

Abstract
Retinal detachment (RD) describes a separation of the neurosensory retina from the retinal pigmented epithelium (RPE). The RPE is essential for normal function of the light sensitive neurons, the photoreceptors. Detachment of the retina from the RPE creates a physical gap that is filled with extracellular fluid. RD initiates cellular and molecular adverse events that affect both the neurosensory retina and the RPE since the physiological exchange of ions and metabolites is severely perturbed. The consequence for vision is related to the duration of the detachment since a rapid reapposition of the two tissues results in the restoration of vision 1. The treatment of RD is exclusively surgical. Removal of vitreous gel (vitrectomy) is followed by the removal non essential part of the retina around the detached area to favor retinal detachment. The removed retinal specimens are res nullius (nothing) and consequently normally discarded. To recover RNA from these surgical specimens, we developed the procedure jouRNAl that allows RNA conservation during the transfer from the surgical block to the laboratory. We also standardized a protocol to purify RNA by cesium chloride ultracentrifugation to assure that the purified RNAs are suitable for global gene expression analysis. The quality of the RNA was validated both by RT-PCR and microarray analysis. Analysis of the data shows a simultaneous involvement of inflammation and photoreceptor degeneration during RD.
Keywords: Genetics, Issue 78, Medicine, Molecular Biology, Biomedical Engineering, Anatomy, Physiology, Neurobiology, Neuroscience, Biochemistry, Genomics, Transcriptomics, Ophthalmology, Eye Diseases, neuroscience, retina, human tissue specimens, RNA purification, microarray analysis, microarray, jouRNAl, RNA, DNA, purification, gel electrophoresis, sequencing, clinical applications, clinical techniques
Introduction
The main therapeutic objective in retinal detachment (RD) is to find a way to limit photoreceptor cell damage and retinal inflammation resulting from the separation of the photoreceptors from the retinal pigmented epithelial cells. During RD, RPE cells are activated, migrate, dedifferentiate, and proliferate at the surface of the detached retina, exerting contractile forces leading to complications. Transcriptomics analysis of RD is a way to identify target genes with modified expression following RD and hence future therapeutic molecules that could improve final visual outcome in combination with surgery. It is well known ribonucleic acid (RNA) is not stable as is deoxyribonucleic acid (DNA), the latter being used extensively for genetic studies, its stability had permitted the sequencing of the Neanderthal genome from paleontological samples of more than 30,000 years old 2. Transcription of DNA of the genes from the genome into messenger RNA is the major process in gene expression, and the mRNA is labile in order to constitute a signal. RNAs are very rapidly degraded by RNAse enzymes which terminate the signal. When tissues are isolated from an organism, the RNAs are very often degraded before the expression is studied in a research laboratory. Degraded RNA is not suited for analysis gene expression. Because the laboratory staff cannot participate in surgery, we developed a procedure that is easy and only requires that the surgeon to recover the tissues into an appropriate RNAse-free solution. The RNA of the tissues is stable and can be analyzed without any sign of degradation after 72 hr at room temperature in this solution. The RNAs from specimens are purified by a standardized method involving an ultracentrifugation on a cesium chloride gradient after having been transferred to the laboratory 3. Then, quality of the RNAs are assessed by agarose gel electrophoresis and by RT-PCR. The RNA purification protocol has the advantage of separating the molecules according to their density that is different for DNA and RNA and assures that the RNA is not contaminated by a DNA molecules that will generate artifactual signals in gene expression studies. In addition, the transfer RNAs (tRNAs), which are quantitatively the most abundant RNA within a cell, are separated according to the same physical property from the ribosomal RNA (rRNAs) and the messenger RNAs (mRNAs), those two last one being the end product of the purification process. The removal of tRNA from the preparation is useful since most of the microarray analytical protocols involved the use of reverse transcriptases and RNA polymerases which are inhibited by tRNA 4-6. The purified RNA from surgical specimens are labeled using standard protocol and hybridized to a microarray chip and the results are analyzed using two complementary methods, the false discovery rate method, and using a novel method based on mutual information and visualized on the web-based server Retinobase 7,8.
Protocol
1. jouRNAl: Procedure to Recover the Specimens from the Surgical Block
In the lab
Get an importation contract from express shipping company.
Fill 10 (or 25) shipping forms with the postal address of the laboratory indicating the contact person in the laboratory (Phone number and Email address).
Prepare 10 (or 25) samples forms numbered 1 to 10 (or 25). These forms include dedicated spaces to inform on a) anonymous identification of the patient, b) the date of the surgery and c) any additional remarks the surgeon would like to add. Prepare 10 (or 25) padded envelopes (150 x 210 mm).
Prepared using RNAse-free reagents 25 (or 65) ml of 6 M guanidine chloride in diethylpyrocarbonate (DEPC)-treated H2O (GHCl).
Fill 10 or 25 numbered 5 ml sterile polyethylene round bottom tubes with 2.4 ml of GHCl solution.
Used argon gas to fill the top part of these tubes, than press tightly on the cap to prevent the oxidation of the GHCl solution over several years of storage in the surgical cabinet.
Introduce every 5 ml tubes in a 50 ml sterile polypropylene conical bottom tube with screw cap. Use a piece of clean paper tissue to hold the 5 ml tube into the 50 ml tube, close the tube.
Place the 50 ml tubes on a polystyrene rack and the rack into a cardboard box with the padded envelopes, the shipping forms, the samples forms.
Paste the instructions (see : 1.12-1.19) inside of the cover of the cardboard box to facilitate their reading.
Send the cardboard box by mail to the contact person in the hospital.
In the hospital
Bring the cardboard box from the surgical cabinet to the surgical room. Caution note: if the procedure involves an immune compromised patient, the cardboard should not be brought to the surgical room.
During the surgery, place the retinal specimen into numbered 5 ml sterile polypropylene filled with GHCl solution. Close tightly the tube.
Return the tube briefly.
Place the tube on the blood tube rocker for 10 min.
Fill in the accompanying sample numbered form.
Report the identification number onto the corresponding numbered 50 ml tube.
Introduce the 5 ml tube with the surgical specimen into the 50 ml tube, replace paper tissue and screw the tube.
Introduce the tube and the filled sample form into it in one of the accompanying padded envelopes. Close it.
Call the express shipping company for pick up.
2. RNA Purification
In the lab
Homogenize the specimen in its 5 ml tube polypropylene with GHCl solution with a homogenizer for 1 min while moving the tube up and down.
Add 270 μl of 2 M potassium acetate pH 5.0. Shake vigorously for 10 min by placing the tube on a shaker in its horizontal position (420 min-1).
Centrifuge 10 min at 5,000 rpm (6,500 x g) at 20 °C.
Aspirate smoothly the soluble fraction without disturbing the pellet.
Transfer into a 14 ml sterile tube.
Add 5.3 ml of 100 mM Tris pH 8.0, N-Lauroylsarcosine 1%.
Add 3.2 g of cesium chloride (CsCl) and mix the tube by vortexing.
Add 1.8 ml of CsCl/ ethylenediaminetetraacetic acid (EDTA) in a sterile 11 ml polyallomer centrifuge tube.
With a 10 ml sterile Pasteur pipette, transfer the RNA solution onto 1.8 ml CsCl/EDTA by sliding slowly on the edge of the tube to avoid disturbing the density cushion.
Place the tubes (a second tube containing the buffers without retina if necessary) into the rotor.
Centrifuge 24 hr at 32,000 rpm (225,000 x g) at 20 °C.
Remove the superior part of the solution with a sterile Pasteur pipette, and discard it.
Remove gradually while checking the moment when the DNA (viscous) is aspirated with a second sterile Pasteur pipette, and discard it.
Remove the remaining solution taking care not to release the RNA pellet with a third sterile Pasteur pipette.
Section the bottom of the tube with a scalpel flame-sterilized, then put the remaining part of the tube it upside down on a sterile gaze.
Reverse the tube and rinse delicately with 160 μl of GHCl.
Let the pellet dry for 10 min.
Resuspend the pellet in 150 μl of (10 mM Tris pH 7.5 - 1 mM EDTA - 0.1% SDS).
Transfer the solution into a 2 ml sterile microcentrifuge tube, then harvest the residual pellet with 30 μl of (10 mM Tris pH 7.5 - 1 mM EDTA - 0.1% SDS).
Add 150 μl of (10 mM Tris pH 7.5 - 1 mM EDTA).
Add 30 μl of 3 M sodium acetate pH 5.0, vortex the tube.
Add 900 μl of ethanol 100% (-20 °C), vortex the tube.
Place the tube 30 min in melting ice.
Centrifuge the tube 30 min at 15,000 rpm at 4 °C.
Aspirate delicately the soluble fraction, and discard it.
Add 500 μl 70% ethanol (RT), vortex the tube.
Centrifuge the tube 20 min at 15,000 rpm (18,000 x g) at 4 °C.
Repeat the rinsing step (70% ethanol).
Centrifuge briefly and eliminate the remaining ethanol with a P200 pipette.
Let the pellet air dry for 10 min.
Resuspend the pellet in 50 μl DEPC-treated H2O.
Mix vigorously by vortexing.
Incubate 15 min at 45 °C in a water bath.
3. RNA Analysis by Gel Electrophoresis
Pour an agarose gel inside a chemical hood.
In a sterile 1.5 ml microcentrifuge tube, add 2 μl of RNA to be analyzed and 6.4 μl of sample prep buffer.
In a second 1.5 ml tube, add 3 μl of RNA standards and 9.6 μl of sample prep buffer.
Heat the tubes 15 min at 65 °C, put them on ice.
Add 1 μl ethidium bromide (EB) loading buffer without dye in the RNA sample tube and 1 μl EB loading buffer with dye in the RNA standards tube.
Run the gel under 80 - 100 V Inside a chemical hood in running buffer, until one of the dyes (bromophenol blue) reaches 2/3 of the bottom of the gel.
Rinse the gel twice 15 min with 250 ml de DEPC-treated 2x SSC.
Take a digitalized image under UV illumination.
Calculate the ratio between the upper band (23S rRNA) and the lower band (16S rRNA).
4. Final Purification of the RNA
Adjust the volume of the RNA sample to 100 μl with DEPC-treated H2O (if necessary).
Add 100 μl of Acid phenol (1 ml phenol saturated in DEPC-treated H2O + 130 μl of 50 mM of sodium acetate, pH 5.2), vortex vigorously.
Centrifuge the 10 min at 13,000 rpm (15,000 x g) at room temperature.
Recover carefully and transfer the aqueous phase (upper phase) into a novel a sterile 1.5 ml microcentrifuge tube.
Add 10 μl of 3 M sodium acetate, pH 5.2 and 250 μl of ethanol 100% (-20 °C).
Incubate the tube 30 min into melting ice.
Centrifuge the tube 30 min at 14,000 rpm (18,000 x g) at 4 °C.
Aspirate carefully the soluble fraction, and discard it.
Add 500 μl of 70% ethanol (room temperature), and vortex the tube.
Centrifuge the tube 20 min at 14,000 rpm (18,000 x g) at 4 °C.
Repeat the rinsing step (70% ethanol).
Centrifuge briefly and eliminate the remaining ethanol with a P200 pipette.
Let air dry the pellet for 10 min.
Resuspend the pellet into 50 μl of DEPC-treated H2O.
Incubate then tube 15 min at 45 °C.
Measure the absorbance (Abs) at 260 nm and 280 nm on 2 μl of the RNA sample. Calculated the ratio Abs260/Abs280.
Store the RNA sample at -80 °C (stable for several years).
5. Solutions and Cleaning Procedures
Potassium acetate 2 M, pH 5.0: Dissolve 19.6 g Potassium acetate into 70 ml of DEPC-treated H2O. Clean the pH meter probe by soaking it into with 1M NaOH for 15 min, followed by 5 rinses with DEPC-treated H2O. Adjust to pH 5.0 with acetic acid then adjust volume to 100 ml with DEPC-treated H2O.
Tris-HCl 100 mM, pH 8.0, N-Lauroylsarcosine 1%: Dissolve 2 g of N-Lauroylsarcosine (under a hood) in to 40 ml of 0.5 M Tris-base pH 8.0. Adjust volume to 200 ml with DEPC-treated H2O.) CsCl/EDTA: 96 g of CsCl in 50 ml 10 mM EDTA pH 7.5 prepared with DEPC-treated H2O. Autoclave and adjust volume to 100 ml with DEPC-treated H2O. Check for pH<7.5.
Agarose gel 1%: Put 1 g of agarose in 62 ml DEPC-treated H2O. Boil in a microwave oven. Add 18 ml of 12.3 M formaldehyde, 20 ml of 5x MOPS. Pour inside a chemical hood.
Sample prep Buffer: To be prepared extemporary. Mix inside a chemical hood 8 μl of 5x MOPS, 16 μl of 12.3 M formaldehyde and 40 μl of formamide.
EB loading buffer (without dye): To be prepared extemporary. Add 4 μl 10 mg/ml ethidium bromide into 20 μl of 80% glycerol prepared with DEPC-treated H2O.
EB loading buffer (with dye): To be prepared extemporary. Add 2 μl 10 mg/ml ethidium bromide into 10 μl of 80% glycerol containing 0.25% xylen cyanol, 0.25% bromophenol blue with DEPC-treated H2O.
Running buffer: To 60 ml 5x MOPS, add 54 ml of 12.3 M formaldehyde and 186 ml of DEPC-treated H2O.
DEPC-treated H2O: Incubate distilled water with 0.1% v/v diethylpyrocarbonate for 2 hr at 37 °C with stirring. Sterilize by Autoclave.
Cleaning the homogenizer: Add 25 ml of cleaning solution (RBS) to 1 L of DEPC-treated H2O, and immerge the axis 1 min with stirring. Incubate 2 hr at 60 °C. Rinse thoroughly with DEPC-treated H2O Wrap the instrument into aluminum and put into the instrument box. Wrap the box. Sterilize by Autoclave.
Cleaning the electrophoresis device: Wash the apparatus, the tray and the 15-wells comb. Incubate 15 min in 3% hydrogen peroxide. Rinse once with 100% ethanol. Air dry.
Cleaning the glass dishes: Clean the glass dishes RBS 2% all the night, then rinse with distilled water before sterilization at 200 °C.
Representative Results
The procedure jouRNAl (Figure 1) allows us recover specimens of retina from the surgical block to purified RNA, and to analyze the transcriptome of retinal detachment. Retinal detachment results in the upregulation of the monocyte chemotactic MCP1 gene CCL2, and in the decrease in expression of the rod photoreceptor transducing gene GNAT1, the short wave cone opsin OPN1SW, and the homeogene CRX (Figure 2). The induction of CCL2 is resulting from inflammation 9, while the concomitant reduction of GNAT1, OPN1SW and CRX is the result of photoreceptor degeneration, both rods and cones. The loss of cones may result from the loss of expression of NXNL1, which encodes for a Rod-derived Cone Viability Factor 7,10, or its paralogue RdCVF2, which is encoded by the NXNL2 gene. Surprisingly, the NXNL2 messenger exists in two different versions. Version 1 (NM_001161625.1) is a coding sequence derived from phylogenic analysis but has not been previously reported to be expressed 11, while version 2 (NM_145283.2), for which several ESTs has been identified is an abnormal mRNA that excludes the second exon of the gene and contains a alternative second exon, containing a repetitive Alu sequence, located more than 40 kb in the 3' direction (Figure 3a). Using RNA purified from Human retina, we can now reported that the two versions of the NXNL2 mRNA are expressed (Figure 3b).
 Figure 1. Picture of the cardboard box containing the material provided by jouRNAl.
Figure 1. Picture of the cardboard box containing the material provided by jouRNAl.
 Figure 2. Representation of the expression of a subset of genes using Retinobase. For the genes displayed in these radar graphs, CCL2, GNAT1, OPN1SW, and CRX, the right part of the figure corresponds to RNA from specimens of retinal detachments (RD1-18), while the left part (NR1-18) are RNA from age-matched controls prepared using post-mortem retinas. The radar graph method is described in 8.
Figure 2. Representation of the expression of a subset of genes using Retinobase. For the genes displayed in these radar graphs, CCL2, GNAT1, OPN1SW, and CRX, the right part of the figure corresponds to RNA from specimens of retinal detachments (RD1-18), while the left part (NR1-18) are RNA from age-matched controls prepared using post-mortem retinas. The radar graph method is described in 8.
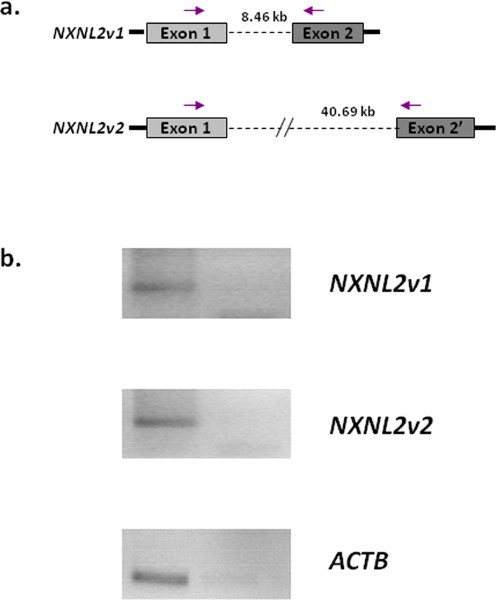 Figure 3. Expression of the two version of the NXNL2 gene in the retina. a. Schematic representation of the NXNL2 gene on chromosome 9. NXNL2v1 has two exons that are predicted by multiple alignment and phylogenic analysis. NXNL2v2 is missing that second exon and includes an alternative exon 2', located >40 kb in the 3' direction. The arrows show the position of the primer used. b. RT-PCR showing the expression of both NXNL2v1 and NXNL2v2 in the retina. The right lanes correspond to reaction in the absence of reverse transcriptase. ACTB, cytoplasmic actin. Primers used: NXNL2v1: 5'-GCATGAGCTGAGGAAGAGGT-3', 5'-CTCA AACGGAGAAATTCTGGA-3', NXNLv2: 5'-TCTGCACCCCCACGTTTATT-3', 5'-AGGGCCTCCT TTTCCATCTA-3'.
Figure 3. Expression of the two version of the NXNL2 gene in the retina. a. Schematic representation of the NXNL2 gene on chromosome 9. NXNL2v1 has two exons that are predicted by multiple alignment and phylogenic analysis. NXNL2v2 is missing that second exon and includes an alternative exon 2', located >40 kb in the 3' direction. The arrows show the position of the primer used. b. RT-PCR showing the expression of both NXNL2v1 and NXNL2v2 in the retina. The right lanes correspond to reaction in the absence of reverse transcriptase. ACTB, cytoplasmic actin. Primers used: NXNL2v1: 5'-GCATGAGCTGAGGAAGAGGT-3', 5'-CTCA AACGGAGAAATTCTGGA-3', NXNLv2: 5'-TCTGCACCCCCACGTTTATT-3', 5'-AGGGCCTCCT TTTCCATCTA-3'.
Discussion
The development of a procedure for tissue recovery from the surgical block has been essential to the transcriptome analysis of retinal detachment. One should notice that this type of surgery is practiced in emergency and that the ophthalmologists operating have little time to participate in a biological research program when they operate. This retinectomy is also performed stochastically in each service, so that the easier way to reach statistical numbers is to work with a network. In such network, the standardization of the tissues collection is essential the success of the biological analysis. By providing a material, very easy to use and precise instructions, that can be stored at room temperature in a surgery cabinet, close to the surgical block, we have encouraged the surgeons to participate in our study.
In addition, the standardization of the purification of the RNA was achieved to get the best of these precious clinical specimens. The collections of pure RNAs can be stored for years at -80 °C and would be used for further studies as novel genomics technologies emerge. The protocol is provided for retinal surgical specimen but can also be used successfully to isolate RNA from rodent retina after dissection. If the RNA are degraded, a) check the pH of the CsCl/EDTA solution. b) make all the solutions from unused chemicals. The main limitation of the technique is the quantity of starting material which should correspond to at least 50,000 cells.
What distinguishes the method used here from most commercial reagents provided to isolate total RNA is the degree of purity. RNA is depleted of any DNA contamination which eliminates the need of using DNAse treatment that can be damaging to any further procedure. In addition, the total RNA preparations are here depleted in tRNA, that are known to be potent inhibitors of RNA polymerases that are commonly used in the amplification step before hybridization to microarray chips. We have observed that the probes synthesized from these RNA preparations have a very high specific activity. The degree of purity of the RNA prepared following the method described here is very well suited for microarray hybridization, but also for constructing cDNA libraries of high quality and for RNA sequencing as we have observed. The laboratory should be RNAse free. The pH of the CsCl/EDTA should be acidic to avoid the degradation of the RNA by alkaline lysis. The density of the CsCl/EDTA should be carefully verified in order not to be too high and to prevent the sedimentation of RNA.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Sacha Reichman and Dominique Santiard-Baron for their help in editing the RNA purification protocol.
References
- Mitry D, Charteris DG, et al. The epidemiology of rhegmatogenous retinal detachment: geographical variation and clinical associations. The British Journal of Ophthalmology. 2010;94(6):678. doi: 10.1136/bjo.2009.157727. [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glisin V, Crkvenjakov R, et al. Ribonucleic acid isolated by cesium chloride centrifugation. Biochemistry. 1974;13(12):2633. doi: 10.1021/bi00709a025. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN, von Zastrow ME, et al. Amplified RNA synthesized from limited quantities of heterogeneous cDNA. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(5):1663. doi: 10.1073/pnas.87.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalieri LF, Yamaura IE. coli tRNAs as inhibitors of viral reverse transcription in vitro. Nucleic Acids Research. 1975;2(12):2315. doi: 10.1093/nar/2.12.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawadogo M. On the inhibition of yeast RNA polymerases A and B by tRNA and alpha-amanitin. Biochemical and biophysical research communications. 1981;98(1):261. doi: 10.1016/0006-291x(81)91897-0. [DOI] [PubMed] [Google Scholar]
- Delyfer MN, Raffelsberger W, et al. Transcriptomic analysis of human retinal detachment reveals both inflammatory response and photoreceptor death. PloS ONE. 2011;6(12):e28791. doi: 10.1371/journal.pone.0028791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalathur RK, Gagniere N, et al. RETINOBASE: a web database, data mining and analysis platform for gene expression data on retina. BMC Genomics. 2008;9:208. doi: 10.1186/1471-2164-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Hisatomi T, et al. Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(7):2425. doi: 10.1073/pnas.0608167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveillard T, Sahel JA. Rod-derived cone viability factor for treating blinding diseases: from clinic to redox signaling. Science Translational Medicine. 2010;2(26):26ps16. doi: 10.1126/scitranslmed.3000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmel F, Leveillard T, et al. Rod-derived Cone Viability Factor-2 is a novel bifunctional-thioredoxin-like protein with therapeutic potential. BMC Molecular Biology. 2007;8:74. doi: 10.1186/1471-2199-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]