

Abstract
3D DNA FISH has become a major tool for analyzing three-dimensional organization of the nucleus, and several variations of the technique have been published. In this article we describe a protocol which has been optimized for robustness, reproducibility, and ease of use. Brightly fluorescent directly labeled probes are generated by nick-translation with amino-allyldUTP followed by chemical coupling of the dye. 3D DNA FISH is performed using a freeze-thaw step for cell permeabilization and a heating step for simultaneous denaturation of probe and nuclear DNA. The protocol is applicable to a range of cell types and a variety of probes (BACs, plasmids, fosmids, or Whole Chromosome Paints) and allows for high-throughput automated imaging. With this method we routinely investigate nuclear localization of up to three chromosomal regions.
Keywords: Genetics, Issue 78, Molecular Biology, Biochemistry, Cellular Biology, Genomics, Epigenetics, Cell Nucleus, Fluorescence, In Situ Hybridization, FISH, 3D DNA FISH, fluorescence in situ hybridization, nuclear structure, fluorescently labeled probes, visualization, imaging, DNA, chromosomes, sequencing, probes, assay
Introduction
DNA fluorescence in situ hybridization (DNA FISH) allows the three-dimensional visualization of individual gene loci, subchromosomal domains and even entire chromosomes during all stages of the cell cycle. 2D FISH is used for metaphase studies while 3D FISH has been extensively used to probe the relationship between the spatial organization of the genome and its function during interphase (1,2 and references therein). Historically, co-association studies were performed by investigating tens to hundreds of individual loci by FISH. More recently powerful high throughput 3C-based techniques such as 4C and Hi-C have been developed3, allowing the investigation of molecular cross-talk between many thousands of different loci. While 3C-based techniques and DNA FISH can be complementary methods, they do not necessarily answer the same questions. 3C based methods provide an ensemble readout of mixed cell populations, resulting in a probability for co-associations. In contrast, while low in throughput, FISH based techniques offer the possibility to analyze spatial arrangements of loci or chromosomes in individual cells according to their developmental or cell cycle stages. Hence, FISH will continue to be an important tool for probing nuclear structure-function relationships.
There are two major considerations in performing successful 3D FISH experiments. These are; 1. obtaining optimally labeled probes and 2. choice of cellular treatments, including fixation, pre- and post-hybridization steps, to preserve nuclear morphology as much as possible while making DNA sufficiently accessible for probe hybridization. Efficient probe labeling is critically important for FISH. Traditionally, nick-translation has been used to introduce either hapten or fluorophore-conjugated nucleotides4. Similarly, commercial nick-translation kits are available for direct hapten or fluorophore incorporation, but also for two-step labeling using aminoallyl nucleotides and amine-reactive dyes. The latter renders dye incorporation more efficient by giving DNA polymerase a less bulky molecule to work with. More recently, kits for the non-enzymatic labeling of DNA have been developed which exploit coordinative binding of platinum to nucleic acids. FISH probes can even be purchased already labeled5. While kits and commercially manufactured probes no doubt give ease of use, they are considerably more expensive than buying the individual components and producing probes in-house. We optimized a low cost nick translation protocol in order to directly label many different BAC probes in multiple colors. We discovered that obtaining highly pure BAC DNA is critical and results in a requirement for only 10-20 ng of probe per FISH slide, compared to 10 - 20-fold more when impure template DNA is used, resulting in major cost- and time-savings. The use of amino-allyldUTP allows flexible labeling of probes with available amine-reactive dyes (e.g. Alexa Fluor or Cy-dyes) or haptens (e.g. biotin, digoxigenin). Hapten-labeled probes are detected by fluorophore-conjugated antibodies or streptavidin to amplify and visualize the signal. Bright directly labeled probes generally show less background staining and avoid over-amplification of signals, hence resulting in a more accurate representation of nuclear localization and locus morphology.
There are several different FISH techniques that use different fixatives, pre-treatments, and post-hybridization washes but these generally fall into the following categories: glutaraldehyde fixation with NaOH denaturation, formaldehyde fixation with HCl denaturation, and formaldehyde fixation with heat denaturation6-9. Each of these has advantages and disadvantages. Glutaraldehyde fixation results in good nuclear structural preservation, but requires treatment with reducing agents to minimize the resulting autofluorescence, and NaOH treatment needs to be carefully controlled to balance sufficient DNA denaturation and potential damage to nuclear structure6. Formaldehyde fixation is less strong, giving an increased likelihood of perturbations of nuclear architecture, but also typically giving stronger and more reliable signals and lower autofluorescence9. HCl treatment depurinates DNA and strips out proteins, providing good access to the DNA for the probes, but may also introduce DNA breaks. Heating physically separates the two DNA strands resulting in good target hybridization and strong signals but can cause some perturbation of nuclear structure8. The degree to which each of these techniques affects protein epitopes varies widely6,9 , resulting in the requirement to experimentally determine for each protein the optimal protocol to use in immuno-FISH experiments.
While there is no 'perfect' DNA FISH technique, they can all be useful if well controlled. Our goal was to optimize a protocol for robust and reproducible DNA FISH to investigate spatial positioning of multiple loci in a variety of cell types10, focusing on the heating method for most reliable signals. With this and the use of an automated imaging system we aimed at increasing the throughput for single cell analysis of nuclear arrangements.
Protocol
1. Generating Directly Labeled DNA Probes by Nick Translation
Note: Directly labeled probes made from BACs (100-250 kb) consistently produce bright signals. If smaller probes are required, use fosmids (40-50 kb) or even plasmids containing 5-10 kb inserts. Identify BAC or fosmid clones corresponding to specific genes using Ensemble or UCSC genome browsers. Prepare high quality BAC DNA by repeated precipitation or use commercially available kits. The less the preparation is contaminated with bacterial genomic DNA, the less probe is required per slide. Perform labeling in two steps: nick translation introducing aminoallyl-dUTP and chemical coupling of an amine-reactive dye.
1.1. Nick Translation
During nick translation, DNase I is used to create single-strand breaks. DNA Polymerase I elongates the 3' ends of these 'nicks', replacing existing nucleotides with new ones, thereby 'translating' the nick and thus providing the opportunity to incorporate labeled nucleotides. Aminoallyl-dUTP is chosen because of efficient incorporation by DNA Polymerase I and its potential for later chemical coupling to amine-reactive dyes or haptens. It is critical to achieve the right balance between DNase I nicking and DNA polymerase I translation; too much DNase I will result in excessive DNA digestion giving low yield and short fragment sizes, too little will not produce enough nicks for the polymerase to initiate translation, giving large fragment size and poor incorporation of aminoallyl-dUTP. The following protocol works well but it may be necessary to titrate the DNAse I from different batches or different manufacturers.
Set up labeling reaction on ice and incubate for 2 hr at 16 °C. This should produce 0.5-1 μg nick-translated DNA and can be scaled up. Dilute DNase I 1:30 in DNase I buffer (i.e. 0.3 U/μl).
| Component | Concentration of stock | Volume or amount |
| BAC DNA | 5-10 μg | |
| NTB buffer | 10x | 5 μl |
| DTT | 0.1 M | 5 μl |
| dNTP mix | 10x | 5 μl |
| Aminoallyl-dUTP | 0.5 mM | 6 μl |
| DNA Polymerase I | 10 U/μl | 1 μl |
| DNase I | 10 U/μl | 1 μl (of a 1:30 dilution) |
| H2O | to 50 μl |
Run 1 μl on a 2% agarose gel to check the size of the labeled fragments. While you run the gel, keep the reaction on ice. Successful nick translation will result in a smear with the bulk of the fragments running between 150 bp and 700 bp with some larger fragments at ~1 kb (CRITICAL STEP: see Representative Results). If needed, add another 1 μl of a fresh 1:30 DNase I dilution and incubate at 16 °C for 15-30 min. Incubation time will vary according to the amount and quality of BAC DNA.
Inactivate DNase I by heating to 75 °C for 10 min.
Clean-up amine-modified DNA using a PCR purification kit. Elute in 100 μl H2O.
Ethanol precipitate DNA by adding 10 μl 3 M NaOAc (pH 5.2) and 275 μl ethanol. Leave at -20 °C for at least 1 hr or overnight. Spin at maximum speed for 30 min at 4 °C. Wash pellet with 500 μl 70% ethanol and allow to air dry. This step removes trace amines which will interfere with the labeling reaction. Resuspend pellet in 6 μl H2O per 1x initial reaction.
Use 1 μl to determine the concentration of the amine-modified DNA by microvolume UV spectroscopy.
1.2. Coupling of the Fluorescent Dye
Fluorescent labeling of the probe is achieved by chemical coupling of the dye. Alexa Fluor succinimidyl ester dyes react with the amines of the amino-allyldUTP modified DNA to form a covalent bond, thereby producing directly labeled probes. Alexa Fluor 488, 555, and 647 dyes have been successful for this process.
Adjust 4 μg of amino-allyl modified DNA to a volume of 5 μl, heat to 95 °C for 5 min, snap cool on ice, and add 3 μl of 0.2 M NaHCO3.
Dissolve one aliquot of amine reactive dye in 2 μl anhydrous DMSO at room temperature. Add 8 μl modified DNA with NaHCO3, vortex, pulse spin and incubate at room temperature in the dark for 1 hr.
Note: The amine reactive dyes can be used to label more than one probe based on the following scheme. Since only 10-20 ng of probe is used per slide labeling, 1 μg gives enough probe for 50-100 slides. It is also possible to store amine-modified DNA for future labeling reactions at -20 °C.
| DNA amount | DNA volume | 0.2 M NaHCO3 | Volume of dye | Total | Enough for |
| 4 x 1 μg | 1.25 μl each | 0.75 μl each | 0.5 μl each | 2.5 μl (1/4) | 50-100 slides each |
| 3 x 1.35 μg | 1.67 μl each | 1 μl each | 0.67 μl each | 3.34 μl (1/3) | 65-130 slide each |
| 2 x 2 μg | 2.5 μl each | 1.5 μl each | 1 μl each | 5 μl (1/2) | 100-200 slides each |
| 1 x 4 μg | 5 μl | 3 μl | 2 μl | 10 μl (full) | 200-400 slides |
Add 90 μl H2O and purify labeled probe using a PCR purification kit. Elute in 100 μl 10 mMTris-Cl (pH 8.5).
Determine probe concentration and labeling efficiency by microvolume full spectrum spectroscopy (see Figure 2) using Beer-Lambert equations (Instructions are given in the datasheet accompanying the Alexa Fluor reactive dyes or can be found online). Probes containing 3-6 dyes per 100 bp work well. Probes with lower degrees of incorporation may give weak FISH signals. Alternatively, run 5 μl on a 2% agarose gel without DNA stain and check the incorporation of the fluorescent dye using a phosphoimager. Post-stain the gel with ethidium bromide or DNA staining alternatives and compare labeled DNA to total DNA.
2. DNA FISH
Via the following steps, cells of interest are fixed to slides and permeabilized. Cellular DNA and directly labeled probes are then denatured together on a hot plate and hybridized overnight in a light-tight humidified chamber.
2.1. Day 1
- Probe Precipitation Note: Precipitate probes at some point during Day 1. This can comfortably be performed in parallel to the treatment of the cells and does not have to be done at the start of the day.
- Mix 10-20 ng directly labeled probe, 6 μl C0t-1 DNA (6 μg), 1 μl single stranded DNA from salmon testes (9.7 μg) and adjust volume to 100 μl with water.
- Add 10 μl 3 M NaOAc and 275 μl EtOH, and mix by vortexing. Precipitate for at least 1 hr at -20 °C.
- Spin in microfuge at maximum speed for 30 min at 4 °C. Wash pellet with 70 % EtOH and spin again. Dry pellet and resuspend in 5 μl deionized formamide. Resuspend for 30 min at 37 °C while shaking at 1000 rpm protected from light (place foil over the thermomixer).
- Add 5 μl of dextran sulphate mix and shake for a further 10 min at 37 °C again protected from light. Pipette up and down just before pipetting onto coverslip. Note: For chromosome painting combined with BAC probes, whole chromosome painting probes which are supplied in ready-to-use hybridization mix are a convenient option. Resuspend precipitated BAC probe (with C0t1 DNA and single stranded DNA from salmon testes) in 10 μl chromosome paint mix by pipetting up and down. Incubate at 37 °C while shaking at 300-500 rpm for 10 min. Use 10 μl of chromosome paint/ BAC probe mix per spot.
Attaching Cells to Slides Note: The cells propensity to adhere to slides greatly varies with cell type. For each cell type, determine the optimal settling time and cell density empirically. For consistent results, cells should be in a single cell suspension. For ease of use circle the area the cells will be spotted on with a hydrophobic pen, although for cells resuspended in PBS this is not strictly necessary. A hydrophobic barrier prevents the suspension from spreading on the slide, keeps up to three samples per slide nicely separated and allows for the use of very small volumes of antibody solution in immuno-FISH. Also, the spotting area can be kept to a minimum if cell numbers are low.
- Mouse fetal liver cells
- Collect each E13.5 mouse fetal liver in approximately 1.5 ml of PBS and resuspend by pipetting up and down. Spin in microfuge at 3,000 rpm for 2 min. Repeat washes three times. At the last wash, strain cells through a 70 μm sieve. Resuspend cells from one liver in 160 μl PBS and use 50 μ per spot. Leave cells to settle for 2 min at room temperature.
Mouse ES Cells 1. Mouse ES cells will not stick readily to the slide. On a slide, circle the area the cells will be spotted on with a hydrophobic pen. Prepare a suspension of 20,000-50,000 cells per 80-100 μl in normal culture medium, pipette into circle and leave to attach for approximately 3 hr in a humidified incubator. Note: Alternatively, ES cells can be cultured on slides, but will then form colonies which might hamper automated imaging.
Fixed Cells 1. Some experiments require the use of fixed cells which do not attach to slides by themselves. Gentle spinning at 300 rpm for 3 min using a cytocentrifuge preserves the three dimensional structure of the cells and ensures that cells do not fall off.
| Cell type | Suspension in | Settling time | Comment |
| Mouse fetal liver | PBS | 2 min | On bench |
| Mouse ES | DMEM with supplements | 4 hr | 37 °C in humidified incubator |
| Mouse lymphocytes | PBS | 2 min | On bench |
| Mouse P20 germ cells | Fix in solution | Cytospin | On bench |
To fix cells, gently submerge slide in 4% PFA (CAUTION: perform in fume hood) for 10 min. Fix cells with the slide flat in a tray for best retention of cells (e.g. 50 ml 4% PFA in the lid of a tip box). Note: This step does not apply to cells fixed prior to attachment to slides.
For all subsequent steps, use 50 ml or 100 ml Coplin jars. Take care not to scrape cells off when moving slides between jars.
Quench in 0.1 M Tris-Cl, pH 7.4 for 10 min at RT.
Permeabilize cells in 0.1% saponin/0.1% Triton X-100 in PBS for 10 min at RT.
Wash twice in PBS for 5 min at RT.
Incubate for at least 20 min in 20% glycerol/ PBS at RT. Note: At this point, slides can be stored in 50% glycerol/ PBS at -20 °C for at least several weeks. It has been observed that storage for at least a day results in an increase in signal strength9. Recalibrate in room temperature 20% glycerol/PBS before proceeding. Note: This is a convenient point to start precipitating the probes (see 2.1.1).
3x freeze/ thawing in liquid nitrogen. Submerge one slide at a time in liquid nitrogen using a small dewar. Withdraw after a few seconds and a characteristic popping sound, and place on paper towel to defrost. Wait for opaque frozen glycerol to disappear then freeze again. Up to 15 slides can be done comfortably in rotation for three freeze/thaw cycles.
Wash twice in PBS for 5 min at RT.
Incubate in 0.1 M HCl for 30 min at RT.
Wash in PBS for 5 min at RT.
Permeabilize in 0.5% saponin/0.5% Triton X-100/PBS for 30 min at RT.
Wash twice in PBS for 5 min at RT.
Equilibrate in 50% formamide/2x SSC for at least 10 min at RT.
Pipette probe onto a coverslip. Remove slide from the jar and dry off any excess liquid around the cell spot(s) with a paper towel. As the formamide dries quickly, take care not to dry out cells. For slides with two or three cell spots, apply 10 μl of probe per spot onto a 22 mm x 50 mm coverslip corresponding to spot location on the slide. Invert coverslip onto slide over spot(s). Seal with rubber cement and allow this to dry completely. Protect from light at all subsequent stages.
Heat slides to 78 °C for precisely 2 min on a hot plate (e.g. an inverted heating block, CRITICAL STEP: see Discussion). Place a cover (cardboard box or similar) over the hot plate to protect from light during denaturation.
Incubate overnight at 37 °C in a light-tight humidified chamber. To humidify, place paper towels in a light-tight box and dampen with water.
2.2 Day 2
Prepare Coplin jars with solutions at the right temperature for subsequent wash steps. Protect slides from light as much as possible during all subsequent steps. A coffee can makes a good light-tight cover for Coplin jars.
Peel off rubber cement and place slide in 2x SSC until coverslips loosen and slide off.
Wash in 50% formamide/2x SSC for 15 min at 45 °C. Place lid on waterbath to protect from light.
Wash in 0.2x SSC for 15 min at 63 °C.
Wash in 2x SSC for 5 min at 45 °C.
Wash in 2x SSC for 5 min at RT.
Wash in PBS for 5 min at RT.
Stain with DAPI (5 μg/ml in 2x SSC) for 2 min in a Coplin jar at RT.
Note: DAPI solution can be stored at 4 °C in the dark for 2 months.
Destain in PBS for 5 min at RT.
To mount the coverslip, pipette mounting medium onto a coverslip. Use 10 μl per cell spot, using 22 mm x 22 mm coverslips for one spot, and 22 mm x 50 mm coverslips for two to three spots. Dry off PBS around the cell spot as much as possible but take care not to dry out the cells. Invert coverslip onto slide, and seal with nail varnish.
Representative Results
The protocol described here (Figure 1 for overview) has been used to great effect to combine DNA FISH with high-throughput image capture for analysis of genomic organization. The generation of good quality probes is critical to success (see Discussion). Figure 2 demonstrates two important quality controls for FISH probes. After nick-translation, a smear of DNA should be visible with the bulk of the fragments running between 150 and 700 bp (Figure 2A). After chemical coupling, incorporation of the fluorescent dye can be gauged by spectroscopic analysis of the probe (Figure 2B).
When using good probes, following the DNA FISH protocol usually results in bright DNA FISH signals on low background. We have successfully used this protocol on a variety of different cell types, and with either epi-fluorescence or confocal microscopes. Automated imaging using an epi-fluorescence microscope requires sharp, easily identifiable FISH signals with little background (Figure 3), while confocal microscopy requires more intense signals. Figure 4A shows representative processed images. Nuclear coordinates of FISH signals can be obtained, and spatial relations of genomic regions can be computed (for an example see Figure 4B). We have also used image stacks obtained by confocal microscopy for 3D modeling of FISH signals within their chromosome territories (Movie 1).
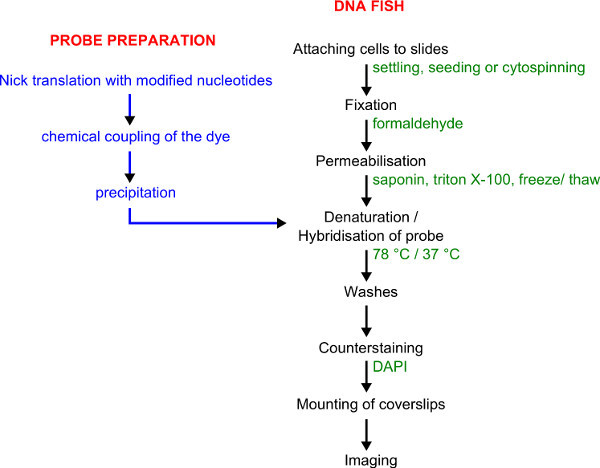 Figure 1. Workflow for probe labeling and DNA FISH. Probe preparation is shown in blue. DNA FISH procedure is shown in black with details given in green.
Figure 1. Workflow for probe labeling and DNA FISH. Probe preparation is shown in blue. DNA FISH procedure is shown in black with details given in green.
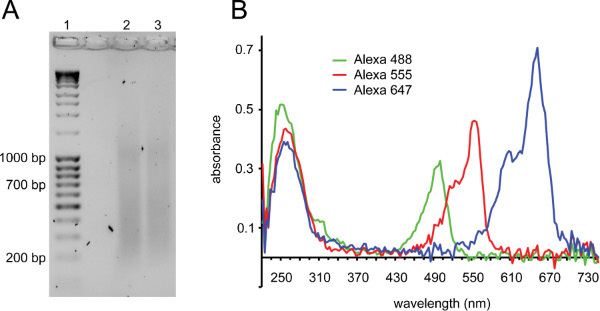 Figure 2. Labeling probes by nick-translation and quality control of labeled probes. A) Gel showing ideal smear before heating to inactivate DNase I (lane 1: DNA ladder, lane 2 and 3: BACs after nick-translation). Fragments should be in the 150-700 bp range. An additional smear at ~1 kb is frequently observed with successful nick-translation reactions. B) Microvolume full spectrum analysis of labeled probes. A DNA peak at 260 nm is observed with a second peak corresponding to the absorbance wavelength of the fluorophore the probe is labeled with: Alexa Fluor 488: 495 nm, Alexa Fluor 555: 555 nm, Alexa Fluor 647: 650 nm. Shown are three probes with good incorporation: fluorophore absorbance peaks are a similar height or higher than the DNA peaks. Alexa Fluor 647 typically has higher absorbance than Alexa Fluor 555, which in turn has higher absorbance than Alexa Fluor 488. Alexa Fluor 488 also absorbs some light at 260 nm, hence the first peak for this probe is higher than the others. Probes with poor incorporation show fluorophore absorbance 2-3-fold lower than the DNA absorbance. Probes with a more than 2-fold higher fluorophore peak than DNA peak typically contain unconjugated dye that may cause background in the FISH.
Figure 2. Labeling probes by nick-translation and quality control of labeled probes. A) Gel showing ideal smear before heating to inactivate DNase I (lane 1: DNA ladder, lane 2 and 3: BACs after nick-translation). Fragments should be in the 150-700 bp range. An additional smear at ~1 kb is frequently observed with successful nick-translation reactions. B) Microvolume full spectrum analysis of labeled probes. A DNA peak at 260 nm is observed with a second peak corresponding to the absorbance wavelength of the fluorophore the probe is labeled with: Alexa Fluor 488: 495 nm, Alexa Fluor 555: 555 nm, Alexa Fluor 647: 650 nm. Shown are three probes with good incorporation: fluorophore absorbance peaks are a similar height or higher than the DNA peaks. Alexa Fluor 647 typically has higher absorbance than Alexa Fluor 555, which in turn has higher absorbance than Alexa Fluor 488. Alexa Fluor 488 also absorbs some light at 260 nm, hence the first peak for this probe is higher than the others. Probes with poor incorporation show fluorophore absorbance 2-3-fold lower than the DNA absorbance. Probes with a more than 2-fold higher fluorophore peak than DNA peak typically contain unconjugated dye that may cause background in the FISH.
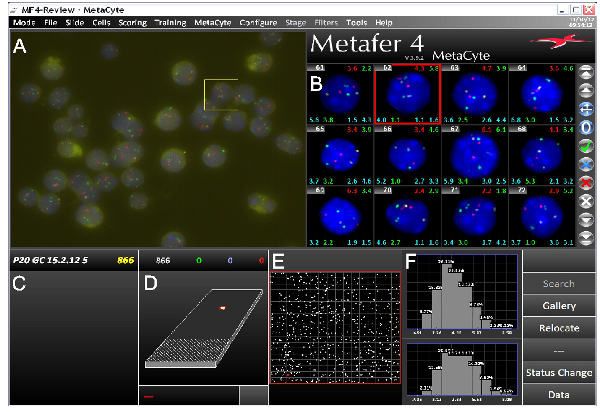 Figure 3. Screenshot from the MetaCyte 3D FISH imaging software showing a typical three-color FISH experiment. The screen is divided into several parts. A) Raw field-of-view (FOV) image captured by the Metafer/MetaCyte slide scanning system. B) Gallery of images of identified nuclei that have undergone image processing and segmentation for FISH signals. Various types of data can be displayed on each gallery image. In this example cell number is shown top left with various interallelic and interlocus distances top right and to either side below each image. C) Slide name with number of nuclei identified in the analysis. D) Depiction of the slide with the area scanned by the system. E) Enlargement of the scanned area. White spots represent identified nuclei per FOV. F) Processed data. In this example maximal interallelic distances between green signals (top) and red signals (bottom) are shown. Data shown is from mouse P20 germ cells. Probes are located on different chromosomes, covering Hbb and Hba genes and a histone cluster. Click here to view larger figure.
Figure 3. Screenshot from the MetaCyte 3D FISH imaging software showing a typical three-color FISH experiment. The screen is divided into several parts. A) Raw field-of-view (FOV) image captured by the Metafer/MetaCyte slide scanning system. B) Gallery of images of identified nuclei that have undergone image processing and segmentation for FISH signals. Various types of data can be displayed on each gallery image. In this example cell number is shown top left with various interallelic and interlocus distances top right and to either side below each image. C) Slide name with number of nuclei identified in the analysis. D) Depiction of the slide with the area scanned by the system. E) Enlargement of the scanned area. White spots represent identified nuclei per FOV. F) Processed data. In this example maximal interallelic distances between green signals (top) and red signals (bottom) are shown. Data shown is from mouse P20 germ cells. Probes are located on different chromosomes, covering Hbb and Hba genes and a histone cluster. Click here to view larger figure.
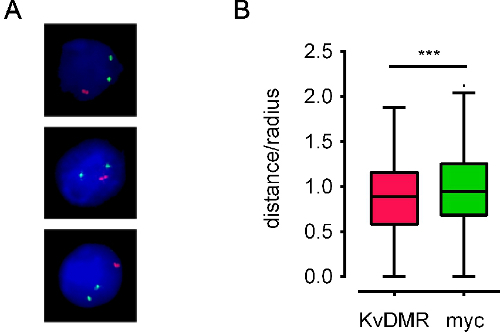 Figure 4. Examples of processed images and derived positional data . A) Processed images for E13.5 mouse fetal liver cells stained with BAC probes located on different chromosomes covering Mycand Kcnq1 genes (termed myc and KvDMR, colored green and red, respectively). B)Tukey box-whisker plots representing the interallelic distance distribution for the same loci in 600 nuclei. Interallelic distance is plotted as a fraction of the nucleus' radius (distance/radius) to account for variations in nuclear size. ES cell nuclei have a radius of around 5 μm. Red probes are significantly closer together than green probes (***: p<0.01, unpaired t-test). Data from10.
Figure 4. Examples of processed images and derived positional data . A) Processed images for E13.5 mouse fetal liver cells stained with BAC probes located on different chromosomes covering Mycand Kcnq1 genes (termed myc and KvDMR, colored green and red, respectively). B)Tukey box-whisker plots representing the interallelic distance distribution for the same loci in 600 nuclei. Interallelic distance is plotted as a fraction of the nucleus' radius (distance/radius) to account for variations in nuclear size. ES cell nuclei have a radius of around 5 μm. Red probes are significantly closer together than green probes (***: p<0.01, unpaired t-test). Data from10.
Movie 1. 3D modeling of two loci within their chromosome territory. Two BAC probes located close to either end of mouse chromosome 7 were labeled (red and far red, (pseudo-colored white)) and hybridized together with a whole chromosome paint (green) to mouse ES cells. Click here to view movie.
Discussion
3D DNA FISH has become a widely recognized tool to analyze spatial arrangements in the nucleus. While FISH provides visual and direct results, as with every technique, one needs to be aware of its limitations. DNA FISH faces the inherent problem that relatively harsh treatments are needed to make chromatin accessible for FISH probes which ultimately disrupts nuclear structure- the very thing that is to be analyzed. Several strategies have been pursued to juggle accessibility and structure preservation. In the protocol described here, chromosomal DNA is made accessible by freeze-thaw permeabilization of cells and heat denaturation prior to probe hybridization. Solovei, et al., (2002) analyzed structural changes after similar treatment and found that the average displacement of chromatin domains was 300 nm which limits the resolution of structural analysis to roughly 1 Mb regions in the genome8. While this is not high resolution, it is a reasonable scale for analyzing nuclear position.
A more practical consideration for FISH analysis is that image acquisition and measurements of nuclear distances are time consuming processes which limit the number of nuclei that can be analyzed. In order to study infrequent events we aimed to do high throughput automated imaging. Then, with suitable controls in place, sufficient numbers can be compared to allow for robust statistical analysis of spatial relations. We routinely analyze 600 nuclei per data point for which we determine the 3D coordinates of all FISH signals within the nuclear volume. These data require very little storage space and allow for analysis of inter- and intra-allelic distances, or radial positions at any later point. Moreover, automated processing can be done in a researcher blind way which greatly reduces the likelihood of unconscious bias.
To enable this kind of high throughput analysis, our goal was to establish a fast and efficient way of performing DNA FISH. We found the protocol described here to be extremely robust, consistently producing bright signals with low background. It worked for all cell types we tried provided we adapted the step of attaching the cells to the slide. Moreover, with one exception, all BACs we used could be turned into bright probes. We have also successfully detected a number of nuclear proteins by antibody staining after DNA FISH. While for some proteins this works well, we recommend comparison with traditional immunofluorescence (IF) to ensure consistency of the antibody staining pattern between IF and DNA immuno FISH (compare 11).
Generally, we found that probes labeled in different colors produced the same results. However, the following aspects should be considered when choosing the labeling dye. First, the color of the fluorophore needs to be well detectable by the microscope used. Second, the wavelength of the light emitted by the fluorophores should be sufficiently different to avoid bleed through into the other channel. This will depend on the filters in the microscope. And third, at least in our hands, some colors produce bright signals more consistently than others. We have always used DAPI (blue) as a counterstain which makes Alexa Fluor 555 (red), 488 (green), and 647 (far red) good choices for labeling probes. With our imaging system, we found Alexa Fluor 555 (red) to produce the brightest signals, followed by Alexa Fluor 488 (green). Alexa Fluor 647 (far red) has the disadvantage that it cannot be detected by the human eye and therefore no signals can be seen when looking down the microscope. Thus, if doing two-color FISH, we recommend the combination of red and green dyes.
Two principal problems may arise: cells washing off the slides and weak signals. How well cells adhere to the slide is hugely variable between cell types, and the best protocol to settle/seed the cells needs to be determined. We have successfully used either positively charged or Poly-L-lysine coated slides (bought ready to use), but found cells generally adhered better to Poly-L-lysine coated slides. We also found that when cells were too dense entire areas would peel off as a sheet, whereas automated imaging was hampered if cells were too sparse. Thus, if the sample is not too precious, different cell numbers should be seeded to determine the ideal density. If cells are particularly prone to wash off, a hydrophobic barrier pen can be used, and FISH steps can be performed by carefully pipetting solutions directly onto the slide. Pipetting also drastically reduces the volumes of reagents needed, but takes considerably longer when doing multiple slides.
Weak signals are usually due to poor probes, and it is well worth investing time into making good ones. Clean BAC DNA should be used as starting material which can be obtained by repeated precipitation or commercially available kits. Contamination with bacterial genomic DNA negatively affects probe quality and careful handling during cell lysis and filtering of the cell lysate is required to keep it to a minimum. It is, however, not necessary to use an exonuclease step to digest linear DNA, as this greatly reduces yield. Efficient labeling of the probe is crucial. The most important quality control for this step is checking the fragment size after nick translation (see Representative Results). A 'good' smear will almost always result in a brightly colored probe. However, we have found that occasionally a certain BAC cannot be processed into a good probe, and a different BAC needs to be chosen. Another important step is heat denaturation. If the temperature is too low, the DNA will be only partially denatured and probe hybridization will be impaired. If the temperature is too high, nuclear structure will be perturbed more than necessary. In our hands, 78 °C on an inverted hot block is the ideal compromise, but this may vary depending on the respective hot block used.
We have had good results using Vectashield mounting medium, but found that SlowFade Gold has less background and preserves the fluorescent signal for longer. We do not recommend the use of hard-set mounting medium as we found this to affect 3D structure and to form air bubbles over time.
In conclusion, brightly fluorescent directly labeled probes together with the DNA FISH protocol described here offer an efficient solution for robust and rapid analysis of nuclear architecture which is applicable to a wide variety of biological questions.
Disclosures
Production of this article was partially sponsored by Carl Zeiss and MetaSystems.
Acknowledgments
This work was funded by the BBSRC and the Wellcome Trust. We would like to thank Simon Walker for assistance with imaging and Felix Krueger for help with bioinformatic analysis.
References
- Chakalova L, Debrand E, Mitchell JA, Osborne CS, Fraser P. Replication and transcription: shaping the landscape of the genome. Nat. Rev. Genet. 2005;6:669–677. doi: 10.1038/nrg1673. [DOI] [PubMed] [Google Scholar]
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell. 1016;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular Cloning: A laboratory Manual. Cold Spring Harbor, New York, USA: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Boyle S, Rodesch MJ, Halvensleben HA, Jeddeloh JA, Bickmore WA. Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosome Res. 2011;19:901–909. doi: 10.1007/s10577-011-9245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K. Visualizing nuclear proteins together with transcribed and inactive genes in structurally preserved cells. Methods. 2002;26:10–18. doi: 10.1016/S1046-2023(02)00003-8. [DOI] [PubMed] [Google Scholar]
- Hewitt SL, Yin B, et al. RAG-1 and ATM coordinate monoallelic recombination and nuclear positioning of immunoglobulin loci. Nat. Immunol. 2009;10:655–664. doi: 10.1038/ni.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Cavallo A, et al. Spatial preservation of nuclear chromatin architecture during three-dimensional fluorescence in situ hybridization (3D-FISH. Experimental Cell Research. 2002;276:10–23. doi: 10.1006/excr.2002.5513. [DOI] [PubMed] [Google Scholar]
- Solovei I. FISH on three-dimensionally preserved nuclei. In: Beatty B, Mai S, Squire J, editors. FISH: A Practical Approach. Oxford, UK: Oxford University Press; 2002. [Google Scholar]
- Krueger C, King MR, et al. Pairing of homologous regions in the mouse genome is associated with transcription but not imprinting status. PLoS One. 2012;7:e38983. doi: 10.1371/journal.pone.0038983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovei I, Cremer M. 3D-FISH on Cultured Cells Combined with Immunostaining. In: Bridger JM, Volpi EV, editors. Fluorescence in situ hybridization (FISH): Protocols and Applications. New York, New York, USA: Humana Press; 2010. [Google Scholar]