

Abstract
Among methods to study protein-protein interaction inside cells, Bimolecular Fluorescence Complementation (BiFC) is relatively simple and sensitive. BiFC is based on the production of fluorescence using two non-fluorescent fragments of a fluorescent protein (Venus, a Yellow Fluorescent Protein variant, is used here). Non-fluorescent Venus fragments (VN and VC) are fused to two interacting proteins (in this case, AKAP-Lbc and PDE4D3), yielding fluorescence due to VN-AKAP-Lbc-VC-PDE4D3 interaction and the formation of a functional fluorescent protein inside cells.
BiFC provides information on the subcellular localization of protein complexes and the strength of protein interactions based on fluorescence intensity. However, BiFC analysis using microscopy to quantify the strength of protein-protein interaction is time-consuming and somewhat subjective due to heterogeneity in protein expression and interaction. By coupling flow cytometric analysis with BiFC methodology, the fluorescent BiFC protein-protein interaction signal can be accurately measured for a large quantity of cells in a short time. Here, we demonstrate an application of this methodology to map regions in PDE4D3 that are required for the interaction with AKAP-Lbc. This high throughput methodology can be applied to screening factors that regulate protein-protein interaction.
Keywords: Molecular Biology, Issue 78, Biochemistry, Cellular Biology, Genetics, Pharmacology, Proteins, Flow Cytometry, Bimolecular Fluorescence Complementation, BiFC, quantative analysis, protein-protein interaction, Förster resonance energy transfer, FRET, Bioluminescence Resonance Energy Transfer, BRET, protein, cell, transfection, fluorescence, microscopy
Introduction
650,000 protein-protein interactions are estimated to exist in the human interactome, playing critical roles in maintaining normal cell functions 1,2. Besides co-immunoprecipitation (co-IP), the gold standard to study protein-protein interaction from cell lysate, several protein fragment complementation assays (PCA) have been developed to improve sensitivity in detecting protein-protein interaction inside cells 3. Techniques include Förster resonance energy transfer (FRET), Bioluminescence Resonance Energy Transfer (BRET), and Bimolecular Fluorescence Complementation (BiFC) 4,5. BiFC is based on the facilitated association of two fragments of a fluorescent protein (here we use Venus; a YFP variant) that are each fused to a potential interacting protein partner (in this example we use AKAP-Lbc and PDE4D3). Interaction of the two proteins of interest inside cells results in functional fluorescence, which can be visualized by fluorescence microscopy using either live or fixed cells 6,7,8. Compared to other PCAs, BiFC is sensitive and less technically challenging, with potential to study the cellular localization in live cells. A major drawback of this technique however is that once formed, the fluorescent protein complex cannot be reversed. Therefore it is not a good method to study dynamic protein-protein interaction. In this paper, we use BiFC to map the interaction sites of PDE4D3 with AKAP-Lbc by monitoring the fluorescence intensities of Venus. Compared to traditional analysis using fluorescence microscopy, which is time-consuming and labor-intensive (if not carried out using automated high throughput machinery), flow cytometry provides a straightforward quantitative analysis of thousands of cells in a heterogeneous population over a short time 9,10. Here, by carrying out a side-by-side comparison of flow cytometric-BiFC analysis and traditional co-IP, we demonstrate that the two methods provide comparable data, however, flow cytometric BiFC analysis is less time consuming and uses less material which may be more useful when cells of interest are limited.
Protocol
1. Cell Transfection
- Plate cells the day before transfection, plating fewer cells for immunofluorescence.
- For immunofluorescence: in a 6-well plate, coat glass cover slips (1 per well) with 0.02% gelatin at 37 °C for at least 4 hr, wash once with medium, and for each well, plate 1 x 105 HEK 293T cells in 2 ml complete growth medium [Dulbecco's modified Eagle's media (DMEM) plus 10% FBS].
- For flow cytometric and Western blot analyses: plate 1.2 x 105 HEK 293T cells/well in 2 ml complete growth medium in 6-well plate (0.3 x 105 HEK 293T cells in 0.5 ml complete growth medium per well in 24-well plate).
- Prepare DNA for transfection according to manufacture's protocol: Effectene (Qiagen) is used for immunofluorescence. TransIT-LT1 (Mirus) is used for flow cytometric and Western blot analyses. Amount of DNA used is listed below.
24-well plate (ng) 6-well plate (ng) CFP-vector 10 50 VNN-AKAP-Lbc 200 1000 VCN-PDE4D3-FL(full-length PDE4D3) 2.5, 5, 10, 20, 40, 80 100 VCN-PDE4D3-UCR1(a PDE4D3 N-terminal fragment containing the UCR1 domain) 100 VCN-PDE4D3-UCR2+CAT (a PDE4D3 C-terminal fragment containing the UCR2 and catalytic domains) 100 - Add indicated amount of plasmid DNA + 4 μl enhancer to 100 μl buffer EC, mix, and incubate for 5 min at room temperature.
- Add 5 μl Effectene, mix, and incubate for 10 min at room temperature. Add drop-wise to cells.
- Add indicated amount of plasmid DNA to 250 μl/50 μl of Opti-MEM I serum-free medium in a sterile tube.
- Add TransIT-LT1 reagent to the diluted DNA mixture and pipette gently to mix. (TransIT-LT1 Reagent:DNA ratio is 3 μl of TransIT-LT1 reagent per 1 μg of DNA).
- Incubate for 30 min at room temperature and add drop-wise to cells.
2. Cell Preparation for Flow Cytometry, Western Blot, and Immunofluorescence
24 hr post transfection, check fluorescence under epifluorescence microscope before harvesting cells.
- Cell preparation for flow cytometric analysis:
- Wash cells with 0.5 ml/2 ml ice-cold PBS for 24-well/6-well plates.
- Detach cells using 50 μl/250 μl 0.05% trypsin.
- Neutralize trypsin with 200 μl/1 ml DMEM medium.
- Collect 250 μl cells (for both 24-well and 6-well plate) by centrifugation at 500 x g for 5 min, use the remaining cells (1 ml) for Western blot analysis.
- Resuspend cells in 250 μl FACS buffer [PBS + 0.5% (w/v) BSA + 2 mM EDTA], store on ice for flow cytometric analysis. Cells can also be fixed in 1% paraformaldehyde (in PBS) if flow cytometry analysis will not be immediate.
- Cell preparation for Western blot analysis: carried out in parallel with cell preparation for flow cytometry analysis.
- Wash cells 1x with 2 ml ice-cold PBS, lyse cells by adding 100 μl of cell lysis buffer [10 mM sodium phosphate buffer, pH 6.95, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 1% Triton X-100].
- Collect cell lysate by centrifugation for 10 min at 21,000 x g at 4 °C.
- Quantify protein concentration by Bradford protein assay, load equal amounts of protein per lane for SDS-PAGE, and transfer to nitrocellulose for immunoblotting.
- Cell preparation for immunofluorescence.
- Rinse cells 3x with 2 ml PBS, fix cells in 1 ml of 3.7% paraformaldehyde (in PBS) for 10 min at room temperature, then wash 3x with 2 ml PBS for 5 min.
- Mount cover slip on a glass slide, using mounting medium. Seal the coverslip edges using nail polish if necessary.
- Store the slides at 4 °C prior to examination.
3. Flow Cytometry and Immunofluorescence Microscopy
- Flow cytometry is performed using a Beckman Coulter Cyan ADP, equipped with 488 nm, 405 nm, and 642 nm solid-state lasers. Summit software is used for acquisition and analysis.
- Calibrate flow cytometer using standard beads.
- Plot Forward scatter (FSC)/Sideward scatter (SSC) on a linear scale for gating live cell population.
- Plot FSC/voltage pulse width for gating non-aggregated live cell population.
- Plot count/FL6 (CFP fluorescence, 405 nm) on a log scale for gating non-aggregated live CFP-positive cells.
- Plot count/FL1 (YFP fluorescence, 488 nm) on a log scale for BiFC intensity.
- Run YFP-CFP- double negative samples, adjust FSC, SSC, FL1 and FL6 photo multiplier tube (PMT) voltage to set the threshold for each signal.
- Run single positive (both YFP+ and CFP+) samples for future off-line compensation
- Acquire at least 10,000 gated events (non-aggregated live CFP+ cells) for analysis.
Immunofluorescence microscopy is performed using a Zeiss LSM 510 confocal microscope. Note: it is important to keep all settings (such as the zoom, pinhole, detector gain, amplifier offset, frame size, scan speed, scan average, and laser power) consistent between samples for proper comparison of all data.
4. Data Analysis (Performed Using Summit Software)
Set up the analysis protocol similar to that for acquisition with proper gating: Gate 1 in FSC/SSC dot plot for live cells. Gate 2 in FSC/pulse width of Gate 1 for non-aggregated live cells. Gate 3 in count/CFP of Gate 3 for non-aggregated CFP+ live cells.
Compensate YFP and CFP signal using YFP+ and CFP+ single positive cells.
Re-determine cut-off lines for CFP/YFP-positive cells.
Export YFP mean fluorescence intensity (MFI) of CFP+ cells to Excel for data-plotting.
Normalize YFP MFI to expression levels of AKAP-Lbc, PDE4D3, and loading control α-tubulin, as determined by Western blot.
Representative Results
AKAP-Lbc and PDE4D3 constructs (Figure 1B) were fused to VN and VC fragments, respectively. Interaction of AKAP-Lbc and PDE4D3 results in functional YFP fluorescence (Figure 1A). Here we use the BiFC method to map AKAP-Lbc-binding sites in PDE4D3. Upstream conserved region 1 (UCR1), upstream conserved region 2 (UCR2) and catalytic region (CAT) are conserved among PDE4 family proteins, therefore, full length (FL), UCR1 and UCR2 plus CAT were used for screening the binding site of PDE4D3 to AKAP-Lbc. The expression of VN-AKAP-Lbc and VC-PDE4D3-fragments in HEK 293T cells results in fluorescence with different intensity (Figure 1C). BiFC indicates that both UCR1 and UCR2+CAT contribute to the interaction of PDE4D3 with AKAP-Lbc. Greater fluorescence intensity resulting from AKAP-Lbc-UCR1 interaction, when compared to AKAP-Lbc-UCR2+CAT, indicates that PDE-UCR1 binds to AKAP-Lbc better than PDE-UCR2+CAT.
The heterogeneous expression of BiFC constructs and CFP in transfected cell requires analysis of a large number of cells for precise interpretation of results. However, traditional analysis by immunofluorescence microscopy is labor-intensive and subjective. This motivated us to take advantage of flow cytometry, to develop a method to rapidly analyze the protein-protein interaction in a large sample population. Initially, we carried out experiments to confirm that BiFC can be used to quantify the intensity of AKAP-Lbc and PDE4D3 interaction. HEK 293T cells were transfected with a low amount of CFP (10 ng), VN-AKAP-Lbc (200 ng), and increasing amounts of VC-PDE4D3 (2.5 ng, 5 ng, 10 ng, 20 ng, 40 ng, 80 ng). Dead cells and cell debris were excluded from analysis using FSC vs SSC double dot (Figure 2A), and cell aggregates were excluded by using SSC vs Pulse Width double dot (Figure 2B). Cells with BiFC signal or CFP expression alone were used to determine the compensation and cut-off for cells without fluorescence (Figures 2C-2F). CFP vector was co-transfected with BiFC constructs and CFP expression was used as a positive marker to indicate successful transfection. Only CFP+ cells were used for measurement of BiFC fluorescence. Transfection of increasing amounts of PDE4D3 resulted in increased Venus (YFP) BiFC signal (Figure 3A), while CFP signal remained unchanged (Figure 3B). Expression of VN-AKAP-Lbc and increasing amounts of VC-PDE4D3 was confirmed by Western blot (Figure 3C). Overall, increased PDE expression resulted in a linear increase of BiFC fluorescence correlating to PDE expression levels (Figure 3D). These results validated the use of flow cytometry to quantify AKAP-Lbc-PDE4D3 interaction by measuring the mean fluorescence intensity (MFI) of BiFC. We therefore used this methodology to map AKAP-Lbc binding sites within PDE4D3 using VN-AKAP-Lbc and VC-PDE4D3 constructs. Consistent with our observations in Figure 1C, expression of VN-AKAP-Lbc and VC-PDE4D3-UCR2+CAT resulted in lower BiFC signal when compared to VC-PDE4D3-FL and VC-PDE4D3-UCR1 (Figure 4B). Similar protein expression was observed in all flow samples (Figure 4C) and mean fluorescence was normalized to the relative expression levels of AKAP-Lbc, PDE4D3, and α-tubulin. Therefore, normalized BiFC MFI indicates that there is no difference in the fluorescence intensity between AKAP-Lbc-PDE4D3-FL and AKAP-Lbc-PDE4D3-UCR1, but much lower signal for AKAP-Lbc-PDE4D3-UCR2+CAT interaction (Figure 4A). These results suggest that there are multiple regions in PDE4D3 that interact with AKAP-Lbc, and that PDE4D3-UCR1 is the primary region of interaction with AKAP-Lbc. This result was also confirmed by co-IP, the gold standard for protein-protein interaction (Figure 5).
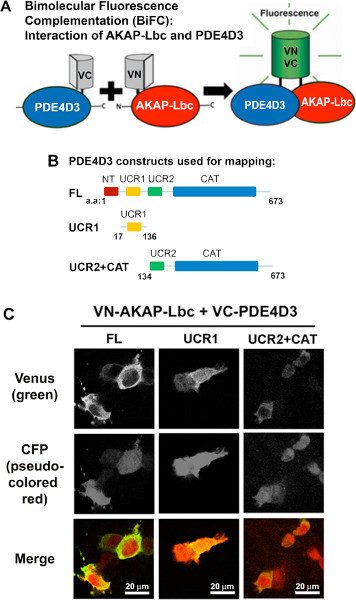 Figure 1. BiFC analysis of AKAP-Lbc-PDE4D3-fragment interaction by immunofluorescence microscopy. A) Schematic representative of Bimolecular Fluorescence Complementation (BiFC). Venus fragments; VN and VC are fused to AKAP-Lbc and PDE4D3 respectively. AKAP-Lbc-PDE4D3 interaction results in functional YFP fluorescence. B) Schematic diagram of PDE4D3 constructs used for mapping. C) Representative BiFC signals of transfected HEK 293T cells. Cells were co-transfected with VN-AKAP-Lbc and VC-PDE4D3 constructs (-FL, -UCR1, -UCR2+CAT) for BiFC analysis. CFP-vector was also co-transfected as a transfection marker. 24 hr post-transfection, cells were fixed for imaging. BiFC signal is shown in green and CFP is shown in pseudocolor red in the merged figures. Click here to view larger figure.
Figure 1. BiFC analysis of AKAP-Lbc-PDE4D3-fragment interaction by immunofluorescence microscopy. A) Schematic representative of Bimolecular Fluorescence Complementation (BiFC). Venus fragments; VN and VC are fused to AKAP-Lbc and PDE4D3 respectively. AKAP-Lbc-PDE4D3 interaction results in functional YFP fluorescence. B) Schematic diagram of PDE4D3 constructs used for mapping. C) Representative BiFC signals of transfected HEK 293T cells. Cells were co-transfected with VN-AKAP-Lbc and VC-PDE4D3 constructs (-FL, -UCR1, -UCR2+CAT) for BiFC analysis. CFP-vector was also co-transfected as a transfection marker. 24 hr post-transfection, cells were fixed for imaging. BiFC signal is shown in green and CFP is shown in pseudocolor red in the merged figures. Click here to view larger figure.
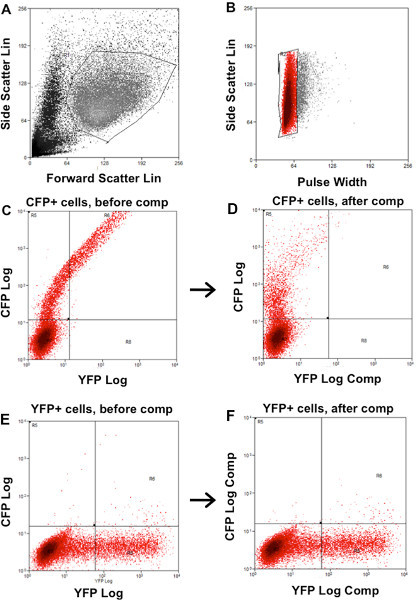 Figure 2. Determination of gates and compensation for FACS analysis. A) FSC vs SSC dot plot to gate live cells and to exclude debris and dead cells. B) Side Scatter vs pulse width dot plot to gate single cells and to exclude aggregates. C) Cells expressing CFP (CFP+ cells) before compensation. D) Cells expressing CFP (CFP+ cells) after YFP compensation. The cut-off line for cells with YFP expression was re-determined. E) cells with BiFC signal (YFP+ cells) before compensation. F) cells with BiFC signal (YFP+ cells) after CFP compensation. The cut-off line for cells with CFP expression was re-determined. Click here to view larger figure.
Figure 2. Determination of gates and compensation for FACS analysis. A) FSC vs SSC dot plot to gate live cells and to exclude debris and dead cells. B) Side Scatter vs pulse width dot plot to gate single cells and to exclude aggregates. C) Cells expressing CFP (CFP+ cells) before compensation. D) Cells expressing CFP (CFP+ cells) after YFP compensation. The cut-off line for cells with YFP expression was re-determined. E) cells with BiFC signal (YFP+ cells) before compensation. F) cells with BiFC signal (YFP+ cells) after CFP compensation. The cut-off line for cells with CFP expression was re-determined. Click here to view larger figure.
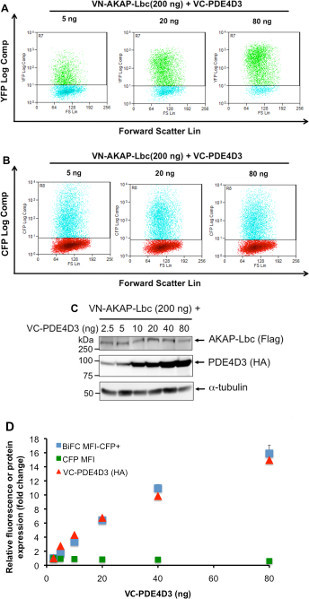 Figure 3. Analysis of AKAP-Lbc-PDE4D3 interaction using BiFC-Flow Cytometry by varying the amount of PDE4D3 expression. A) Increased BiFC signal inside CFP+ cells transfected with increasing amounts of VC-PDE4D3. Cells were transfected with CFP (10 ng), VN-AKAP-Lbc (200 ng) and increasing amounts of VC-PDE4D3 (2.5 ng to 80 ng). Non-aggregated CFP+ live cells were gated and YFP fluorescence is shown as YFP vs Forward Scatter double dot plot. YFP+ cells are shown in green. Representative data from 5 ng, 20 ng, and 80 ng VC-PDE4D3 is shown. B) CFP fluorescence in live cells is shown as CFP vs Forward Scatter double dot plot, indicating consistent CFP fluorescence among all samples. C) Western blot analysis demonstrating increasing PDE4D3 expression with consistent expression of AKAP-Lbc and α-tubulin (used as a loading control). D) Venus (YFP) Mean fluorescence intensity (MFI) of CFP+ cells is correlated with VC-PDE4D3 expression. Fold change of both YFP MFI from CFP+ cells is plotted along with CFP MFI fold change from non-aggregated live cells. Relative expression levels of VC-PDE4D3 determined by Image J analysis of Western blot are also plotted. Click here to view larger figure.
Figure 3. Analysis of AKAP-Lbc-PDE4D3 interaction using BiFC-Flow Cytometry by varying the amount of PDE4D3 expression. A) Increased BiFC signal inside CFP+ cells transfected with increasing amounts of VC-PDE4D3. Cells were transfected with CFP (10 ng), VN-AKAP-Lbc (200 ng) and increasing amounts of VC-PDE4D3 (2.5 ng to 80 ng). Non-aggregated CFP+ live cells were gated and YFP fluorescence is shown as YFP vs Forward Scatter double dot plot. YFP+ cells are shown in green. Representative data from 5 ng, 20 ng, and 80 ng VC-PDE4D3 is shown. B) CFP fluorescence in live cells is shown as CFP vs Forward Scatter double dot plot, indicating consistent CFP fluorescence among all samples. C) Western blot analysis demonstrating increasing PDE4D3 expression with consistent expression of AKAP-Lbc and α-tubulin (used as a loading control). D) Venus (YFP) Mean fluorescence intensity (MFI) of CFP+ cells is correlated with VC-PDE4D3 expression. Fold change of both YFP MFI from CFP+ cells is plotted along with CFP MFI fold change from non-aggregated live cells. Relative expression levels of VC-PDE4D3 determined by Image J analysis of Western blot are also plotted. Click here to view larger figure.
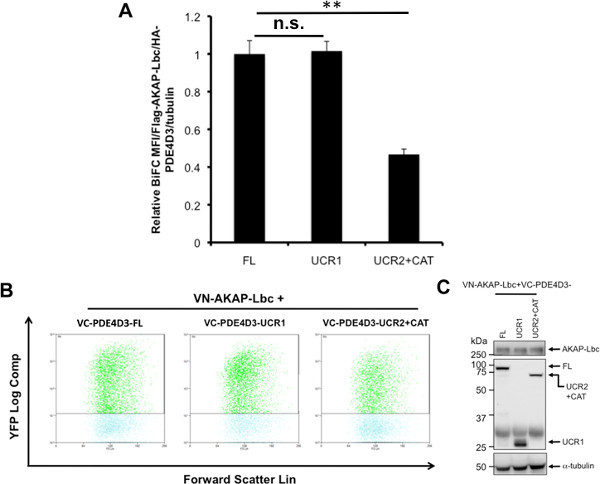 Figure 4. BiFC analysis of AKAP-Lbc interaction with PDE4D3-FL, PDE4D3-UCR1, PDE4D3-UCR2+CAT using Flow Cytometry. A) AKAP-Lbc-PDE4D3-fragment interaction was quantified by Venus-MFI normalized to protein expression (AKAP-Lbc, PDE4D3 fragments and loading control α-tubulin). Difference in BiFC signal were examined by one way analysis of variance (ANOVA) A p value <0.01 is considered very significant (**) while a p value >0.05 is considered not significant (n.s.). B) Representative BiFC signal in transfected HEK 293T cells. HEK 293T cells were co-transfected with CFP, VN-AKAP-Lbc and VC-PDE4D3 constructs (-FL, -UCR1, -UCR2+CAT). Non-aggregated live CFP+ cells were gated, and their BiFC fluorescence is shown in YFP vs Forward Scatter double dot plot. C) Western blot indicating comparable expression of AKAP-Lbc and PDE4D3-fragments. Click here to view larger figure.
Figure 4. BiFC analysis of AKAP-Lbc interaction with PDE4D3-FL, PDE4D3-UCR1, PDE4D3-UCR2+CAT using Flow Cytometry. A) AKAP-Lbc-PDE4D3-fragment interaction was quantified by Venus-MFI normalized to protein expression (AKAP-Lbc, PDE4D3 fragments and loading control α-tubulin). Difference in BiFC signal were examined by one way analysis of variance (ANOVA) A p value <0.01 is considered very significant (**) while a p value >0.05 is considered not significant (n.s.). B) Representative BiFC signal in transfected HEK 293T cells. HEK 293T cells were co-transfected with CFP, VN-AKAP-Lbc and VC-PDE4D3 constructs (-FL, -UCR1, -UCR2+CAT). Non-aggregated live CFP+ cells were gated, and their BiFC fluorescence is shown in YFP vs Forward Scatter double dot plot. C) Western blot indicating comparable expression of AKAP-Lbc and PDE4D3-fragments. Click here to view larger figure.
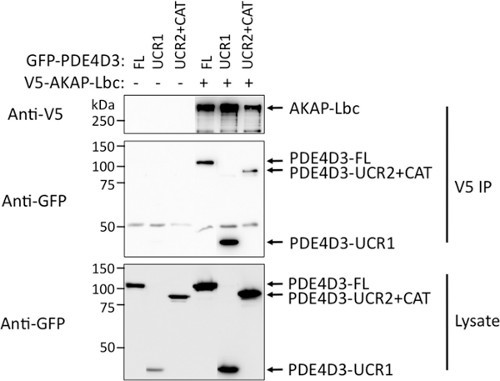 Figure 5. Confirmation of Flow Cytometry analysis by co-immunoprecipitation. HEK 293T cells were co-transfected for expression of V5-AKAP-Lbc and GFP-PDE4D3 constructs (-FL, -UCR1, -UCR2+CAT). 48 hr post-transfection, AKAP-Lbc was immunoprecipitated using V5-agarose beads. Binding of PDE4D3-FL, UCR1 and UCR2+CAT was detected by SDS-PAGE and Western blot using anti-GFP primary antibody. The corresponding PDE4D3 input used for immunoprecipitation is also shown, indicating similar expression for all PDE4D3-fragments.
Figure 5. Confirmation of Flow Cytometry analysis by co-immunoprecipitation. HEK 293T cells were co-transfected for expression of V5-AKAP-Lbc and GFP-PDE4D3 constructs (-FL, -UCR1, -UCR2+CAT). 48 hr post-transfection, AKAP-Lbc was immunoprecipitated using V5-agarose beads. Binding of PDE4D3-FL, UCR1 and UCR2+CAT was detected by SDS-PAGE and Western blot using anti-GFP primary antibody. The corresponding PDE4D3 input used for immunoprecipitation is also shown, indicating similar expression for all PDE4D3-fragments.
Discussion
BiFC is a simple and sensitive method to study protein-protein interaction. This method cannot be used to identify new protein-protein interactions, however, it is especially convenient to confirm protein-protein interaction inside cells and to study functional properties; such as subcellular localization of protein complexes, mapping of protein-protein interaction sites, and for screening of small molecules/peptides that can modulate protein-protein interaction. Because VN and VC fragments are non-fluorescent, background fluorescence is low, thus aiding the sensitivity of this assay. Time is needed to mature/stabilize the VN-protein-VC-protein interaction, thus there is delay to visualizing the protein-protein interaction, and optimal time-points may therefore have to be empirically determined. It should also be noted that VN-protein-VC-protein interaction is not reversible, therefore unfortunately with current Venus BiFC constructs this assay is not suitable to study dynamic kinetics of protein-protein interaction.
Proper controls are important for BiFC analysis. A suitable negative control includes non-interacting proteins or non-specific peptides in the relevant BiFC vector, or if possible, a mutant that disrupts protein-protein interaction. Empty BiFC vector is not a good control as it results in non-specific high background fluorescence. If BiFC is not observed in two interacting proteins, expression should be analyzed. Also, BiFC signal may vary depending on the protein-protein interaction sites. It should be noted that spatial modulation of VN and VC interaction may also affect the fluorescence intensity; therefore it is initially important to carry out BiFC experiments using both C-terminal and N-terminal VN/VC expression constructs. For example, BiFC may not occur if protein-protein interaction blocks the re-association of the Venus fragments. Therefore, multiple combinations of VN and VC constructs should be tested (i.e. both C and N terminal protein fusions).
Fluorescence intensity of BiFC is proportional to the strength of protein-protein interaction, with higher BiFC intensity indicating stronger interaction and a lower fluorescence intensity suggesting weaker interaction. BiFC MFI is therefore used as indicator for protein-protein interaction. For precise analysis of protein-protein interaction by BiFC, it is very important that Western blot analysis is performed to ensure equal expression of different constructs and that no protein degradation is occurring, which may interfere with correct analysis. Lysate from the same samples is therefore preferred for Western blot analysis to determine similar protein expression for all samples analyzed. Additionally, it is critical to determine suitable amount of plasmids for transfection to ensure linear expression and interaction of proteins, and among this range, the interaction of protein correlates with fluorescence intensity of BiFC signal.
Low amount of CFP plasmid is used as a marker for successful transfection, so only CFP positive cells were analyzed here. A different fluorescent protein, such as RFP could be used to minimize the bleed-through between colors11. For Flow Cytometry, single positive cells are needed to be included for compensation. If possible, at least 10,000 CFP positive cells are counted to ensure a good sample size.
In summary, here we demonstrate that coupling BiFC methodology with flow cytometric analysis can be used as a good indicator of protein-protein interaction inside cells. Traditional analysis of BiFC using immunofluorescence microscope is time consuming, and sample size is much lower than that corresponding to flow cytometric analysis. Thus, coupling flow cytometry with BiFC may be advantageous for analysis of protein-protein interaction when transfection efficiency is low. In this report, we took advantage of BiFC-Flow Cytometry to map the interaction sites in PDE4D3 for binding to AKAP-Lbc. This technique could also be extended to screen for molecules or peptides that may disrupt or enhance protein-protein interaction, for drug discovery 12. This method can also be used to study certain physiological events, such as ligand-induced internalization of GPCR's 13.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank the O'Bryan lab at UIC for critical experimental evaluation and discussion. This work was supported by American Heart Association Grant 11SDG5230003 to GKC and National Center for Advancing Translational Science - UIC Center for Clinical and Translational Sciences Grant UL1TR000050.
References
- Collura V, Boissy G. From protein-protein complexes to interactomics. Subcell Biochem. 2007;43:135–183. doi: 10.1007/978-1-4020-5943-8_8. [DOI] [PubMed] [Google Scholar]
- Stumpf MP, et al. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. U.S.A. 2008;105:6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhawat SS, Ghosh I. Split-protein systems: beyond binary protein-protein interactions. Curr. Opin. Chem. Biol. 2011;15:789–797. doi: 10.1016/j.cbpa.2011.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwane S, Kiely PA. Tools used to study how protein complexes are assembled in signaling cascades. Bioeng. Bugs. 2011;2:247–259. doi: 10.4161/bbug.2.5.17844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehler J. New methodologies for measuring protein interactions in vivo and in vitro. Curr. Opin. Struct. Biol. 2005;15:4–14. doi: 10.1016/j.sbi.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006;1:1278–1286. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu YJ, Suarez CD, Hu CD. Visualization of ternary complexes in living cells by using a BiFC-based FRET assay. Nat. Protoc. 2008;3:1693–1702. doi: 10.1038/nprot.2008.157. [DOI] [PubMed] [Google Scholar]
- Wong KA, O'Bryan JP. Bimolecular fluorescence complementation. J. Vis. Exp. 2011. p. e2643. [DOI] [PMC free article] [PubMed]
- Sugarbaker EV, Thornthwaite JT, Temple WT, Ketcham AS. Flow cytometry: general principles and applications to selected studies in tumor biology. Int. Adv. Surg. Oncol. 1979;2:125–153. [PubMed] [Google Scholar]
- Koshy S, Alizadeh P, Timchenko LT, Beeton C. Quantitative measurement of GLUT4 translocation to the plasma membrane by flow cytometry. J. Vis. Exp. 2010. p. e2429. [DOI] [PMC free article] [PubMed]
- Smith CL. Basic confocal microscopy. Curr. Protoc. Mol. Biol. 2008;Chapter 14(Unit 14):11. doi: 10.1002/0471142727.mb1411s81. [DOI] [PubMed] [Google Scholar]
- Westwick JK, Lamerdin JE. Improving drug discovery with contextual assays and cellular systems analysis. Methods Mol. Biol. 2011;756:61–73. doi: 10.1007/978-1-61779-160-4_3. [DOI] [PubMed] [Google Scholar]
- Kilpatrick LE, Holliday ND. Dissecting the pharmacology of G protein-coupled receptor signaling complexes using bimolecular fluorescence complementation. Methods Mol. Biol. 2012;897:109–138. doi: 10.1007/978-1-61779-909-9_6. [DOI] [PubMed] [Google Scholar]