rLCMV) and Candid#1 (rCandid#1) the pCAGGs NP and L from each virus were co-transfected together with their respective human pol-I S and L RNA segments (Figure 3). Generation of recombinant trisegmented LCMV (r3LCMV) and Candid#1 (r3Candid#1) followed a similar protocol but the S segment was split into two different pol-I S plasmids, each encoding a distinctive reporter gene: in one, the NP open reading frame (ORF) was replaced with the Green Fluorescent Protein (GFP) reporter gene (hpol-I S GFP/GP) and, in the other, the viral glycoprotein precursor (GPC) ORF is replaced with the Gaussia luciferase (Gluc) reporter gene (hpol-I S NP/Gluc). A schematic representation of the protocol is illustrated in Figure 4. The following transfection and infection protocol has been established for 6-well-plates.
OptiMEM-Lipofectamine 2000 (LPF2000) mixture: Prepare 250 μl of OptiMEM media and, depending on the virus rescue, 10 - 12 μg of LPF2000 (1 μg/μl) per transfection (Table 1). A ratio of 2.5 μg LPF2000 per μg of DNA is recommended. Incubate for 5 - 10 min at RT. Meanwhile, prepare the plasmid transfection mixture in a separate microcentrifuge tube (step 1.2).
Plasmid DNA transfection mixture: Prepare the plasmid transfection cocktail in a microcentrifuge tube using the recommended amounts for each virus rescue (see Table 1). Bring the final volume to 50 μl with OptiMEM.
OptiMEM-LPF2000-DNA plasmid mixture: Add 250 μl of the OptiMEM-LPF2000 mixture (step 1.1) into the plasmid DNA transfection mixture (step 1.2). Incubate this mixture for 20 - 30 min at RT. Meanwhile prepare and count 1 x 106 Vero cells per transfection reaction. Vero cells will be transfected in suspension.
- Preparation of Vero cells: Vero cells are cultured in 100 mm dishes. Before starting, bring the 1x PBS, DMEM 10% FBS 1% PS media, and trypsin-EDTA mixture to 37 °C.
- Wash cells, twice, with 5 ml of 1x PBS.
- Trypsinize cells with 1 ml of trypsin-EDTA and wait until cells detach (approximately 5 min). Gentle tapping maybe required to fully detach the cells from the dishes.
- Carefully, resuspend the cells in 10 ml of DMEM 10% FBS 1% PS in a 15 ml centrifuge tube.
- Centrifuge the cells for 5 min at 1,000 rpm in a centrifuge.
- Resuspend the cells in 10 ml of DMEM 10% FBS 1% PS and count the cells using a hemocytometer and adjust concentration to 1 x 106 cells/ml. It was observed that 0.5 - 1 x 106 Vero cells per transfection produced the best results.
Mix LPF2000/DNA and cells: After 20 - 30 min room temperature incubation (step 1.3), add 1 ml of Vero cells (1 x 106) (step 1.4) to the OptiMEM-LPF2000-DNA plasmid mixture and incubate for 5 min at room temperature.
Seed LPF2000/DNA-cell mix into 6-well-plates: Add the DNA-LPF2000-Vero mixture (step 1.5) into the wells of the 6-well plate. Gently shake the 6-well-plate and let the transfection incubate overnight in the incubator at 37 °C and 5% CO2.
Change transfection medium: Approximately 16 - 24 hr post-transfection, remove the transfection media, add 2 ml of infection media and incubate transfected cells for an additional 48 hr.
- Cell passage: After 48 hr of incubation in infection media, remove the tissue culture supernatant (TCS) and pass the transfected cells into a 100 mm dish. The TCS rarely contains infectious virus because the transfected cells need additional incubation to generate virus particles. For the cell passage:
- Wash cells, twice, with 2 ml of 1x PBS.
- Trypsinize cells with 0.5 ml of trypsin-EDTA and wait until cells detach.
- Resuspend the cells in 1 ml of DMEM 10% FBS 1% PS in a 1.5 ml microcentrifuge tube.
- Centrifuge the cells for 5 min at 5,000 rpm, 4 °C in a microcentrifuge.
- Carefully resuspend the cells in 1 ml of infection media and transfer the cells to a 100 mm dish. Bring up the total volume in the plate, with infectious media, to 8 ml, a sufficient volume to prevent drying of the cells as well as for concentrating the virus.
Gently shake the 100 mm dish and continue the incubation of Vero cells in the incubator at 37 °C and 5% CO2 for 72 hr.
Collection of TCS: After 72 hr incubation, collect the TCS in a 15 ml centrifuge tube. Remove cell debris by centrifuging for 5 min at 2,500 rpm in a centrifuge. Infectious arenavirus is found in the TCS, so remove from the cell debris pellet and store the TCS at -80 °C.
2. Confirmation of Recombinant Arenavirus Rescue
Detection of a successful rescue of rLCMV and rCandid#1 is achieved via a focus forming unit (FFU) immunofluorescence assay (IFA). For wild-type virus rescue we use antibodies specific for the viral NP. The r3LCMV and r3Candid#1 encode two reporter genes (GFP and Gluc), thus allowing viral detection and titration without the need of antibodies by fluorescence microscopy (GFP) and/or luminescence (Gluc).
- Confirm wild-type recombinant arenavirus rescue: The day before titration, wash Vero cells twice with PBS, trypsinize and prepare 96-well plates to reach 80 - 90% confluence next day (4 x 104 cells/well). Gently shake the plates by hand to get a uniformed distribution of the cells. Culture the cells, overnight, in the 37 °C incubator with 5% CO2. Check the cells under the microscope to confirm a monolayer, before proceeding with the infection:
- Serially dilute (10 fold dilutions) the virus-containing TCS recovered from the transfected cells in the 100 mm dish (step 1.10) in OptiMEM.
- Remove media from the seeded Vero cells, wash twice with 50 μl of 1x PBS, and infect cells with 50 μl of the serially diluted virus (step 2.1). Infect cells for 1.5 hr at 37 °C and 5% CO2.
- After infection, remove the virus inoculum, add 100 μl of infection media/well and incubate the cells for 16 - 18 hr. Viral re-infection may occur after 18 hr incubation, which could result in over-estimation of the viral titers.
- At 16 - 18 hr post-infection, remove the TCS from each well and fix the cells with 4% formaldehyde diluted in 1x PBS, for 15 min at RT.
- Next, remove the fixation solution and permeabilize the cells with 0.1% triton X-100 diluted in 1x PBS, for 10 min at room temperature.
- Aspirate the permeabilization solution, then wash cells 3 times with 1x PBS.
- Block the cells with 2.5% albumin bovine serum (BSA) in 1x PBS (blocking solution) for 1 hr at RT. Alternatively, the cells can be blocked overnight at 4 °C and continued with antibody incubation the following day.
- Meanwhile prepare the primary antibody. Dilute the LCMV- and Candid#1-specific primary antibodies in blocking solution, and then centrifuge the antibody/blocking solution for 15 min at 3,500 rpm. The monoclonal anti-LCMV NP antibody clone 1.1.3 (1:30 dilution) 16 and the monoclonal anti-JUNV NP antibody SA02-BG12 from BEI Resources (1:500 dilution) can be used for the detection of rLCMV and rCandid#1, respectively.
- After a 1 hr incubation, aspirate the blocking solution and add 50 μl of the primary antibody to the cells and incubate for 1 hr at 37 °C.
- At this time, dilute the secondary antibody in blocking solution, and then centrifuge the antibody/blocking solution for 15 min at 3,500 rpm. The polyclonal rabbit anti-mouse IgG FITC secondary antibody for the detection of primary monoclonal antibodies was used to obtain images observed in Figure 5.
- After 1 hr incubation, aspirate the primary antibody, wash 3 times with 1x PBS, and add the secondary antibody for 30 min at 37 °C.
- Following 30 min incubation, aspirate the secondary antibody and wash 3 times with 1x PBS. The cells are now ready to be observed using fluorescence microscopy to both determine the success of viral rescue and titration by counting the focus forming units per ml (FFU/ml).
- r3LCMV and r3Candid#1 viral rescue: Since these viruses express two reporter genes, their rescues can be monitored by fluorescence microscopy to detect GFP expression or Gluc expression in TCS. Rescue can be also confirmed by expression of Gluc in TCS. To titrate the trisegmented viruses, we follow steps 2.1 to 2.1.4, however cells do not need to be fixed to determine a successful viral rescue because of GFP expression. The titration of the virus will be determined by counting the FFU/ml. To determine successful viral rescue by Gluc expression, Gluc expression from TCS can be measured using a Gluc assay kit (Biolux Gaussia Luciferase Assay kit, New England Biolabs). To that end:
- Pipet 100 μl of TCS into a 96-well white plate (Flat bottom microtiter plates, Fisher Scientific).
- Set up the luminometer (LumiCount, Packard Biosciences).
- Add 50 - 100 μl Gluc Assay solution (as recommended by the manufacturer) to each sample and measure the Gluc reporter gene expression with the luminometer. As a negative control, Gluc expression of the TCS from the wild-type viruses infected cells was measured.
3. Passage of Tissue Culture Supernatants
Viral rescue depends on transfection efficiencies. Vero cells have been shown to have lower transfection efficiencies than other cell lines 14 . If virus titers in the TCS are low, infect fresh Vero cells at a multiplicity of infection (MOI) of 0.01 (LCMV) or 0.1 (Candid#1) for 72 hr to amplify the virus.
Representative Results
Successful rescue of a recombinant wild-type arenavirus will be confirmed by the presence of viral antigens using IFA (Figure 5). In the case of recombinant trisegmented viruses, successful viral rescue can be assessed by observing GFP expression by fluorescence microscopy (Figure 6A). Successful rescue will be further confirmed by assessing Gluc expression (Figure 6B). Representative results illustrating the successful rescue of wild-type and trisegmented arenavirus were obtained using the above indicated protocol.
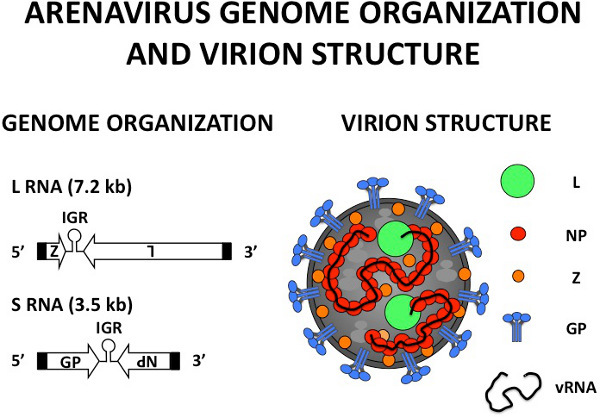 Figure 1. Arenavirus genome organization and virion structure. Arenaviruses are enveloped, negative-sense RNA viruses with bisegmented genomes 3. Each segment uses an ambisense coding strategy to direct the synthesis of two viral proteins in opposite orientation 3. The Large (L) segment (7.2 kb) encodes the RNA-dependent-RNA-polymerase (L) and the small RING finger protein (Z) that has matrix-like functions, including directing the budding process 3. The Small (S) segment (3.5 kb) encodes the glycoprotein precursor (GPC) and the nucleoprotein (NP). GPC is post-translationally processed to produce GP-1 and GP-2, which associate to form the glycoprotein complex (GP) that constitute the spikes on the surface of the virion structure responsible for receptor recognition and cell entry 17, 18 . NP encapsidates the viral RNA (vRNA) genome and together with the L protein form the viral ribonucleoproteins (vRNPs) that are the minimal components for viral replication and transcription 12 . IGR: intergenic region.
Figure 1. Arenavirus genome organization and virion structure. Arenaviruses are enveloped, negative-sense RNA viruses with bisegmented genomes 3. Each segment uses an ambisense coding strategy to direct the synthesis of two viral proteins in opposite orientation 3. The Large (L) segment (7.2 kb) encodes the RNA-dependent-RNA-polymerase (L) and the small RING finger protein (Z) that has matrix-like functions, including directing the budding process 3. The Small (S) segment (3.5 kb) encodes the glycoprotein precursor (GPC) and the nucleoprotein (NP). GPC is post-translationally processed to produce GP-1 and GP-2, which associate to form the glycoprotein complex (GP) that constitute the spikes on the surface of the virion structure responsible for receptor recognition and cell entry 17, 18 . NP encapsidates the viral RNA (vRNA) genome and together with the L protein form the viral ribonucleoproteins (vRNPs) that are the minimal components for viral replication and transcription 12 . IGR: intergenic region.
 Figure 2. Arenavirus rescue plasmids. Diagram of the plasmids used for the generation of recombinant arenavirus in Vero cells. Protein expression pCAGGs plasmid uses the chicken β-actin promoter and the rabbit β-globin polyadenylation (pA) signal sequences to direct the synthesis of viral L and NP 4, 7 . CMV-IE enhancer: cytomegalovirus immediate early enhancer. Intron: chicken β-actin intron sequence. The hpol-I plasmids uses the human polymerase I promoter and murine polymerase terminator sequences to direct the synthesis of the viral RNA segments. Both pCAGGs and hpol-I plasmids are Ampicillin resistant (Ampr).
Figure 2. Arenavirus rescue plasmids. Diagram of the plasmids used for the generation of recombinant arenavirus in Vero cells. Protein expression pCAGGs plasmid uses the chicken β-actin promoter and the rabbit β-globin polyadenylation (pA) signal sequences to direct the synthesis of viral L and NP 4, 7 . CMV-IE enhancer: cytomegalovirus immediate early enhancer. Intron: chicken β-actin intron sequence. The hpol-I plasmids uses the human polymerase I promoter and murine polymerase terminator sequences to direct the synthesis of the viral RNA segments. Both pCAGGs and hpol-I plasmids are Ampicillin resistant (Ampr).
 Figure 3. Wild-type and trisegmented arenavirus rescue. In the black oval are the plasmids required for the generation of recombinant wild-type arenavirus: pCAGGs NP and L, and hpol-I S and L. NP and L are required to initiate viral transcription and replication. The hpol-I S and L direct intracellular synthesis, via pol-I, of S and L antigenomic RNA species. In the red oval are the plasmids required for the generation of trisegmented arenaviruses: pCAGGs NP and L, as well as the hpol-I L plasmid, which are the same as those described for wild-type arenavirus rescue. The hpol-I S is separated into two plasmids: in one plasmid, NP is replaced with GFP (hpol-I S GFP/GP) and in the other plasmid, the GP is replaced by Gluc gene (hpol-I S NP/Gluc).
Figure 3. Wild-type and trisegmented arenavirus rescue. In the black oval are the plasmids required for the generation of recombinant wild-type arenavirus: pCAGGs NP and L, and hpol-I S and L. NP and L are required to initiate viral transcription and replication. The hpol-I S and L direct intracellular synthesis, via pol-I, of S and L antigenomic RNA species. In the red oval are the plasmids required for the generation of trisegmented arenaviruses: pCAGGs NP and L, as well as the hpol-I L plasmid, which are the same as those described for wild-type arenavirus rescue. The hpol-I S is separated into two plasmids: in one plasmid, NP is replaced with GFP (hpol-I S GFP/GP) and in the other plasmid, the GP is replaced by Gluc gene (hpol-I S NP/Gluc).
 Figure 4. Arenavirus rescue protocol. Arenavirus rescue is done in Vero cells using a 6-well plate format. Briefly, Vero cells are transfected in suspension on Day 1 using Lipofectamine 2000. At 24 hr post-transfection, media is replaced with fresh infectious media. Transfected cells are incubated for an additional 48 hr and then scaled-up into a 100 mm dishes (Day 4). Vero cells in the 100 mm dishes are incubated for an additional 72 hr. On Day 7 post-transfection TCS are collected for viral detection by either IFA (rLCMV and rCandid#1) or fluorescence microscopy (r3LCMV and r3Candid#1) following infection in fresh Vero cells. If the viral titers are lower than expected, TCS can be used to infect fresh Vero cells to amplify the virus.
Figure 4. Arenavirus rescue protocol. Arenavirus rescue is done in Vero cells using a 6-well plate format. Briefly, Vero cells are transfected in suspension on Day 1 using Lipofectamine 2000. At 24 hr post-transfection, media is replaced with fresh infectious media. Transfected cells are incubated for an additional 48 hr and then scaled-up into a 100 mm dishes (Day 4). Vero cells in the 100 mm dishes are incubated for an additional 72 hr. On Day 7 post-transfection TCS are collected for viral detection by either IFA (rLCMV and rCandid#1) or fluorescence microscopy (r3LCMV and r3Candid#1) following infection in fresh Vero cells. If the viral titers are lower than expected, TCS can be used to infect fresh Vero cells to amplify the virus.
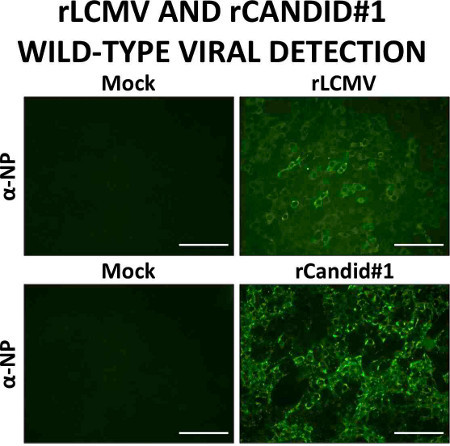 Figure 5. Confirmation of a successful wild-type viral rescue. TCS from cell passage (100 mm dishes) are used to infect fresh Vero cells in 96-well plates. At 24 hr post-infection, cells are fixed, permeabilized, and blocked before staining with LCMV- or Candid#1-specific antibodies. Presence of viral antigens is observed using fluorescence microscopy. Scale bars, 100 μm.
Figure 5. Confirmation of a successful wild-type viral rescue. TCS from cell passage (100 mm dishes) are used to infect fresh Vero cells in 96-well plates. At 24 hr post-infection, cells are fixed, permeabilized, and blocked before staining with LCMV- or Candid#1-specific antibodies. Presence of viral antigens is observed using fluorescence microscopy. Scale bars, 100 μm.
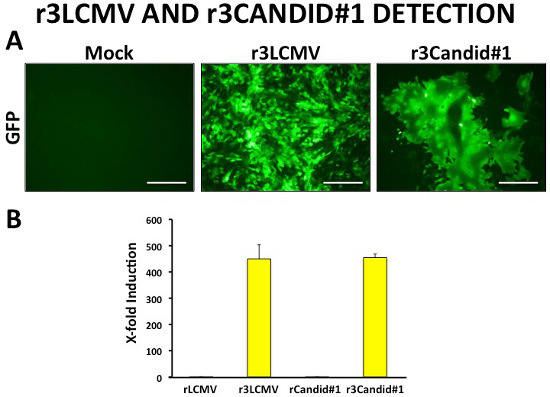 Figure 6. Successful rescue of trisegmented viruses. Recombinant trisegmented LCMV (r3LCMV) and Candid#1 (r3Candid#1) encode both GFP and Gluc. Successful viral rescue can be quickly determined by fluorescence microscopy (A). Gluc reporter gene expression can be used as a secondary confirmation of viral rescue. Fold induction was determined by normalization of Gluc luminescence values to wild-type virus infections (B). Scale bars, 100 μm.
Figure 6. Successful rescue of trisegmented viruses. Recombinant trisegmented LCMV (r3LCMV) and Candid#1 (r3Candid#1) encode both GFP and Gluc. Successful viral rescue can be quickly determined by fluorescence microscopy (A). Gluc reporter gene expression can be used as a secondary confirmation of viral rescue. Fold induction was determined by normalization of Gluc luminescence values to wild-type virus infections (B). Scale bars, 100 μm.
 Table 1. Lipofectamine/plasmid DNA concentration for arenavirus rescue. Recommended DNA and LPF2000 concentrations for generation of recombinant wild-type and trisegmented arenaviruses in Vero cells are indicated.
Table 1. Lipofectamine/plasmid DNA concentration for arenavirus rescue. Recommended DNA and LPF2000 concentrations for generation of recombinant wild-type and trisegmented arenaviruses in Vero cells are indicated.
Discussion
Generation of recombinant arenaviruses using plasmid-based reverse genetics techniques has become a widely used approach for the investigation of many different facets of arenavirus biology. Here we document a crucial improvement to the current system by performing arenavirus rescues in Vero cells, allowing for the potential generation of FDA-approved vaccine candidates against arenaviruses and vaccine vectors against other infectious diseases.
The experimental procedures involved are in general well established and should not pose significant hurdles. There are, however, several factors that should be carefully monitored to ensure a consistent success. Proper maintenance of Vero cells is crucial for a successful viral rescue. Additionally, it is imperative to have good plasmid preparations to generate recombinant arenavirus (see Protocol for proper cell maintenance and plasmid preparation). For the rescue of rLCMV and rCandid#1 wild-type or their trisegmented versions it is recommended three independent transfections for each recombinant virus to increase the likelihood of a successful rescue, as Vero cells are not easily transfected 14 . If more than one recombinant virus rescue is attempted, scale the following steps accordingly to the number of viruses to be rescued. As a negative control, we include a transfection in the absence of pCAGGs NP (-pCAGGs NP).
Since LCMV- and Candid#1-infected cells do not display classic cytopathic effect (CPE), successful viral rescue of wild-type rLCMV and rCandid#1 viruses must be assessed through the detection of infectious progeny, which could be done using a classic plaque assay or via IFA. We recommend the use of the IFA over the classic plaque assay because the former can be completed within 24 hr instead of the 5 - 6 days required for development of plaques. The r3LCMV or r3Candid#1 being rescued encode two reporter genes (GFP and Gluc), thus allowing viral detection and titration without the need of antibodies by fluorescence microscopy (GFP) and/or luminescence (Gluc). As in the rescue of recombinant LCMV and Candid#1 wild-type rescue, an unsuccessful trisegmented virus rescue will result in the lack of fluorescent Vero cells upon inoculation with TCS from transfected cells.
Given that the S and L segments are driven by the human polyermase I promoter, this rescue system can be applied in other human cells. Indeed we have also successfully generated both recombinant wild-type and trisegmented rLCMV and rCandid#1 viruses in 293T human embryonic kidney cells at high efficiency. Additionally, transfection in cell monolayers also resulted in virus rescue, albeit not as efficient as when transfecting Vero and 293T cells in suspension. In regards to the detection of wild-type LCMV and Candid#1 viral rescue, primary antibodies against other viral proteins could also be used. In the case of the r3 viruses, we could detect Gluc by luminescence instead of GFP expression. Alternatively, other reporter genes can be used in place of Gluc and GFP 5 .
Our virus rescue system has proven to be highly efficient in our hands, and here we provide the best transfection conditions. It is highly recommended to use the indicated amounts of plasmids. If virus rescue is performed in other human cell lines, the amount of cells, DNA, and/or LPF2000 should be tested.
The generation of wild-type and trisegmented arenavirus using plasmid-based reverse genetics in Vero cells will allow for studying multiple aspects of the biology of arenavirus as well as for the future development of FDA-approved vaccines and vaccine vectors.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank past and present members in JCT and LM-S laboratories for the development and improvement of the arenavirus reverse genetics techniques and plasmids. We also thank Snezhana Dimitrova for technical support. The monoclonal antibody directed to JUNV NP (SA02-BG12) was obtained from BEI Resources (NIAID Biodefense and Emerging Infections Research Resources Repository). BYHC was supported by Grant Number GM068411 from Institutional Ruth L. Kirschstein National Research Service Award. EO-R is a Fullbright-Conicyt (BIO 2008) and a Rochester Vaccine Fellowship (2012) recipient. EO-R current support is provided by a postdoctoral Fellowship for Diversity & Academic Excellence from the Office for Faculty Development & Diversity at the University of Rochester. Research in LM-S laboratory is funded by the NIH grants RO1 AI077719, R21NS075611-01, R03AI099681-01A1, the NIAID Centers of Excellence for Influenza Research and Surveillance (HHSN266200700008C), and The University of Rochester Center for Biodefense Immune Modeling (HHSN272201000055C).
Research at JCT laboratory was supported by grants RO1 AI047140, RO1 AI077719, and RO1 AI079665 from NIH.
References
- Albarino CG, Bird BH, Chakrabarti AK, Dodd KA, Flint M, Bergeron E, White DM, Nichol ST. The major determinant of attenuation in mice of the Candid1 vaccine for Argentine hemorrhagic fever is located in the G2 glycoprotein transmembrane domain. Journal of Virology. 2011;85:10404–10408. doi: 10.1128/JVI.00856-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton LL. Lymphocytic choriomeningitis virus: a neglected central nervous system pathogen. Clinical Infectious Diseases: an official publication of the Infectious Diseases Society of America. 1996;22:197. doi: 10.1093/clinids/22.1.197. [DOI] [PubMed] [Google Scholar]
- Buchmeier MJ, Peter CJ, de la Torre JC. Fields BN, Knipe DM, Howley PM, editors. Arenaviridae: The viruses and their replication. Fields' Virology. Fifth. 2007. p. 179201827.
- Emonet SE, Urata S, de la Torre JC. Arenavirus reverse genetics: new approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology. 2011;411:416–425. doi: 10.1016/j.virol.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emonet SF, Garidou L, McGavern DB, de la Torre JC. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3473–3478. doi: 10.1073/pnas.0900088106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SA, Graham MB, Kuehnert MJ, Kotton CN, Srinivasan A, Marty FM, Comer JA, Guarner J, Paddock CD, DeMeo DL, et al. Transmission of lymphocytic choriomeningitis virus by organ transplantation. The New England Journal of Medicine. 2006;354:2235–2249. doi: 10.1056/NEJMoa053240. [DOI] [PubMed] [Google Scholar]
- Flatz L, Bergthaler A, de la Torre JC, Pinschewer DD. Recovery of an arenavirus entirely from RNA polymerase I/II-driven cDNA. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:4663–4668. doi: 10.1073/pnas.0600652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther S, Lenz O. Lassa virus. Critical reviews in clinical laboratory sciences. 2004;41:339–390. doi: 10.1080/10408360490497456. [DOI] [PubMed] [Google Scholar]
- Harrison LH, Halsey NA, McKee KT, Peters CJ, Barrera Oro JG, Briggiler AM, Feuillade MR, Maiztegui JI. Clinical case definitions for Argentine hemorrhagic fever. Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America. 1999;28:1091–1094. doi: 10.1086/514749. [DOI] [PubMed] [Google Scholar]
- Isaacson M. Viral hemorrhagic fever hazards for travelers in Africa. Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America. 2001;33:1707–1712. doi: 10.1086/322620. [DOI] [PubMed] [Google Scholar]
- Kerber R, Rieger T, Busch C, Flatz L, Pinschewer DD, Kummerer BM, Gunther S. Cross-species analysis of the replication complex of Old World arenaviruses reveals two nucleoprotein sites involved in L protein function. Journal of Virology. 2011;85:12518–12528. doi: 10.1128/JVI.05091-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. Journal of Virology. 2000;74:3470–3477. doi: 10.1128/jvi.74.8.3470-3477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mets MB, Barton LL, Khan AS, Ksiazek TG. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. American Journal of Ophthalmology. 2000;130:209–215. doi: 10.1016/s0002-9394(00)00570-5. [DOI] [PubMed] [Google Scholar]
- Murakami S, Horimoto T, Yamada S, Kakugawa S, Goto H, Kawaoka Y. Establishment of canine RNA polymerase I-driven reverse genetics for influenza A virus: its application for H5N1 vaccine production. Journal of Virology. 2008;82:1605–1609. doi: 10.1128/JVI.01876-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, et al. A new arenavirus in a cluster of fatal transplant-associated diseases. The New England Journal of Medicine. 2008;358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- Rodrigo WW, de la Torre JC, Martinez-Sobrido L. Use of single-cycle infectious lymphocytic choriomeningitis virus to study hemorrhagic fever arenaviruses. Journal of Virology. 2011;85:1684–1695. doi: 10.1128/JVI.02229-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek JM, Kunz S. Cell entry by human pathogenic arenaviruses. Cellular Microbiology. 2008;10:828–835. doi: 10.1111/j.1462-5822.2007.01113.x. [DOI] [PubMed] [Google Scholar]
- Rojek JM, Sanchez AB, Nguyen NT, de la Torre JC, Kunz S. Different mechanisms of cell entry by human-pathogenic Old World and New World arenaviruses. Journal of Virology. 2008;82:7677–7687. doi: 10.1128/JVI.00560-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez AB, de la Torre JC. Rescue of the prototypic Arenavirus LCMV entirely from plasmid. Virology. 2006;350:370–380. doi: 10.1016/j.virol.2006.01.012. [DOI] [PubMed] [Google Scholar]