

Abstract
The occurrence of protein tyrosine nitration under disease conditions is now firmly established and represents a shift from the signal transducing physiological actions of •NO to oxidative and potentially pathogenic pathways. Tyrosine nitration is mediated by reactive nitrogen species such as peroxynitrite anion (ONOO–) and nitrogen dioxide (•NO2), formed as secondary products of •NO metabolism in the presence of oxidants including superoxide radicals (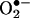 ), hydrogen peroxide (H2O2), and transition metal centers. The precise interplay between •NO and oxidants and the identification of the proximal intermediate(s) responsible for nitration in vivo have been under controversy. Despite the capacity of peroxynitrite to mediate tyrosine nitration in vitro, its role on nitration in vivo has been questioned, and alternative pathways, including the nitrite/H2O2/hemeperoxidase and transition metal-dependent mechanisms, have been proposed. A balanced analysis of existing evidence indicates that (i) different nitration pathways can contribute to tyrosine nitration in vivo, and (ii) most, if not all, nitration pathways involve free radical biochemistry with carbonate radicals (
), hydrogen peroxide (H2O2), and transition metal centers. The precise interplay between •NO and oxidants and the identification of the proximal intermediate(s) responsible for nitration in vivo have been under controversy. Despite the capacity of peroxynitrite to mediate tyrosine nitration in vitro, its role on nitration in vivo has been questioned, and alternative pathways, including the nitrite/H2O2/hemeperoxidase and transition metal-dependent mechanisms, have been proposed. A balanced analysis of existing evidence indicates that (i) different nitration pathways can contribute to tyrosine nitration in vivo, and (ii) most, if not all, nitration pathways involve free radical biochemistry with carbonate radicals (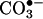 ) and/or oxo–metal complexes oxidizing tyrosine to tyrosyl radical followed by the diffusion-controlled reaction with •NO2 to yield 3-nitrotyrosine. Although protein tyrosine nitration is a low-yield process in vivo, 3-nitrotyrosine has been revealed as a relevant biomarker of •NO-dependent oxidative stress; additionally, site-specific nitration focused on particular protein tyrosines may result in modification of function and promote a biological effect. Tissue distribution and quantitation of protein 3-nitrotyrosine, recognition of the predominant nitration pathways and individual identification of nitrated proteins in disease states open new avenues for the understanding and treatment of human pathologies.
) and/or oxo–metal complexes oxidizing tyrosine to tyrosyl radical followed by the diffusion-controlled reaction with •NO2 to yield 3-nitrotyrosine. Although protein tyrosine nitration is a low-yield process in vivo, 3-nitrotyrosine has been revealed as a relevant biomarker of •NO-dependent oxidative stress; additionally, site-specific nitration focused on particular protein tyrosines may result in modification of function and promote a biological effect. Tissue distribution and quantitation of protein 3-nitrotyrosine, recognition of the predominant nitration pathways and individual identification of nitrated proteins in disease states open new avenues for the understanding and treatment of human pathologies.
Early after the discovery of the signal transducing physiological functions of the free radical nitric oxide (•NO) in the vasculature and nervous system (e.g., vasodilation and neurotransmission), it became evident that •NO could also participate as a cytotoxic effector molecule and/or a pathogenic mediator when produced at high rates by either inflammatory stimuli-induced nitric oxide synthase (iNOS) or overstimulation of the constitutive forms (eNOS and nNOS) (1). Much of •NO-mediated pathogenicity depends on the formation of secondary intermediates such as peroxynitrite anion (ONOO–) and nitrogen dioxide (•NO2) that are typically more reactive and toxic than •NO per se (2). The formation of reactive nitrogen species from •NO requires the presence of oxidants such as superoxide radicals (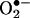 ), hydrogen peroxide (H2O2), and transition metal centers, the concentration of which can be increased either by •NO itself or by the same mediators that up-regulate •NO production. Nitrogen dioxide can also be formed in hydrophobic environments from the reactions of •NO with molecular oxygen, where these species concentrate (3, 4). One of the molecular footprints left by the reactions of reactive nitrogen species with biomolecules is the nitration (i.e., addition of nitro group, –NO2) of protein tyrosine residues to 3-nitrotyrosine. The formation of protein 3-nitrotyrosine was originally addressed in early protein chemistry studies with tetranitromethane aimed at establishing the function of tyrosines in proteins (5). This now-established posttranslational modification attracts considerable interest to biomedical research, because it can alter protein function, is associated to acute and chronic disease states, and can be a predictor of disease risk.
), hydrogen peroxide (H2O2), and transition metal centers, the concentration of which can be increased either by •NO itself or by the same mediators that up-regulate •NO production. Nitrogen dioxide can also be formed in hydrophobic environments from the reactions of •NO with molecular oxygen, where these species concentrate (3, 4). One of the molecular footprints left by the reactions of reactive nitrogen species with biomolecules is the nitration (i.e., addition of nitro group, –NO2) of protein tyrosine residues to 3-nitrotyrosine. The formation of protein 3-nitrotyrosine was originally addressed in early protein chemistry studies with tetranitromethane aimed at establishing the function of tyrosines in proteins (5). This now-established posttranslational modification attracts considerable interest to biomedical research, because it can alter protein function, is associated to acute and chronic disease states, and can be a predictor of disease risk.
Seminal work by Beckman et al. (6) and Ischiropoulos et al. (7) demonstrated the capacity of peroxynitrite to cause protein tyrosine nitration in vitro and established the concept that biologically produced intermediates could promote nitration in vivo (8, 9), as was also suggested in an earlier work by Ohshima et al. (10). However, the role of peroxynitrite as a central species in biological nitration has been more recently questioned (11, 12), and alternative pathways, most notably mechanisms that depend on the formation of •NO2 by the action of hemeperoxidases and/or transition metal complexes on a main product of •NO metabolism, nitrite (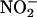 ), have been presented as contributors (12–15).
), have been presented as contributors (12–15).
In addition to nitration, inflammatory conditions promote other oxidative modifications in tyrosine such as chlorination, bromination, and hydroxylation to 3-chloro-, 3-bromo-, or 3-hydroxytyrosine, the detection of which may assist in identifying preferential nitration pathways. The extent of these oxidative modifications in vivo can reach similar values per milligram of tissue protein (e.g., 10–100 pmol/mg) and are comparable to the levels of protein S-nitrosation. It is also important to appreciate that other oxidative processes triggered by reactive nitrogen species such as thiol and methionine oxidation, disruption of iron–sulfur clusters, and oxidation of transition metal centers (2) can, in many cases, be more relevant than nitration in the promotion cell dysfunction/death.
A balanced analysis of all of the existing in vitro and in vivo evidence indicates that more than one pathway can contribute to protein tyrosine nitration. Interestingly, the alternative nitration pathways share common characteristics, because they involve free radical biochemistry with the participation of transient tyrosyl, •NO2, and carbonate (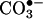 ) radicals and/or oxo–metal complexes.
) radicals and/or oxo–metal complexes.
NO Reaction with Superoxide and the Formation of Peroxynitrite
A relevant debate in the field has been whether peroxynitrite can be produced biologically at levels high enough to play a significant role in •NO-dependent pathology (16, 17). Because peroxynitrite is a transient species with a biological half-life [10–20 ms (18)] even shorter than that of •NO [1–30 s (1)], it cannot be directly measured, and its presence must be inferred from a combination of analytical, pharmacological, and/or genetic approaches (19).† The biological reactions of ·NO with 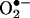 were initially proposed during studies that characterized the chemical nature of the endothelial-derived relaxing factor as •NO (20–22). Indeed, the biological half-life and actions of •NO in the vasculature were prolonged by superoxide dismutase (SOD), the enzyme that eliminates
were initially proposed during studies that characterized the chemical nature of the endothelial-derived relaxing factor as •NO (20–22). Indeed, the biological half-life and actions of •NO in the vasculature were prolonged by superoxide dismutase (SOD), the enzyme that eliminates 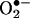 at diffusion-controlled rates, and were decreased by enhanced
at diffusion-controlled rates, and were decreased by enhanced 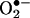 formation by redox-cycling molecules or high oxygen tensions. Thus, early data firmly established the existence of •NO and
formation by redox-cycling molecules or high oxygen tensions. Thus, early data firmly established the existence of •NO and 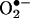 interactions, although at the time the reaction was perceived as one to just limit the biological half-life of •NO to yield relatively inert nitrate (
interactions, although at the time the reaction was perceived as one to just limit the biological half-life of •NO to yield relatively inert nitrate (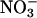 ).
).
The rate constant of the reaction of 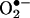 with •NO is larger (∼1010 M–1·s–1) than with SOD (1–2 × 109 M–1·s–1), and therefore •NO sometimes outcompetes SOD for
with •NO is larger (∼1010 M–1·s–1) than with SOD (1–2 × 109 M–1·s–1), and therefore •NO sometimes outcompetes SOD for 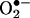 . The reaction of •NO with
. The reaction of •NO with 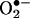 leads to the diffusion-controlled formation of peroxynitrite anion (ONOO–) (Eq. 1; for a review, see ref. 2):
leads to the diffusion-controlled formation of peroxynitrite anion (ONOO–) (Eq. 1; for a review, see ref. 2):
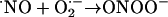 |
[1] |
The influence of SOD on the half-life of •NO and the formation of peroxynitrite under various fluxes of •NO and 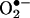 (23) and in different vascular tissue layers (24) have been recently analyzed. Being
(23) and in different vascular tissue layers (24) have been recently analyzed. Being 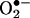 , one of the factors controlling the half-life, for instance, of vascular- (20–22) and inflammatory cell- (25) derived •NO, the subsequent formation of peroxynitrite is obligatory.‡ However, •NO and
, one of the factors controlling the half-life, for instance, of vascular- (20–22) and inflammatory cell- (25) derived •NO, the subsequent formation of peroxynitrite is obligatory.‡ However, •NO and 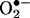 reactions do not necessarily result in tissue oxidative injury and in some cases can even be cytoprotective (30, 31). Indeed, low levels of peroxynitrite could be detoxified by enzymatic and nonenzymatic systems (32–35) and then, direct toxic effects of either •NO or
reactions do not necessarily result in tissue oxidative injury and in some cases can even be cytoprotective (30, 31). Indeed, low levels of peroxynitrite could be detoxified by enzymatic and nonenzymatic systems (32–35) and then, direct toxic effects of either •NO or 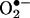 neutralized. Formation of peroxynitrite might also play subtle roles in signal transduction processes (36–39). However, identification of oxidation and nitration products in conjunction with pharmacological and/or genetic approaches has substantiated direct toxicity of peroxynitrite in pathophysiologically relevant conditions (see ref. 19 and references therein and below). Peroxynitrite is a strong one- and two-electron oxidant and can also evolve to •OH, •NO2, and
neutralized. Formation of peroxynitrite might also play subtle roles in signal transduction processes (36–39). However, identification of oxidation and nitration products in conjunction with pharmacological and/or genetic approaches has substantiated direct toxicity of peroxynitrite in pathophysiologically relevant conditions (see ref. 19 and references therein and below). Peroxynitrite is a strong one- and two-electron oxidant and can also evolve to •OH, •NO2, and 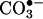 radicals. Peroxynitrite does not react directly with tyrosine (2) but oxidizes and nitrates it through its radical products. Revisions on the biological chemistry of peroxynitrite and its derived radicals have been published recently (2, 19, 40).
radicals. Peroxynitrite does not react directly with tyrosine (2) but oxidizes and nitrates it through its radical products. Revisions on the biological chemistry of peroxynitrite and its derived radicals have been published recently (2, 19, 40).
The Transition Metal-Dependent Formation of Strong Oxidants and Nitrogen Dioxide
Elevations of redox active metals systems such as iron (Fe), copper (Cu), and manganese (Mn) contribute to oxidative damage in disease states (12, 14). Also, inflammatory conditions trigger the release of leukocyte peroxidases, including myeloperoxidase (MPO) and eosinophil peroxidase (EPO), which play central roles in tissue oxidant formation (13, 15, 41). Oxidative stress promotes metal mobilization from proteins by mechanisms that include  oxidation of labile iron–sulfur clusters [e.g., aconitase and other dehydratases (42)], redox-dependent metal release from storage or transport proteins (transferrin, ferritin, and ceruloplasmin), oxidative modifications of heme proteins that result in heme release and/or degradation (myoglobin and cytochrome c), and histidine oxidation with disruption of metal coordination sites (i.e., Cu-Zn SOD), among others. Metals released from proteins are trapped by low molecular-weight (LMW) chelators such as adenine nucleotides (ATP3-and ADP2-), tri- and dicarboxylic acids (citrate, isocitrate, and piruvate), or phosphate or bind to macromolecules and in conjunction with free hemin (12, 14) become more readily accessible to undesired redox biochemistry (43). Typically, in the reductive environment of tissues, these metal complexes become reduced, and in particular ferrous iron can react with hydrogen peroxide to form variable amounts of free •OH and oxoferryl species [Fenton reaction (43); Eq. 2]:
oxidation of labile iron–sulfur clusters [e.g., aconitase and other dehydratases (42)], redox-dependent metal release from storage or transport proteins (transferrin, ferritin, and ceruloplasmin), oxidative modifications of heme proteins that result in heme release and/or degradation (myoglobin and cytochrome c), and histidine oxidation with disruption of metal coordination sites (i.e., Cu-Zn SOD), among others. Metals released from proteins are trapped by low molecular-weight (LMW) chelators such as adenine nucleotides (ATP3-and ADP2-), tri- and dicarboxylic acids (citrate, isocitrate, and piruvate), or phosphate or bind to macromolecules and in conjunction with free hemin (12, 14) become more readily accessible to undesired redox biochemistry (43). Typically, in the reductive environment of tissues, these metal complexes become reduced, and in particular ferrous iron can react with hydrogen peroxide to form variable amounts of free •OH and oxoferryl species [Fenton reaction (43); Eq. 2]:
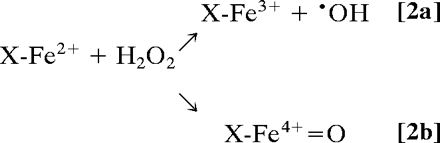 |
The large reactivity of free •OH with most biomolecules and its minimal diffusion distance (three to four molecular diameters) makes it unselective for protein modification unless site-specifically formed within metal-binding sites; oxo–iron complexes live longer and are more selective in substrate/target reactions. Oxidized transition metals also react with H2O2 to form secondary oxidizing species, e.g., ferric heme reaction with H2O2 yields an oxoferryl intermediate plus a porphyrin radical cation (14).
Oxo–metal complexes, in turn, favor formation of •NO2. Indeed, 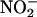 evolves to •NO2 via a one-electron oxidation. For instance, MPO (and EPO) can oxidize
evolves to •NO2 via a one-electron oxidation. For instance, MPO (and EPO) can oxidize 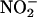 by compounds I (k = 2 × 106 M–1·s–1) (44) and II (k = 5.5 × 102 M–1·s–1) oxo–iron intermediates (14, 15) (Eqs. 3–5):
by compounds I (k = 2 × 106 M–1·s–1) (44) and II (k = 5.5 × 102 M–1·s–1) oxo–iron intermediates (14, 15) (Eqs. 3–5):
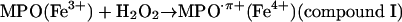 |
[3] |
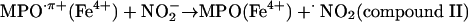 |
[4] |
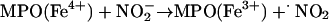 |
[5] |
Other hemeproteins, free hemin, and LMW complexes can undergo similar reactions to produce •NO2, although presumably at lower rates (12).
The reactions of peroxynitrite with oxidized transition metal centers including MPO (45) can sometimes concomitantly yield oxo–metal complexes and •NO2. Indeed, the O—O bond in ONOO– is significantly weakened by association with Lewis acids such as H+, CO2, or transition metal centers (MenX), which favor its homolysis (ref. 46; Eq. 6):
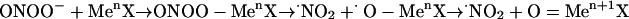 |
[6] |
Although the H+- or CO2-catalyzed homolysis results in radical yields of ≈30% and 35%, respectively, oxidant yield from metals (Eq. 6) can, in some cases, reach up to 100% (6, 46). Thus, there is positive feedback among redox active metals and oxidants and •NO to facilitate the production of the mediators of nitration.
Iron Regulatory Proteins, •NO, and Oxidant Stress
NO and oxidants influence the biochemical mechanisms that regulate cell iron homeostasis and that involve iron regulatory proteins 1 and 2 (IRP-1/2). IRPs play pivotal roles in assuring the maintenance of iron in the LMW pool at levels sufficient for the biosynthesis of heme- and nonheme proteins but not exceeding amounts that may impose a risk of metal-dependent oxidative damage (47). IRP-1 in its inactive state contains a cubane 4Fe-4S cluster with a labile iron (Feα) and displays aconitase activity. Total disassembly of the cluster to the apoprotein yields a fully active IRP-1, which in turn promotes down-regulation of ferritin and up-regulation of the transferrin receptor with an overall increase of LMW iron. Mild oxidative stress and/or •NO levels lead to IRP-1 activation, whereas over-production of reactive oxygen species or peroxynitrite extends their action beyond reversible cluster disassembly and produces irreversible inactivation via oxidation of critical thiols. IRP-2 does not contain the iron–sulfur cluster, and its activity is controlled by level of expression and oxidant-stimulated proteolytic degradation.
The oxidant-dependent inactivation of IRPs may be viewed as a negative feedback to inhibit the amplification of oxidative damage but, in fact, it disrupts the reversible mechanisms for controlling iron homeostasis and may render the cell more vulnerable. Also, an increase of the labile iron pool by •NO may favor, on initiation of oxidant stress, nitration reactions in target cells before the homeostatic responses for iron sequestration at the protein expression level are achieved. Interestingly, during inflammatory conditions, IRP-1 is found to be not only inactivated but also nitrated (48). The role of IRPs in the regulation of iron-dependent nitration reactions remains to be elucidated.
The Free Radical Pathways to Tyrosine Nitration
The very existence of 3-nitrotyrosine in vivo supports free radical biochemistry (Fig. 1). In fact, the two most widely invoked mechanisms of biological nitration, namely the peroxynitrite and the hemeperoxidase pathways, lead to the concomitant formation of tyrosyl radicals and •NO2, which combine at diffusion-controlled rates to form 3-nitrotyrosine (Fig. 1).
Fig. 1.

The free radical pathways of tyrosine nitration.
The oxidants leading to tyrosyl radical are either 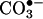 or oxo–metal complexes and, to a lesser extent, •OH. Importantly, •NO2 alone is inefficient in promoting nitration, because it must react first with tyrosine to yield tyrosyl radical, a reaction that is slow compared to other processes that •NO2 undergoes, for example, with thiols. The dimerization of tyrosyl radicals to 3,3′-dityrosine competes with the formation of 3-nitrotyrosine. However, protein tyrosyl radicals can be stabilized, with intra- and intermolecular dimerization limited due to spatial and diffusional constraints, both in aqueous and hydrophobic compartments, in which case their reaction with •NO2 is favored. Another competing pathway is the formation of 3-hydroxytyrosine, which can be mediated by •OH and oxo–metal complexes (15). An alternative radical mechanism for tyrosine nitration involves the reaction of a tyrosyl radical with •NO to form 3-nitrosotyrosine followed by a sequential two-electron oxidation to 3-nitrotyrosine via tyrosine iminoxyl radical; this mechanism may operate in transition metal-containing proteins that can readily oxidize 3-nitrosotyrosine before the latter can reversibly generate tyrosyl radicals and •NO, such as in the case of prostaglandin H synthase-2 (49).
or oxo–metal complexes and, to a lesser extent, •OH. Importantly, •NO2 alone is inefficient in promoting nitration, because it must react first with tyrosine to yield tyrosyl radical, a reaction that is slow compared to other processes that •NO2 undergoes, for example, with thiols. The dimerization of tyrosyl radicals to 3,3′-dityrosine competes with the formation of 3-nitrotyrosine. However, protein tyrosyl radicals can be stabilized, with intra- and intermolecular dimerization limited due to spatial and diffusional constraints, both in aqueous and hydrophobic compartments, in which case their reaction with •NO2 is favored. Another competing pathway is the formation of 3-hydroxytyrosine, which can be mediated by •OH and oxo–metal complexes (15). An alternative radical mechanism for tyrosine nitration involves the reaction of a tyrosyl radical with •NO to form 3-nitrosotyrosine followed by a sequential two-electron oxidation to 3-nitrotyrosine via tyrosine iminoxyl radical; this mechanism may operate in transition metal-containing proteins that can readily oxidize 3-nitrosotyrosine before the latter can reversibly generate tyrosyl radicals and •NO, such as in the case of prostaglandin H synthase-2 (49).
Is Electrophilic Aromatic Nitration a Biological Mechanism?
The reactions of peroxynitrite with transition metal centers may promote tyrosine nitration by a free radical-independent chemistry, namely electrophilic aromatic nitration (6, 7). In this mechanism, peroxynitrite would first form a complex with the transition metal to yield a polarized carrier of nitronium cation (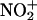 ), which may then decompose to free
), which may then decompose to free 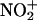 by heterolysis (Eq. 7):
by heterolysis (Eq. 7):
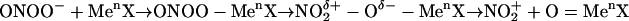 |
[7] |
Then, the polarized carrier or 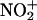 itself could attack tyrosine as a two-electron acceptor to yield a nitroarenium ion intermediate, which then evolves to 3-nitrotyrosine and a proton (50) (Eqs. 8–10):
itself could attack tyrosine as a two-electron acceptor to yield a nitroarenium ion intermediate, which then evolves to 3-nitrotyrosine and a proton (50) (Eqs. 8–10):
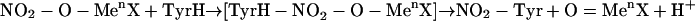 |
[8] |
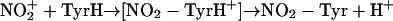 |
[9] |
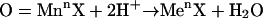 |
[10] |
The half-life of 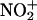 is too short in aqueous systems because of its fast reaction with H2O and subsequent decay to
is too short in aqueous systems because of its fast reaction with H2O and subsequent decay to 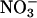 , and therefore the metal-bound
, and therefore the metal-bound 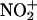 complex would be the proximal reactant (Eq. 8). This mechanism of nitration§ may operate for some metal complexes such as in the peroxynitrite-dependent nitrations of Tyr-108 of bovine CuZn SOD (6) or Tyr-34 of human MnSOD (51), although current data cannot rule out a free radical mechanism.
complex would be the proximal reactant (Eq. 8). This mechanism of nitration§ may operate for some metal complexes such as in the peroxynitrite-dependent nitrations of Tyr-108 of bovine CuZn SOD (6) or Tyr-34 of human MnSOD (51), although current data cannot rule out a free radical mechanism.
Electrophilic aromatic nitration can also occur in vitro by the action of nitryl chloride (NO2Cl), formed from the relatively slow reaction of MPO-derived hypochlorous acid with 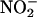 . However, the role of NO2Cl as a nitrating molecule in vivo is highly unlikely (52). In summary, present evidence cannot yet confirm that electrophilic aromatic nitration is a biologically relevant mechanism.
. However, the role of NO2Cl as a nitrating molecule in vivo is highly unlikely (52). In summary, present evidence cannot yet confirm that electrophilic aromatic nitration is a biologically relevant mechanism.
The Limited Efficiency of the Nitration Reactions in Biology
Biological nitration yields are low, and, under inflammatory conditions, one to five 3-nitrotyrosine residues per 10,000 tyrosine residues (100–500 μmol/mol) are detected (15, 53). This is due to a multiplicity of processes (reactions, diffusion) that the precursors of nitrating species (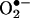 , H2O2, •NO) and the proximal intermediates of nitration (•NO,
, H2O2, •NO) and the proximal intermediates of nitration (•NO, 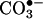 , oxo–metal complexes) can undergo, to repair mechanisms of the tyrosyl radical such as reduction by glutathione and, presumably, by metabolism of 3-nitrotyrosine.¶
, oxo–metal complexes) can undergo, to repair mechanisms of the tyrosyl radical such as reduction by glutathione and, presumably, by metabolism of 3-nitrotyrosine.¶
The idea of peroxynitrite as a mediator of biological nitration was challenged by observations demonstrating that the oxidative biochemistry of peroxynitrite could be strongly modulated by the relative fluxes of •NO and 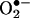 (56–58). Indeed, in homogenous systems, reasonable nitration yields are obtained only at an optimal
(56–58). Indeed, in homogenous systems, reasonable nitration yields are obtained only at an optimal 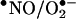 flux ratio ∼ one (11, 58), because excess of either •NO or
flux ratio ∼ one (11, 58), because excess of either •NO or 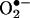 reacts with tyrosyl radicals and •NO2 and deactivates the process. Also, under low radical fluxes, the direct reaction of tyrosine with •NO2 competes with the recombination reaction of •NO2 with the tyrosyl radical. The observations demonstrated that free radical processes triggered by pure peroxynitrite may result in outcomes different than those initiated by the simultaneous production of •NO and
reacts with tyrosyl radicals and •NO2 and deactivates the process. Also, under low radical fluxes, the direct reaction of tyrosine with •NO2 competes with the recombination reaction of •NO2 with the tyrosyl radical. The observations demonstrated that free radical processes triggered by pure peroxynitrite may result in outcomes different than those initiated by the simultaneous production of •NO and 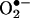 (11, 58) do not reconcile well with solid biological evidence (see below), supporting peroxynitrite-mediated nitration in vivo. This apparent conflict can be solved by appreciating the role of two key factors present in cells and tissues: first, SOD, which significantly lessens increases in the steadystate levels of
(11, 58) do not reconcile well with solid biological evidence (see below), supporting peroxynitrite-mediated nitration in vivo. This apparent conflict can be solved by appreciating the role of two key factors present in cells and tissues: first, SOD, which significantly lessens increases in the steadystate levels of 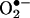 , despite large variations of
, despite large variations of 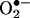 production rates; and second, the rapid transmembrane diffusion and/or cell consumption of •NO that operates as a drain of excess •NO; thus, augmentation of either •NO or
production rates; and second, the rapid transmembrane diffusion and/or cell consumption of •NO that operates as a drain of excess •NO; thus, augmentation of either •NO or 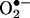 fluxes in vivo will serve to trap more of the partner radical, which otherwise is being consumed by other processes instead of accumulating and interfering with the nitration process. Experimental confounding factors have also obscured this discussion, including, in some works, the presence of uric acid (see below) and the lack of consideration of the diffusional properties of extracellular peroxynitrite and
fluxes in vivo will serve to trap more of the partner radical, which otherwise is being consumed by other processes instead of accumulating and interfering with the nitration process. Experimental confounding factors have also obscured this discussion, including, in some works, the presence of uric acid (see below) and the lack of consideration of the diffusional properties of extracellular peroxynitrite and 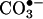 . Thus, a modest nitration but one that is responsive to the increase on the absolute •NO and/or
. Thus, a modest nitration but one that is responsive to the increase on the absolute •NO and/or 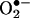 production rates can be generated by peroxynitrite in vivo. As examples at the cell level, the role of endogenous peroxynitrite in protein nitration has been substantiated by thorough studies on motoneurons (59) and smooth muscle cells (60) undergoing biologically relevant challenges.
production rates can be generated by peroxynitrite in vivo. As examples at the cell level, the role of endogenous peroxynitrite in protein nitration has been substantiated by thorough studies on motoneurons (59) and smooth muscle cells (60) undergoing biologically relevant challenges.
In inflammatory cells, the hemeperoxidase nitration pathway (MPO, EPO) appears to more important extra- than intracellularly, because intracellular peroxidases may have limited supply of 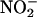 (61, 62). However, after polymorphonuclear cell degranulation, MPO can be taken up by nonphagocytic cells, and a strong codistribution of MPO and 3-nitrotyrosine is observed in different inflammatory processes (63). Excess •NO can also modulate the hemeperoxidase-mediated nitration; indeed, •NO readily reacts with resting state MPO as well as with compounds I and II, and therefore peroxidases may serve as catalytic sinks of •NO and influence nitration yields (64).
(61, 62). However, after polymorphonuclear cell degranulation, MPO can be taken up by nonphagocytic cells, and a strong codistribution of MPO and 3-nitrotyrosine is observed in different inflammatory processes (63). Excess •NO can also modulate the hemeperoxidase-mediated nitration; indeed, •NO readily reacts with resting state MPO as well as with compounds I and II, and therefore peroxidases may serve as catalytic sinks of •NO and influence nitration yields (64).
The Biological Significance of Protein Tyrosine Nitration
The small fraction of nitrated protein in the context of total tissue protein questions its possible biological relevance. It is noteworthy to indicate, however, that (i) a relatively limited number of proteins are preferential targets of nitration, and (ii) within these proteins, only one or a few specific tyrosines can be nitrated (65); because the species participating in nitration have short diffusion distances and site-specific nitration occurs at metal- or hemeperoxidase-binding sites, nitration reactions can be concentrated on proteins, cell, or tissue compartments as revealed by immunochemical (with antibodies against protein 3-nitrotyrosine) and proteomic-based methods. Thus, although there is a dilutional effect in quantitating total nitrated protein by analytical techniques in tissues, nitration can be focused on specific tyrosines and potentially result in modification, loss, or gain of function.
Addition of a –NO2 group to tyrosine lowers the pKa of its phenolic –OH by 2–3 units and adds a bulky substituent; if placed in relevant tyrosines, nitration can alter protein function and conformation, impose steric restrictions, and also inhibit tyrosine phosphorylation. However, to have biological significance, a loss-of-function modification requires a large fraction of protein to become nitrated at specific critical tyrosines and result in 3-nitrotyrosine to tyrosine ratios in the range of 0.1–1.0; documentation of these large ratios for a given protein in vivo is scanty, and it is doubtful that many proteins will undergo such an extent of nitration. An alternative scenario is a gain-of-function modification, in which case a small fraction of nitrated protein can elicit a substantive biological signal. This attractive concept has been shown in a few proteins such as cytochrome c, which acquires a strong peroxidase activity after nitration (66), in nitrated fibrinogen, which accelerates clot formation (67) and in protein kinase Cε, which becomes activated and translocates on nitration (68).
A notable example of loss of enzyme activity linked to nitration in vivo is the mitochondrial enzyme MnSOD. This protein is nitrated by peroxynitrite in Tyr-34 by a Mn-catalyzed process, which leads to enzyme inactivation. Nitrated and inactivated MnSOD is found in acute and chronic inflammatory processes both in animal models and human diseases (69). Interestingly, MnSOD nitration via the hemeperoxidase/•NO2 pathway does not lead to significant inactivation (70), suggesting that nitration occurs at tyrosines that are more superficial than the metal-adjacent Tyr-34, and that the association of nitration plus inactivation of MnSOD may serve to unravel the chemical nature of the nitrating species. A recent quantitation of the extent of MnSOD nitration in rat kidneys during angiotensin II infusion provided support for the hypothesis of nitration as a direct cause of enzyme inactivation (71). Similarly, the vascular enzyme prostacyclin synthase (PGI2) synthase becomes site-specifically nitrated by peroxynitrite at heme-adjacent Tyr-430 in a process catalyzed by the active site heme-thiolate involving transient ferrylspecies (72, 73). Note that the subcellular distribution of PGI2 synthase in vascular endothelium, colocalized with eNOS at the caveola, may further contribute for it to be a sensitive and critical target of peroxynitrite.∥ Recent experiments indicate that inflammation of artery walls results in rapid PGI2 synthase nitration by a peroxynitrite-dependent mechanism (73). Examples of MnSOD and PGI2 synthase affirm the concept that transition metal catalysis provides selectivity and efficiency to peroxynitrite as a biological nitrating agent. Finally, it is relevant to note that actin, which can constitute 5% or more of cell protein, is heavily nitrated in sickle cell disease and that the extents of nitration observed in tissues are sufficient to induce defective cytoskeletal polymerization (53).
The clinical relevance of protein tyrosine nitration has been recently underscored by the observation of a strong association between protein 3-nitrotyrosine levels and coronary artery disease risk (75). Circulating levels of protein 3-nitrotyrosine may serve as a biomarker to assess atherosclerosis risk as well as to monitor the vasculoprotective action of drugs. Interestingly, circulating MPO levels may also serve as predictor of cardiovascular risk (76). Thus, enhanced •NO consumption by either vascular wall/inflammatory cell-derived 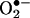 and/or MPO provides a common link between decreases in •NO bioavailability and increases in protein tyrosine nitration.
and/or MPO provides a common link between decreases in •NO bioavailability and increases in protein tyrosine nitration.
Mitochondrial Nitration and Its Relevance to Cell Death
Mitochondria have been recognized as critical sources and targets of nitrating species (reviewed in ref. 77). Although the formation of •NO mainly occurs at extramitochondrial sites, the facile diffusion of •NO to mitochondria and its combination with mitochondrial-derived 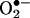 results in the formation of peroxynitrite, which accounts for much of the disruption of mitochondrial metabolism initially attributed to direct actions of •NO and to the nitration of mitochondrial proteins. A cytochrome c-dependent peroxidase-like mechanism of nitration may also operate (70). Mitochondrial proteins are nitrated in vitro and in vivo, including MnSOD, aconitase, cytochrome c, voltage-dependent anion channel, ATPase, and succinyl-CoA oxoacid-CoA transferase (77, 78). The nitration/inactivation of MnSOD operates as a positive loop for enhanced intramitochondrial peroxynitrite formation, which in turn triggers apoptotic signaling of cell death, in part by the thiol oxidation-dependent assembly of the permeability transition pore (77). Detection of nitrated mitochondrial proteins and even nitrated cytochrome c in the cytosol may serve to reflect extents of mitochondrial •NO-dependent oxidative stress (66, 77, 78). Importantly, the enhanced peroxidatic activity of nitrated cytochrome c (66, 77) may further contribute to oxidative damage.
results in the formation of peroxynitrite, which accounts for much of the disruption of mitochondrial metabolism initially attributed to direct actions of •NO and to the nitration of mitochondrial proteins. A cytochrome c-dependent peroxidase-like mechanism of nitration may also operate (70). Mitochondrial proteins are nitrated in vitro and in vivo, including MnSOD, aconitase, cytochrome c, voltage-dependent anion channel, ATPase, and succinyl-CoA oxoacid-CoA transferase (77, 78). The nitration/inactivation of MnSOD operates as a positive loop for enhanced intramitochondrial peroxynitrite formation, which in turn triggers apoptotic signaling of cell death, in part by the thiol oxidation-dependent assembly of the permeability transition pore (77). Detection of nitrated mitochondrial proteins and even nitrated cytochrome c in the cytosol may serve to reflect extents of mitochondrial •NO-dependent oxidative stress (66, 77, 78). Importantly, the enhanced peroxidatic activity of nitrated cytochrome c (66, 77) may further contribute to oxidative damage.
Red Blood Cell Hemoglobin as a Sink of Nitrating Species
Not all hemeproteins promote peroxynitrite-dependent nitration under biologically relevant conditions, because some reduce it by two electrons to 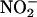 or isomerize it to
or isomerize it to 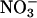 (79–82). In this context, oxyhemoglobin (oxyHb), present at 5 mM in red blood cells, serves as a sink to capture intravascularly formed nitrating species. Indeed, a fraction of intravascularly formed peroxynitrite could diffuse into red blood cells before enacting target molecule reactions with plasma components (83) and could undergo a fast reaction with oxyHb, which results in its isomerization to
(79–82). In this context, oxyhemoglobin (oxyHb), present at 5 mM in red blood cells, serves as a sink to capture intravascularly formed nitrating species. Indeed, a fraction of intravascularly formed peroxynitrite could diffuse into red blood cells before enacting target molecule reactions with plasma components (83) and could undergo a fast reaction with oxyHb, which results in its isomerization to 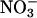 , without significant formation of nitrating species (82). Hemoglobin nitration is observed only under excess peroxynitrite over hemoglobin. Because •NO and
, without significant formation of nitrating species (82). Hemoglobin nitration is observed only under excess peroxynitrite over hemoglobin. Because •NO and 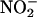 are also oxidized by oxyHb to rather unreactive
are also oxidized by oxyHb to rather unreactive 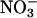 oxyHb may ultimately serve to inhibit the intravascular formation of •NO2. A similar inhibitory role on the formation of nitrating species may be played by deoxyhemoglobin, which reduces
oxyHb may ultimately serve to inhibit the intravascular formation of •NO2. A similar inhibitory role on the formation of nitrating species may be played by deoxyhemoglobin, which reduces 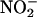 to •NO (84) and, presumably, peroxynitrite to
to •NO (84) and, presumably, peroxynitrite to 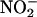 .
.
Lessons from Pharmacology
Pharmacological experiments have been instrumental in defining the biological mechanisms of nitration. Inhibition of NOS consistently leads to inhibition of nitration, and the sources of 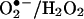 can be discriminated with inhibitors of NAD(P)H oxidases, xanthine oxidase, or respiratory chain uncouplers, among others. If the mechanism is peroxynitrite-dependent, it should be inhibited SOD (native, modified, or liposome-entrapped) or SOD-mimics, as long as the enzyme is located in the same compartment where
can be discriminated with inhibitors of NAD(P)H oxidases, xanthine oxidase, or respiratory chain uncouplers, among others. If the mechanism is peroxynitrite-dependent, it should be inhibited SOD (native, modified, or liposome-entrapped) or SOD-mimics, as long as the enzyme is located in the same compartment where 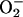 is formed, and is unaffected by MPO inhibitors and catalase (60, 85). In cases where SOD is not close enough to
is formed, and is unaffected by MPO inhibitors and catalase (60, 85). In cases where SOD is not close enough to 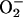 sources, it can enhance peroxynitrite-dependent nitration (7, 8). On the other hand, hemeperoxidase-dependent pathways can be stimulated by SOD, inhibited by catalase and MPO inhibitors, as observed in H2O2-producing inflammatory cells in the presence of
sources, it can enhance peroxynitrite-dependent nitration (7, 8). On the other hand, hemeperoxidase-dependent pathways can be stimulated by SOD, inhibited by catalase and MPO inhibitors, as observed in H2O2-producing inflammatory cells in the presence of 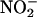 (13). The participation of LMW iron in the nitration reactions can be assessed by metal chelators, which block Fenton chemistry, although experiments in this line are scarce. Importantly, desferrioxamine can also directly trap •NO2 and
(13). The participation of LMW iron in the nitration reactions can be assessed by metal chelators, which block Fenton chemistry, although experiments in this line are scarce. Importantly, desferrioxamine can also directly trap •NO2 and 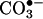 and prevent tyrosine nitration (86).
and prevent tyrosine nitration (86).
Uric acid inhibits nitration reactions and has been used as a scavenger of peroxynitrite in vitro and in vivo (87). Although uric acid does not react at appreciable rates with peroxynitrite (2), it readily reacts with peroxynitrite-derived radicals and oxo–metal complexes. Unfortunately, the presence of uric acid has confounded the analysis of nitration yields by peroxynitrite in various reports when xanthine/xanthine oxidase has been used as a source of 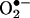 and can be easily overcome with the use of alternative substrates (88). LMW porphyrin complexes with Mn and Fe have SOD-mimic and peroxynitrite-reductase activities and react quickly with
and can be easily overcome with the use of alternative substrates (88). LMW porphyrin complexes with Mn and Fe have SOD-mimic and peroxynitrite-reductase activities and react quickly with 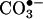 (89). They have been successfully used to inhibit •NO-dependent injury and nitration in cell and animal models of inflammation (90, 91). The redox and kinetic properties of these SOD-mimics and peroxynitrite decomposition catalysts can be modulated by changes in the porphyrin substituents, which can also affect its cellular/tissue distribution, aspects that require further research.
(89). They have been successfully used to inhibit •NO-dependent injury and nitration in cell and animal models of inflammation (90, 91). The redox and kinetic properties of these SOD-mimics and peroxynitrite decomposition catalysts can be modulated by changes in the porphyrin substituents, which can also affect its cellular/tissue distribution, aspects that require further research.
Lessons from Genetics: Knockouts, Transgenics, and Human Deficiency Syndromes
Knockout and transgenic animals and genetically engineered cells of the enzymes that participate in the formation or elimination of nitrating species are available. The preferential nitration pathways under various pathologically relevant conditions have been assessed by genetic approaches that involve SODs, namely overexpression of MnSOD or CuZnSOD or heterozygous MnSOD knockouts and MPO and EPO knockouts. MnSOD or CuZnSOD overexpressors decreased •NO-dependent injury and nitration in neurotoxicity and vascular injury models (92, 93).** On the contrary, heterozygous MnSOD knockouts resulted in larger extents of brain nitration in a model of neurotoxicity, consistent with enhanced mitochondrial formation and reactions of peroxynitrite (94).
MPO and EPO knockouts have evidenced the peroxidase-dependent nitration pathway in models of inflammation (15), but the relative contribution was highly model-dependent. The inability of MPO from human neutrophils to cause nitration in some models may result from lack of H2O2 substrate, because all 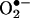 can react with •NO to form peroxynitrite (25) and/or because the nitration capabilities of hemeperoxidases may vary according to their access to
can react with •NO to form peroxynitrite (25) and/or because the nitration capabilities of hemeperoxidases may vary according to their access to 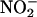 and peroxynitrite (61, 62, 95). In this context, release and sequestration of neutrophil-derived MPO in specific tissue compartments during inflammation are important aspects of MPO distribution and nitration capacity in vivo (63). However, results involving MPO in mice should be cautiously extrapolated to human inflammatory disease; indeed, surprisingly, whereas MPO levels positively correlate with cardiovascular risk in humans (76), MPO knockout mice develop atherosclerosis more rapidly than WT (96). MPO or EPO overexpresses are not available yet, but in the future they may also provide clues to the role of MPO and EPO in pathology. Enhanced removal of H2O2 by stable transfection of cells with catalase can also be useful to assess the contribution of the hemeperoxidase pathway (60). The hemeperoxidase-independent nitration observed in MPO and EPO knockouts (15) may be due, in addition to peroxynitrite, to the LMW metal-dependent pathway (12). The recent development of IRPs knockout mice (97) offers opportunities to study inflammatory models and nitration in the context of altered iron homeostasis.
and peroxynitrite (61, 62, 95). In this context, release and sequestration of neutrophil-derived MPO in specific tissue compartments during inflammation are important aspects of MPO distribution and nitration capacity in vivo (63). However, results involving MPO in mice should be cautiously extrapolated to human inflammatory disease; indeed, surprisingly, whereas MPO levels positively correlate with cardiovascular risk in humans (76), MPO knockout mice develop atherosclerosis more rapidly than WT (96). MPO or EPO overexpresses are not available yet, but in the future they may also provide clues to the role of MPO and EPO in pathology. Enhanced removal of H2O2 by stable transfection of cells with catalase can also be useful to assess the contribution of the hemeperoxidase pathway (60). The hemeperoxidase-independent nitration observed in MPO and EPO knockouts (15) may be due, in addition to peroxynitrite, to the LMW metal-dependent pathway (12). The recent development of IRPs knockout mice (97) offers opportunities to study inflammatory models and nitration in the context of altered iron homeostasis.
Circulating immune system cells (i.e., monocytes and neutrophils) from patients suffering from genetic deficiencies of NADPH oxidase (chronic granulomatous disease) or MPO (MPO deficiency) have altered interactions of •NO with oxidants (25, 62). Future studies on cells and tissues from these patients may offer important opportunities to understand the mechanisms of biological protein nitration in humans.
Conclusion and Perspectives
The analysis presented herein shows that biological protein tyrosine nitration†† mainly occurs through free radical pathways, and that there is an interplay involving excess •NO, oxidants, and transition metal centers. The individual or combined contribution of the peroxynitrite and hemeperoxidase/transition metal-dependent pathways to biological nitration, the detection of nitrated proteins with altered function in vivo, and the data arising from pharmacological and genetic approaches support the significance of nitration as a biochemical process linked to •NO-dependent pathophysiology. Approaches directed at inhibiting the oxidative modifications caused by reactive nitrogen species, including protein tyrosine nitration, open new avenues for the treatment of inflammatory, vascular, and neurodegenerative diseases.
Acknowledgments
I thank Drs. Stanley L. Hazen, Harry Ischiropoulos, Bruce A. Freeman, Joe S. Beckman, Irwin Fridovich, Jonathan S. Stamler, Jean Claude Drapier, and Ronald P. Mason for valuable comments, and my coinvestigators in Montevideo for contributions to the elucidation of various aspects presented in the manuscript. This work was supported by the Howard Hughes Medical Institute, Fogarty–National Institutes of Health, the John Simon Guggenheim Memorial Foundation, and Universidad de la República.
Abbreviations: NOS, NO synthase; MPO, myeloperoxidase; EPO, eosinophil peroxidase; SOD, superoxide dismutase; LMW, low molecular weight.
Footnotes
The debate on the biological production and measurement of peroxynitrite is analogous to a previous one on 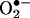 that arose after the discovery of SOD by J. M. McCord and I. Fridovich. The recurring discussions during the first decade led I. Fridovich and associates to invest large research efforts into fully (and redundantly) establishing the concept that
that arose after the discovery of SOD by J. M. McCord and I. Fridovich. The recurring discussions during the first decade led I. Fridovich and associates to invest large research efforts into fully (and redundantly) establishing the concept that 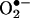 was a biologically relevant reactive intermediate.
was a biologically relevant reactive intermediate.
Under some circumstances, SOD could augment the bioavailability of •NO by 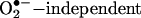 mechanisms such as (i) the SOD-mediated oxidation of nitroxyl anion (NO–) to •NO (26);. and (ii) the SOD-mediated reduction of S-nitrosothiols (27). These alternative mechanisms for the actions of SOD could mainly operate intracellularly (28). The immediate vascular responses to extracellularly added SOD are due to the prevention of the oxidative inactivation of readily diffusible •NO.
mechanisms such as (i) the SOD-mediated oxidation of nitroxyl anion (NO–) to •NO (26);. and (ii) the SOD-mediated reduction of S-nitrosothiols (27). These alternative mechanisms for the actions of SOD could mainly operate intracellularly (28). The immediate vascular responses to extracellularly added SOD are due to the prevention of the oxidative inactivation of readily diffusible •NO. 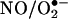 interactions in the vasculature are viewed as contributions to the development of vascular disease states, including atherogenesis, hypertension, and hyperglycemia (29).
interactions in the vasculature are viewed as contributions to the development of vascular disease states, including atherogenesis, hypertension, and hyperglycemia (29).
In the electrophilic aromatic nitration mechanism, there is no net redox change in the metal center (Eqs. 7–10). However, direct and unambiguous spectroscopic observation is sometimes difficult due to low steady-state concentrations and/or the absorption of peroxynitrite–metal complexes; additionally, there is no formation of •NO2.
Nitrated proteins can also undergo enhanced proteolytic degradation (54), and reports have suggested that a denitrase activity may be present in vivo (55).
Some metabolic conditions favor the role of NOS as a significant source of peroxynitrite (74). NOS, in turn, can be nitrated and potentially inactivated both in vitro and in vivo.
On the contrary, expression of amyotrophic lateral sclerosis-associated mutants of CuZnSOD leads to increased motoneuron nitration and apoptosis (59), possibly due to a toxic gain of function of SOD that promotes peroxynitrite formation and nitration reactions.
Biological nitration is not restricted only to tyrosine nitration but may also result in nitration of tryptophan residues, as well as DNA bases, sugars, and lipids that can result in altered or new biological activities.
References
- 1.Ignarro, L. J. (1990) Annu. Rev. Pharmacol. Toxicol. 30, 535–560. [DOI] [PubMed] [Google Scholar]
- 2.Radi, R., Denicola, A., Alvarez, B., Ferrer-Sueta, G. & Rubbo, H. (2000) in Nitric Oxide, ed. Ignarro, L. (Academic, San Diego), pp. 57–82.10733873
- 3.Liu, X., Miller, M. J., Joshi, M. S., Thomas, D. D. & Lancaster, J. R., Jr. (1998) Proc. Natl. Acad. Sci. USA 95, 2175–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Denicola, A., Batthyany, C., Lissi, E., Freeman, B. A., Rubbo, H. & Radi, R. (2002) J. Biol. Chem. 277, 932–936. [DOI] [PubMed] [Google Scholar]
- 5.Sokolovsky, M., Riordan, J. F. & Vallee, B. L. (1966) Biochemistry 5, 3582–3589. [DOI] [PubMed] [Google Scholar]
- 6.Beckman, J. S., Ischiropoulos, H., Zhu, L., van der Woerd, M., Smith, C., Chen, J., Harrison, J., Martin, J. C. & Tsai, M. (1992) Arch. Biochem. Biophys. 298, 438–445. [DOI] [PubMed] [Google Scholar]
- 7.Ischiropoulos, H., Zhu, L., Chen, J., Tsai, M., Martin, J. C., Smith, C. D. & Beckman, J. S. (1992) Arch. Biochem. Biophys. 298, 431–437. [DOI] [PubMed] [Google Scholar]
- 8.Ischiropoulos, H., Zhu, L. & Beckman, J. S. (1992) Arch. Biochem. Biophys. 298, 446–451. [DOI] [PubMed] [Google Scholar]
- 9.Beckmann, J. S., Ye, Y. Z., Anderson, P. G., Chen, J., Accavitti, M. A., Tarpey, M. M. & White, C. R. (1994) Biol. Chem. Hoppe–Seyler 375, 81–88. [DOI] [PubMed] [Google Scholar]
- 10.Ohshima, H., Friesen, M., Brouet, I. & Bartsch, H. (1990) Food Chem. Toxicol. 28, 647–652. [DOI] [PubMed] [Google Scholar]
- 11.Pfeiffer, S., Schmidt, K. & Mayer, B. (2000) J. Biol. Chem. 275, 6346–6352. [DOI] [PubMed] [Google Scholar]
- 12.Thomas, D. D., Espey, M. G., Vitek, M. P., Miranda, K. M. & Wink, D. A. (2002) Proc. Natl. Acad. Sci. USA 99, 12691–12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eiserich, J. P., Hristova, M., Cross, C. E., Jones, A. D., Freeman, B. A., Halliwell, B. & van der Vliet, A. (1998) Nature 391, 393–397. [DOI] [PubMed] [Google Scholar]
- 14.Bian, K., Gao, Z., Weisbrodt, N. & Murad, F. (2003) Proc. Natl. Acad. Sci. USA 100, 5712–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brennan, M. L., Wu, W., Fu, X., Shen, Z., Song, W., Frost, H., Vadseth, C., Narine, L., Lenkiewicz, E., Borchers, M. T., et al. (2002) J. Biol. Chem. 277, 17415–17427. [DOI] [PubMed] [Google Scholar]
- 16.Fukuto, J. & Ignarro, L. (1997) Acc. Chem. Res. 30, 149–152. [Google Scholar]
- 17.Eu, J. P., Liu, L., Zeng, M. & Stamler, J. S. (2000) Biochemistry 39, 1040–1047. [DOI] [PubMed] [Google Scholar]
- 18.Denicola, A., Souza, J. M. & Radi, R. (1998) Proc. Natl. Acad. Sci. USA 95, 3566–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radi, R., Peluffo, G., Alvarez, M. N., Naviliat, M. & Cayota, A. (2001) Free Radical Biol. Med. 30, 463–488. [DOI] [PubMed] [Google Scholar]
- 20.Gryglewski, R. J., Palmer, R. M. & Moncada, S. (1986) Nature 320, 454–456. [DOI] [PubMed] [Google Scholar]
- 21.Rubanyi, G. M. & Vanhoutte, P. M. (1986) Am. J. Physiol. 250, H822–H827. [DOI] [PubMed] [Google Scholar]
- 22.Ignarro, L. J., Byrns, R. E., Buga, G. M., Wood, K. S. & Chaudhuri, G. (1988) J. Pharmacol. Exp. Ther. 244, 181–189. [PubMed] [Google Scholar]
- 23.Liochev, S. I. & Fridovich, I. (2002) Free Radical Biol. Med. 33, 137–141. [DOI] [PubMed] [Google Scholar]
- 24.Buerk, D. G., Lamkin-Kennard, K. & Jaron, D. (2003) Free Radical Biol. Med. 34, 1488–1503. [DOI] [PubMed] [Google Scholar]
- 25.Clark, S. R., Coffey, M. J., Maclean, R. M., Collins, P. W., Lewis, M. J., Cross, A. R. & O'Donnell, V. B. (2002) J. Immunol. 169, 5889–5896. [DOI] [PubMed] [Google Scholar]
- 26.Murphy, M. E. & Sies, H. (1991) Proc. Natl. Acad. Sci. USA 88, 10860–10864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jourd'heuil D., Laroux, F. S., Miles, A. M., Wink, D. A. & Grisham, M. B. (1999) Arch. Biochem. Biophys. 361, 323–330. [DOI] [PubMed] [Google Scholar]
- 28.Hobbs, A. J., Fukuto, J. M. & Ignarro, L. (1994) Proc. Natl. Acad. Sci. USA 91, 10992–10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sowers, J. R. (2002) N. Engl. J. Med. 346, 1999–2001. [DOI] [PubMed] [Google Scholar]
- 30.Gutierrez, H. H., Nieves, B., Chumley, P., Rivera, A. & Freeman, B. A. (1996) Free Radical Biol. Med. 21, 43–52. [DOI] [PubMed] [Google Scholar]
- 31.Sarti, P., Avigliano, L., Gorlach, A. & Brune, B. (2002) Cell Death Differ. 9, 1160–1162. [DOI] [PubMed] [Google Scholar]
- 32.Radi, R., Beckman, J. S., Bush, K. M. & Freeman, B. A. (1991) J. Biol. Chem. 266, 4244–4250. [PubMed] [Google Scholar]
- 33.Trujillo, M. & Radi, R. (2002) Arch. Biochem. Biophys. 397, 91–98. [DOI] [PubMed] [Google Scholar]
- 34.Arteel, G. E., Briviba, K. & Sies, H. (1999) FEBS Lett. 445, 226–230. [DOI] [PubMed] [Google Scholar]
- 35.Bryk, R., Griffin, P. & Nathan, C. (2000) Nature 407, 211–215. [DOI] [PubMed] [Google Scholar]
- 36.Brito, C., Naviliat, M., Tiscornia, A. C., Vuillier, F., Gualco, G., Dighiero, G., Radi, R. & Cayota, A. M. (1999) J. Immunol. 162, 3356–3366. [PubMed] [Google Scholar]
- 37.Go, Y. M., Patel, R. P., Maland, M. C., Park, H., Beckman, J. S., Darley-Usmar, V. M. & Jo, H. (1999) Am. J. Physiol. 277, H1647–H1653. [DOI] [PubMed] [Google Scholar]
- 38.Mihm, M. J., Wattanapitayakul, S. K., Piao, S. F., Hoyt, D. G. & Bauer, J. A. (2003) Biochem. Pharmacol. 65, 1189–1197. [DOI] [PubMed] [Google Scholar]
- 39.Cosentino, F., Eto, M., De Paolis, P., van der Loo, B., Bachschmid, M., Ullrich, V., Kouroedov, A., Delli Gatti, C., Joch, H., Volpe, M., et al. (2003) Circulation 107, 1017–1023. [DOI] [PubMed] [Google Scholar]
- 40.Augusto, O., Bonini, M. G., Amanso, A. M., Linares, E., Santos, C. C. & De Menezes, S. L. (2002) Free Radical Biol. Med. 32, 841–859. [DOI] [PubMed] [Google Scholar]
- 41.Hazen, S. L., Gaut, J. P., Hsu, F. F., Crowley, J. R., d'Avignon, A. & Heinecke, J. W. (1997) J. Biol. Chem. 272, 16990–16998. [DOI] [PubMed] [Google Scholar]
- 42.Fridovich, I. (1997) J. Biol. Chem. 272, 18515–18517. [DOI] [PubMed] [Google Scholar]
- 43.Yamazaki, I. & Piette, L. H. (1990) J. Biol. Chem. 265, 13589–13594. [PubMed] [Google Scholar]
- 44.Burner, U., Furtmuller, P. G., Kettle, A. J., Koppenol, W. H. & Obinger, C. (2000) J. Biol. Chem. 275, 20597–20601. [DOI] [PubMed] [Google Scholar]
- 45.Sampson, J. B., Rosen, H. & Beckman, J. S. (1996) Methods Enzymol. 269, 210–218. [DOI] [PubMed] [Google Scholar]
- 46.Ferrer-Sueta, G., Quijano, C., Alvarez, B. & Radi, R. (2002) Methods Enzymol. 349, 23–37. [DOI] [PubMed] [Google Scholar]
- 47.Bouton, C. & Drapier, J. C. (2003) Sci. STKE 182, pe17. [DOI] [PubMed] [Google Scholar]
- 48.Soum, E., Brazzolotto, X., Goussias, C., Bouton, C., Moulis, J. M., Mattioli, T. A. & Drapier, J. C. (2003) Biochemistry 42, 7648–7654. [DOI] [PubMed] [Google Scholar]
- 49.Gunther, M. R., Hsi, L. C., Curtis, J. F., Gierse, J. K., Marnett, L. J., Eling, T. E. & Mason, R. P. (1997) J. Biol. Chem. 272, 17086–17090. [DOI] [PubMed] [Google Scholar]
- 50.Esteves, P. M., De M Carneiro, J. W., Cardoso, S. P., Barbosa, A. G., Laali, K. K., Rasul, G., Prakash, G. K. & Olah, G. A. (2003) J. Am. Chem. Soc. 125, 4836–4849. [DOI] [PubMed] [Google Scholar]
- 51.Quijano, C., Hernandez-Saavedra, D., Castro, L., McCord, J. M., Freeman, B. A. & Radi, R. (2001) J. Biol. Chem. 276, 11631–11638. [DOI] [PubMed] [Google Scholar]
- 52.Whiteman, M., Siau, J. L. & Halliwell, B. (2003) J. Biol. Chem. 278, 8380–8384. [DOI] [PubMed] [Google Scholar]
- 53.Aslan, M., Ryan, T. M., Townes, T. M., Coward, L., Kirk, M. C., Barnes, S., Alexander, C. B., Rosenfeld, S. S. & Freeman, B. A. (2003) J. Biol. Chem. 278, 4194–4204. [DOI] [PubMed] [Google Scholar]
- 54.Grune, T., Blasig, I. E., Sitte, N., Roloff, B., Haseloff, R. & Davies, K. J. (1998) J. Biol. Chem. 273, 10857–10862. [DOI] [PubMed] [Google Scholar]
- 55.Irie, Y., Saeki, M., Kamisaki, Y., Martin, E. & Murad, F. (2003) Proc. Natl. Acad. Sci. USA 100, 5634–5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubbo, H., Radi, R., Trujillo, M., Telleri, R., Kalyanaraman, B., Barnes, S., Kirk, M. & Freeman, B. A. (1994) J. Biol. Chem. 269, 26066–26075. [PubMed] [Google Scholar]
- 57.Miles, A. M., Bohle, D. S., Glassbrenner, P. A., Hansert, B., Wink, D. A. & Grisham, M. B. (1996) J. Biol. Chem. 271, 40–47. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein, S., Czapski, G., Lind, J. & Merenyi, G. (2000) J. Biol. Chem. 275, 3031–3036. [DOI] [PubMed] [Google Scholar]
- 59.Estevez, A. G., Crow, J. P., Sampson, J. B., Reiter, C., Zhuang, Y., Richardson, G. J., Tarpey, M. M., Barbeito, L. & Beckman, J. S. (1999) Science 286, 2498–2500. [DOI] [PubMed] [Google Scholar]
- 60.Fries, D. M., Paxinou, E., Themistocleous, M., Swanberg, E., Griendling, K. K., Salvemini, D., Slot, J. W., Heijnen, H. F., Hazen, S. L. & Ischiropoulos, H. (2003) J. Biol. Chem. 278, 22901–22907. [DOI] [PubMed] [Google Scholar]
- 61.Jiang, Q. & Hurst, J. K. (1997) J. Biol. Chem. 272, 32767–32772. [DOI] [PubMed] [Google Scholar]
- 62.Rosen, H., Crowley, J. R. & Heinecke, J. W. (2002) J. Biol. Chem. 277, 30463–30468. [DOI] [PubMed] [Google Scholar]
- 63.Baldus, S., Eiserich, J. P., Brennan, M. L., Jackson, R. M., Alexander, C. B. & Freeman, B. A. (2002) Free Radical Biol. Med. 33, 1010. [DOI] [PubMed] [Google Scholar]
- 64.Hazen, S. L., Zhang, R., Shen, Z., Wu, W., Podrez, E. A., MacPherson, J. C., Schmitt, D., Mitra, S. N., Mukhopadhyay, C., Chen, Y., et al. (1999) Circ. Res. 85, 950–958. [DOI] [PubMed] [Google Scholar]
- 65.Souza, J. M., Daikhin, E., Yudkoff, M., Raman, C. S. & Ischiropoulos, H. (1999) Arch. Biochem. Biophys. 371, 169–178. [DOI] [PubMed] [Google Scholar]
- 66.Cassina, A., Hodara, R., Souza, J., Thomson, L., Castro, L., Ischiropoulos, H., Freeman, B. A. & Radi, R. (2000) J. Biol. Chem. 275, 21409–21415. [DOI] [PubMed] [Google Scholar]
- 67.Vadseth, C., Souza, J. M., Thomson, L., Seagraves, A., Nagaswami, C., Scheiner, T., Torbet, J., Vilaire, G., Bennett, J. S., Murciano, J. C., et al. (2003) J. Biol. Chem., jbc/M306101200v1. [DOI] [PubMed]
- 68.Balafanova, Z., Bolli, R., Zhang, J., Zheng, Y., Pass, J. M., Bhatnagar, A., Tang, X.-L., Wang, O., Cardwell, E. & Ping, P. (2002) J. Biol. Chem. 277, 15021–15027. [DOI] [PubMed] [Google Scholar]
- 69.MacMillan-Crow, L. A., Crow, J. P., Kerby, J. D., Beckman, J. S. & Thompson, J. A. (1996) Proc. Natl. Acad. Sci. USA 93, 11853–11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castro, L., Eiserich, J. P., Sweeney, S., Radi, R. & Freeman, B. A. (2004) Arch. Biochem. Biophys. 421, 99–107. [DOI] [PubMed] [Google Scholar]
- 71.Guo, W., Adachi, T., Matsui, R., Xu, S., Jiang, B., Zou, M. H., Kirber, M., Lieberthal, W. & Cohen, R. A. (2003) Am. J. Physiol. 285, H1396–H1403. [DOI] [PubMed] [Google Scholar]
- 72.Schmidt, P., Youhnovski, N., Daiber, A., Balan, A., Arsic, M., Bachschmid, M., Przybylski, M. & Ullrich, V. (2003) J. Biol. Chem. 278, 12813–12819. [DOI] [PubMed] [Google Scholar]
- 73.Bachschmid, M., Thurau, S., Zou, M. H. & Ullrich, V. (2003) FASEB J. 17, 914–916. [DOI] [PubMed] [Google Scholar]
- 74.Stuehr, D., Pou, S. & Rosen, G. M. (2001) J. Biol. Chem. 276, 14533–14536. [DOI] [PubMed] [Google Scholar]
- 75.Shishehbor, M. H., Aviles, R. J., Brennan, M. L., Fu, X., Goormastic, M., Pearce, G. L., Gokce, N., Keaney, J. F., Jr., Penn, M. S., Sprecher, D. L., et al. (2003) J. Am. Med. Assoc. 289, 1675–1680. [DOI] [PubMed] [Google Scholar]
- 76.Brennan, M. L., Penn, M. S., Van Lente, F., Nambi, V., Shishehbor, M. H., Aviles, R. J., Goormastic, M., Pepoy, M. L., McErlean, E. S., Topol, E. J., et al. (2003) N. Engl. J. Med. 349, 1595–1604. [DOI] [PubMed] [Google Scholar]
- 77.Radi, R., Cassina, A., Hodara, R., Quijano, C. & Castro, L. (2002) Free Radical Biol. Med. 33, 1451–1464. [DOI] [PubMed] [Google Scholar]
- 78.Turko, I. V., Li, L., Aulak, K. S., Stuehr, D. J., Chang, J. Y. & Murad, F. (2003) J. Biol. Chem. 278, 33972–33977. [DOI] [PubMed] [Google Scholar]
- 79.Pearce, L. L., Pitt, B. R. & Peterson, J. (1999) J. Biol. Chem. 274, 35763–35767. [DOI] [PubMed] [Google Scholar]
- 80.Pietraforte, D., Salzano, A. M., Scorza, G., Marino, G. & Minetti, M. (2001) Biochemistry 40, 15300–15309. [DOI] [PubMed] [Google Scholar]
- 81.Herold, S., Shivashankar, K. & Mehl, M. (2002) Biochemistry 41, 13460–13472. [DOI] [PubMed] [Google Scholar]
- 82.Romero, N., Radi, R., Linares, E., Augusto, O., Detweiler, C. D., Mason, R. P. & Denicola, A. (2003) J. Biol. Chem. 278, 44049–44057. [DOI] [PubMed] [Google Scholar]
- 83.Romero, N., Denicola, A., Souza, J. M. & Radi, R. (1999) Arch. Biochem. Biophys. 368, 23–30. [DOI] [PubMed] [Google Scholar]
- 84.Cosby, K., Partovi, K. S., Crawford, J. H., Patel, R. P., Reiter, C. D., Martyr, S., Yang, B. K., Waclawiw, M. A., Zalos, G., Xu, X., et al. (2003) Nat. Med. 9, 1498–1505. [DOI] [PubMed] [Google Scholar]
- 85.Estevez, A. G., Sampson, J. B., Zhuang, Y., Spear, N., Richardson, G. J., Crow, J. P., Tarpey, M. M., Barbeito, L. & Beckman, J. S. (2000) Free Radical Biol. Med. 28, 437–446. [DOI] [PubMed] [Google Scholar]
- 86.Bartesaghi, S., Trujillo, M., Denicola, A., Folkes, L., Wardman, P. & Radi, R. (2004) Free Radical Biol. Med. 36, 471–483. [DOI] [PubMed] [Google Scholar]
- 87.Scott, G. S. & Hooper, D. C. (2001) Med. Hypotheses 56, 95–100. [DOI] [PubMed] [Google Scholar]
- 88.Trujillo, M., Alvarez, M. N., Peluffo, G., Freeman, B. A. & Radi, R. (1998) J. Biol. Chem. 273, 7828–7834. [DOI] [PubMed] [Google Scholar]
- 89.Ferrer-Sueta, G., Vitturi, D., Batinic-Haberle, I., Fridovich, I., Goldstein, S., Czapski, G. & Radi, R. (2003) J. Biol. Chem. 278, 27432–27438. [DOI] [PubMed] [Google Scholar]
- 90.Muscoli, C., Cuzzocrea, S., Riley, D. P., Zweier, J. L., Thiemermann, C., Wang, Z. Q. & Salvemini, D. (2003) Br. J. Pharmacol. 140, 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pacher, P., Liaudet, L., Bai, P., Mabley, J. G., Kaminski, P. M., Virag, L., Deb, A., Szabo, E., Ungvari, Z., Wolin, M. S., Groves, J. T. & Szabo, C. (2003) Circulation 107, 896–904. [DOI] [PubMed] [Google Scholar]
- 92.Keller, J. N., Kindy, M. S., Holtsberg, F. W., St Clair, D. K., Yen, H. C., Germeyer, A., Steiner, S. M., Bruce-Keller, A. J., Hutchins, J. B. & Mattson, M. P. (1998) J. Neurosci. 18, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang, H. D., Johns, D. G., Xu, S. & Cohen, R. A. (2002) Am. J. Physiol. 282, H1697–H1702. [DOI] [PubMed] [Google Scholar]
- 94.Kim, G. W. & Chan, P. H. J (2002) Cereb. Blood Flow Metab. 22, 798–809. [DOI] [PubMed] [Google Scholar]
- 95.Gaut, J. P., Byun, J., Tran, H. D., Lauber, W. M., Carroll, J. A., Hotchkiss, R. S., Belaaouaj, A. & Heinecke, J. W. (2002) J. Clin. Invest. 109, 1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brennan, M. L. Anderson, M. M., Shih, D. M., Qu, X. D., Wang, X., Mehta, A. C., Lim, L. L., Shi, W., Hazen, S. L., Jacob, J. S., et al. (2001) J. Clin. Invest. 107, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.LaVaute, T., Smith, S., Cooperman, S., Iwai, K., Land, W., Meyron-Holtz, E., Drake, S. K., Miller, G., Abu-Asab, M., Tsokos, M., et al. (2001) Nat. Genet. 27, 209–214. [DOI] [PubMed] [Google Scholar]