

Abstract
It has been traditionally difficult to measure the thermodynamic stability of membrane proteins because fully reversible protocols for complete folding these proteins were not available. Knowledge of the thermodynamic stability of membrane proteins is desirable not only from a fundamental theoretical standpoint, but is also of enormous practical interest for the rational design of membrane proteins and for optimizing conditions for their structure determination by crystallography or NMR. Here, we describe the design of a fully reversible system to study equilibrium folding of the outer membrane protein A from Escherichia coli in lipid bilayers. Folding is shown to be two-state under appropriate conditions permitting data analysis with a classical folding model developed for soluble proteins. The resulting free energy and m value, i.e., a measure of cooperativity, of unfolding are 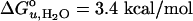 and m = 1.1 kcal/mol M–1, respectively, in a reference bilayer composed of palmitoyl-oleoyl-phosphatidylcholine (C16:0C18:1PC) and palmitoyloleoyl-phosphatidylglycerol (C16:0C18:1PG). These values are strong functions of the lipid bilayer environment. By systematic variation of lipid headgroup and chain composition, we show that elastic bilayer forces such as curvature stress and hydrophobic mismatch modulate the free energy and cooperativity of folding of this and perhaps many other membrane proteins.
and m = 1.1 kcal/mol M–1, respectively, in a reference bilayer composed of palmitoyl-oleoyl-phosphatidylcholine (C16:0C18:1PC) and palmitoyloleoyl-phosphatidylglycerol (C16:0C18:1PG). These values are strong functions of the lipid bilayer environment. By systematic variation of lipid headgroup and chain composition, we show that elastic bilayer forces such as curvature stress and hydrophobic mismatch modulate the free energy and cooperativity of folding of this and perhaps many other membrane proteins.
The material elastic properties of biological membranes such as curvature stress and bilayer deformation from hydrophobic mismatch have emerged as important functional modulators of ion channels, receptors, and other integral membrane proteins (1–5). These properties are also expected to modulate the thermodynamic stability of membrane proteins because the membrane environment imposes very different mechanical constraints (6, 7) on membrane proteins than water does on soluble proteins (8). A thorough understanding of the factors that determine the stability of membrane proteins is of fundamental theoretical interest to understand the forces that shape these proteins (9). Accurate assessments of their thermodynamic stability will also aid in the design of more stable membrane proteins for therapeutic applications and for structural studies of this structurally underrepresented class of proteins. For example, overcoming conformational dynamics has been a major breakthrough in the recent determination of the structure of lactose permease (10) and superstable mutants of diacylglycerolkinase have dramatically improved crystallization and NMR conditions (11, 12). Unfortunately, quantitative studies of membrane protein stability have been hampered by difficulties to completely and reversibly unfold these proteins (13, 14). We have now designed a fully reversible system to study equilibrium folding of membrane proteins in lipid bilayers, by using the outer membrane protein A (OmpA) from Escherichia coli as a model. OmpA is an abundant protein of the outer membranes of Gram-negative bacteria, where it serves a structural role and also functions as a phage and colicin receptor. The eight-stranded β-barrel structure of the N-terminal transmembrane domain (residues 1–177) has been solved by x-ray crystallography (15) and NMR (16). Denatured OmpA in solution spontaneously refolds into lipid bilayer membranes after dilution of denaturants (17), and the kinetics of refolding of OmpA have been shown to depend on the composition, thickness, and overall curvature of the lipid bilayer (18). We demonstrate here that OmpA folding into lipid bilayers is a reversible two-state process that can be quantitatively described by the free energy of unfolding and the cooperativity parameter m, which both depend on the mechanical properties of the host lipid bilayer. We also discuss how our approach using a β-barrel model protein may be generalized to analyze the thermodynamic stability of α-helical membrane proteins.
Methods
Protein Expression and Purification. OmpA was isolated and purified in the fully unfolded form in 8 M urea from the outer membranes of E. coli as described (17).
Small Unilamellar Vesicles (SUVs). Lipid stocks (Avanti Polar Lipids) dissolved in chloroform were mixed to yield the desired compositions. Total lipid (12 μmol) was dried on the bottom of a glass test tube under a stream of nitrogen and further in a desiccator under a high vacuum overnight. The lipid films were then dispersed in 10 mM glycine buffer (pH 10.0) containing 2 mM EDTA to a final lipid concentration of 10 mM. The lipid dispersions were sonicated for 50 min by using a Branson ultrasonifier microtip at 50% duty cycle. SUVs were equilibrated over night before use. The average diameter of the SUVs determined by dynamic light scattering (Protein Solutions DynaPro) was 30 nm.
Equilibrium Unfolding and Refolding of OmpA. Concentrated OmpA in urea was diluted ≈100-fold into SUVs and incubated for 3 h at 37.5°C for refolding. The final OmpA concentration and lipid-to-protein molar ratios were 12 μM and 800, respectively, in all experiments. The refolded protein–lipid complex was divided into 20–24 aliquots. Appropriate amounts of a freshly made 10 M urea stock solution and a buffer solution were added to the aliquots, which were further diluted 10-fold for fluorescence or 2.5-fold for SDS/PAGE equilibrium unfolding experiments. These reactions were incubated overnight at 37.5°C. For equilibrium refolding measurements by SDS/PAGE, aliquots of 4 mM SUVs were preequilibrated at appropriate urea concentrations and small amounts of OmpA were added to each sample to yield the same concentrations as in the unfolding experiments. The refolding reactions were incubated overnight at 37.5°C.
SDS/PAGE. The equilibrated samples were loaded on SDS/12.5% PAGE without boiling. The reactions were terminated by 1:1 mixing of the samples with the treatment buffer at room temperature (17).
Fluorescence and CD Spectroscopy. Fluorescence spectra were collected in SPEX Fluoromax or Fluorolog spectrofluorometers. The excitation wavelength was 290 nm and fluorescence was measured in the 300–400 nm wavelength range at a scan rate of 0.15 nm/s using 6-nm slits. Far-UV CD spectroscopy was performed on an Aviv 215 spectropolarimeter. The protein and lipid concentrations were the same as in the SDS/PAGE experiments. All spectra were corrected for light scattering with reference samples of identical composition and concentration, but without protein.
Airfuge Flotation Assay. Discontinuous sucrose gradients with 30% (30 μl), 25% (40 μl), 20% (40 μl), 10% (40 μl) in pH 10.0 glycine buffer containing 6 M urea were carefully prepared in Airfuge tubes from bottom to top. Three different 50-μl samples in 6 M urea and 30% sucrose were placed at the bottom: 2.4 mM SUVs with 0.1 mol% [7-nitro-2–1,3-benzoxadiazol-4-yl] (NBD)-diC18:1PE, 3 μM OmpA, and 3 μM OmpA that had been refolded in 2.4 mM SUVs. The tubes were spun in a Beckman Airfuge at 25 psi for 50 min. Thirty-two-microliter fractions were taken from each tube and diluted with 93 μl of 8 M urea. Each fraction was analyzed for NBD fluorescence by the excitation at 460 nm and emission at 532 nm and for OmpA fluorescence at 290 nm and 352 nm, respectively.
Fitting of Unfolding Curves. Fluorescence spectra were parameterized by calculating an average emission wavelength, 〈λ〉 defined as 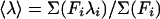 (19). λi and Fi are the wavelength and the corresponding fluorescence intensity at the ith measuring step in the spectrum. The unfolding curves, 〈λ〉 vs. [urea], were fitted to the following form of the two-state model
(19). λi and Fi are the wavelength and the corresponding fluorescence intensity at the ith measuring step in the spectrum. The unfolding curves, 〈λ〉 vs. [urea], were fitted to the following form of the two-state model
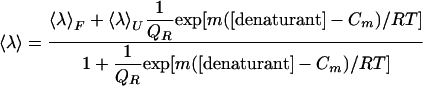 |
[1] |
using Wavemetrics igorpro software. 〈λ〉F and 〈λ〉U are the average emission wavelengths of the folded and unfolded states, respectively, determined from linear extrapolations to 0 M urea of the plateau values of the two states. Cm is the urea concentration where the fractions of folded and unfolded states are equal. QR is the relative ratio of the total fluorescence intensity of the native state to that of the unfolded state and is needed for normalization when one uses 〈λ〉's to represent species concentrations (19). The free energy of unfolding is obtained from the fitted values of Cm and m:
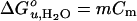 |
[2] |
Results and Discussion
Previous kinetic studies showed that urea-denatured OmpA can be refolded in bilayers of small and large unilamellar vesicles composed of a large variety of lipids and that folding into SUVs is faster than into large unilamellar vesicles (LUVs) (18). For thick bilayers, the insertion kinetics become prohibitively slow in LUVs, but are still experimentally accessible in SUVs. Therefore, we are using here equilibrated SUVs to assess equilibrium folding and to study effects of bilayer mechanics on the thermodynamic stability of OmpA. To test the reversibility of folding, OmpA was unfolded and refolded in SUVs composed of 92.5% palmitoyl-oleoyl-phosphatidylcholine (C16:0C18:1PC) and 7.5% palmitoyl-oleoyl-phosphatidylglycerol (C16:0C18:1PG) by variation of the urea concentration in pH 10 buffer at 37.5°C. To estimate the fraction of folded OmpA, we used the fact that without sample boiling, native OmpA migrates in SDS gels as a 30-kDa form, but as a 35-kDa form when fully or partially denatured (20). Fig. 1A shows that unfolding and refolding reactions produce the same gel migration patterns and, together with many similar experiments under different conditions, indicates that the reaction is at equilibrium. The midpoint of the transition occurs at 3.2 M urea. The denatured state in the presence of lipid vesicles is indistinguishable from the denatured state in 6–8 M urea as shown by Trp fluorescence (Fig. 2A) and CD spectroscopy (Fig. 2B). A total of 7.5% of negatively charged C16:0C18:1PG was included to facilitate complete dissociation of denatured OmpA from the membranes (Fig. 2C). OmpA has a calculated pI of 5.6, and therefore is negatively charged at high pH. Denaturation and dissociation was incomplete in pure C16:0C18:1PC bilayers (data not shown). Full thermodynamic reversibility was observed in the pH range 7.0–10.0, but dissociation of the denatured state from the membrane was favored at higher pH values (data not shown). The bilayer structure itself is maintained in high concentrations of urea as has been demonstrated by x-ray diffraction and [31P]NMR (21, 22). Lipid bilayers are quite permeable to urea (23). Therefore, we do not expect the bilayers to be subject to osmotic pressure or tension under equilibrium condititions.
Fig. 1.

Reversibility and two-state behavior of OmpA folding in lipid bilayer membranes composed of 92.5% C16:0C18:1PC and 7.5% C16:0C18:1PG at pH 10 and 37.5°C. (A) Comparison of equilibrium unfolding and refolding of OmpA monitored by SDS/PAGE without sample boiling. The 30-kDa form represents the native state, and the 35-kDa form represents denatured states. Transition midpoints are indicated with arrows. (B) Comparison of the urea-induced equilibrium unfolding of OmpA in lipid bilayers monitored by SDS/PAGE (open circles) and Trp fluorescence (filled circles). (C) Same as B, but with increasing mol fractions of the short-chain lipid diC12PC. For clarity, successive curves are each shifted on the abscissa by +1 M urea.
Fig. 2.

Characterization of denatured states of OmpA. (A) Fluorescence spectra of native membrane (C16:0C18:1PC/PG, 92:5:7.5)-inserted OmpA (solid line), denatured OmpA obtained by treating native membrane-inserted OmpA with 8 M urea (dashed line), and denatured OmpA in the absence of lipids (dotted line). The emission maxima of the native and denatured states were 333.6 nm and 353.6 nm, respectively. (B) Far-UV CD spectra of native membrane-inserted OmpA (solid line), denatured OmpA obtained by treating native membrane-inserted OmpA with 6 M urea (dashed line), and denatured OmpA in the absence of lipids (dotted line). (C) Airfuge flotation assay to measure degree of membrane-association of denatured OmpA in 6 M urea. NBD-labeled vesicles in the absence (open circles) or presence (filled circles) of denatured OmpA float to the top, and denatured OmpA in the absence (open squares) or presence (filled squares) of vesicles remains at the bottom, indicating separation of denatured OmpA from the vesicles.
Previous kinetic studies indicate that OmpA refolds via several kinetic intermediates in essentially 0 M urea (17, 24). However, this does not imply anything about whether urea-induced equilibrium unfolding is a two-state or multistate process, which is important to know for further analysis of the equilibrium data. To assess whether equilibrium unfolding of OmpA is two- or multistate, unfolding was analyzed at the same lipid-to-protein ratio (800:1) by Trp fluorescence and SDS/PAGE, i.e., two parameters that report on a very early and a very late kinetic phase of the folding reaction, respectively (17, 24). As shown in Fig. 1B, the unfolded fractions obtained from SDS/PAGE and fluorescence coincide as expected for a two-state, but not a multistate, process. Two-state unfolding by solvent denaturation obeys the law
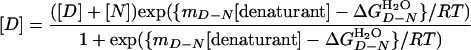 |
[3] |
where [D] and [N] are the concentrations of the denatured and native states, respectively, 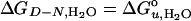 is the free energy of unfolding in water (0 M urea), and mD–N = –dΔGD–N/d[denaturant], which is commonly believed to be independent of the denaturant concentration. mD–N (henceforth abbreviated m) has been linked to the amount of protein area that becomes solvent-exposed upon unfolding and is also a measure of the cooperativity of unfolding (25, 26). When the data are analyzed with this model (see Methods), we obtain best fits for
is the free energy of unfolding in water (0 M urea), and mD–N = –dΔGD–N/d[denaturant], which is commonly believed to be independent of the denaturant concentration. mD–N (henceforth abbreviated m) has been linked to the amount of protein area that becomes solvent-exposed upon unfolding and is also a measure of the cooperativity of unfolding (25, 26). When the data are analyzed with this model (see Methods), we obtain best fits for 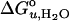 of 3.4 kcal/mol and an m value of 1.1 kcal/mol M–1.
of 3.4 kcal/mol and an m value of 1.1 kcal/mol M–1.
Based on the reversible folding system developed, the effects of lipid composition and bilayer thickness on OmpA stability were investigated by increasing the fraction of the lipids having various molecular structures as guest lipids at a fixed fraction (7.5%) of C16:0C18:1PG. The remaining fraction was filled with the reference lipid C16:0C18:1PC. First, we examined the effect of short saturated acyl chain PCs with chain lengths ranging from 10 to 14 carbons on the stability of OmpA. Representative transition curves for the example of diC12PC measured by SDS/PAGE and fluorescence are displayed in Figs. 1C and 3A. Two-state behavior and reversibility was maintained in all cases. The resulting values of 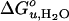 and m decreased proportionally to the amount of the guest lipid fraction in the bilayer (Fig. 4 A and B). This effect was most pronounced for the shortest acyl chain lipid diC10PC and least pronounced for diC14PC.
and m decreased proportionally to the amount of the guest lipid fraction in the bilayer (Fig. 4 A and B). This effect was most pronounced for the shortest acyl chain lipid diC10PC and least pronounced for diC14PC.
Fig. 3.

Representative unfolding curves measured by Trp fluorescence with increasing mol fractions of a short-chain saturated PC (A), a long-chain mono-unsaturated PC (B), a long-chain mono-unsaturated PE (C), and a double-unsaturated PC (D). The C16:0C18:1PG fraction was kept fixed at 7.5% in A–C and at 12.5% in D. The remainder of the bilayer was filled with C16:0C18:1PC. All experiments were carried out at 37.5°C.
Fig. 4.

Dependence of 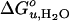 and m-value of OmpA unfolding on lipid composition. Effect of saturated and mono-unsaturated PCs and PEs on
and m-value of OmpA unfolding on lipid composition. Effect of saturated and mono-unsaturated PCs and PEs on 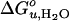 (A) and m (B). Effect of cis-double-unsaturated PCs on
(A) and m (B). Effect of cis-double-unsaturated PCs on 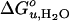 (C) and m (D). All experiments were carried out at 37.5°C.
(C) and m (D). All experiments were carried out at 37.5°C.
When a lipid with longer acyl chains (C18:0C18:1PC) was tested (Fig. 3B), the trend was reversed (Fig. 4 A and B). An even more dramatic increase of 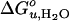 and m was found for another lipid (C16:0C18:1PE) with the same chain composition, but with a smaller polar headgroup than the reference lipid (Fig. 3C). Unsaturated lipids with PE headgroups are cone-shaped and therefore assume inverted hexagonal (HII) phases at higher temperatures (6). The investigated lipid mixtures containing C16:0C18:1PE still form bilayers at 37°C, but with an increased internal lateral pressure caused by intrinsic curvature stress (6, 27). These lipids also have larger area expansion and bending moduli than shorter saturated PCs (28). Including 30% C16:0C18:1PE in reference bilayers, increased
and m was found for another lipid (C16:0C18:1PE) with the same chain composition, but with a smaller polar headgroup than the reference lipid (Fig. 3C). Unsaturated lipids with PE headgroups are cone-shaped and therefore assume inverted hexagonal (HII) phases at higher temperatures (6). The investigated lipid mixtures containing C16:0C18:1PE still form bilayers at 37°C, but with an increased internal lateral pressure caused by intrinsic curvature stress (6, 27). These lipids also have larger area expansion and bending moduli than shorter saturated PCs (28). Including 30% C16:0C18:1PE in reference bilayers, increased 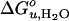 and m by up to 60% (Fig. 4 A and B).
and m by up to 60% (Fig. 4 A and B).
To further test the effect of lipid shape and internal membrane pressure on the thermodynamic stability of OmpA, a series of cis-double-unsaturated lipids (diC14:1PC to diC20:1PC), which induce smaller elastic moduli and larger curvature stresses in lipid bilayers (29), were examined (Fig. 3D). 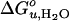 and m increased in the case of all these lipids, but contrary to the saturated lipid series, the lipids with the shortest acyl chains (diC14:1PC) induced the largest increases in
and m increased in the case of all these lipids, but contrary to the saturated lipid series, the lipids with the shortest acyl chains (diC14:1PC) induced the largest increases in 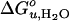 (Fig. 4 C and D).
(Fig. 4 C and D).
The values of 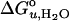 and m of all tested guest lipids exhibited a linear dependence on their mol fraction in the membrane. Therefore, each data set was linearly extrapolated to the point where the mol fraction of the guest lipid reached 100%. The extrapolated values for 100% of each examined lipid were plotted as a function of hydrophobic thickness (30) of the lipid bilayer (Fig. 5). Straight lines were obtained for the saturated and double-unsaturated PC series. The two lipids with a single cis double bond extended the range of the saturated series. The major differences between the two series are that the saturated lipids yielded a positive (337 cal/mol/Å) and the double-unsaturated lipids yielded a negative (–166 cal/mol/Å) slope and larger values of
and m of all tested guest lipids exhibited a linear dependence on their mol fraction in the membrane. Therefore, each data set was linearly extrapolated to the point where the mol fraction of the guest lipid reached 100%. The extrapolated values for 100% of each examined lipid were plotted as a function of hydrophobic thickness (30) of the lipid bilayer (Fig. 5). Straight lines were obtained for the saturated and double-unsaturated PC series. The two lipids with a single cis double bond extended the range of the saturated series. The major differences between the two series are that the saturated lipids yielded a positive (337 cal/mol/Å) and the double-unsaturated lipids yielded a negative (–166 cal/mol/Å) slope and larger values of 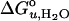 .
.
Fig. 5.

Dependence of 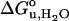 (A) and m-value (B) on the hydrophobic thickness of PC bilayers with saturated and mono-unsaturated acyl chains (filled circles) and cis-double-unsaturated acyl chains (open circles).
(A) and m-value (B) on the hydrophobic thickness of PC bilayers with saturated and mono-unsaturated acyl chains (filled circles) and cis-double-unsaturated acyl chains (open circles).
The free energies of transfer of unfolded OmpA into membranes coupled with folding are rather small (0 to –8 kcal/mol depending on lipid species). This is consistent with theoretical free energy estimates for α-helical (31) and β-barrel (32) membrane proteins from whole-residue hydropathy values taken from the thermodynamically derived Wimley–White (WW) hydropathy scale (31). A simple calculation of the free energies of transfer of the approximately 8 × 10 residues of OmpA that span the membrane using the augmented WW scale yields a value very close to zero (+0.95 kcal/mol) if one takes into account that two Arg–Glu pairs form salt bridges in the interior of the barrel (15). Because the scale only considers transfer of residues from water into the hydrophobic environment, we conclude that –1 to –9 kcal/mol of energy likely comes from folding. This is –13 to –110 cal/mol per residue, if 80 residues fold into the β-barrel. These estimates obviously are extremely crude because they are based on the sums of many differences of large numbers, which may compound small errors into large ones, and because contributions from tertiary structure and fine-tuned lipid interactions, such as those discussed below, are neglected. In view of many such possible complications that are almost impossible to predict, the relatively good agreement between experiment and prediction is remarkable.
To estimate the contribution of the hydrophobic effect to the increase of membrane protein stability with increasing membrane thickness, we calculated the hydrophobic surface area of OmpA from its crystal structure by rolling a probe of 1.4 Å radius over the hydrophobic surface (33). This surface, which should roughly represent the area of the protein/lipid interface, is 2158 Å2. Because our best estimate of the hydrophobic thickness of the OmpA transmembrane domain is 26 Å, the average hydrophobic perimeter is 83 Å. If fully developed, the hydrophobic effect is expected to contribute 25 cal/mol for each Å2 of hydrophobic contact (34). Therefore, 2,075 cal/mol should be gained for each Å of increased membrane thickness. This value is about six times larger than the experimentally found increment. Even if one uses smaller values of 15–20 cal/mol/Å2 as commonly assumed in protein folding (33), it is clear that the hydrophobic effect grossly overestimates the experimental value. There must be opposing forces, such as mechanical energy stored in the membrane due to hydrophobic mismatch at the protein/lipid interface (35). Lipids need to be stretched if too short and compressed if too long compared to the hydrophobic 26 Å of the protein. Both effects cost energy (up to ≈1.7 kcal/mol/Å according to our measurements). Because there is no discontinuity at ≈26 Å, the energy of lipid deformation seems to be symmetric, i.e., the energy costs of lipid compression and stretching are similar.
The 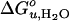 vs. dhydrophobic lines of the saturated and unsaturated lipids meet at ≈30 Å, i.e., at lipids with 20 carbon acyl chains. The increase of
vs. dhydrophobic lines of the saturated and unsaturated lipids meet at ≈30 Å, i.e., at lipids with 20 carbon acyl chains. The increase of 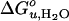 of the shorter unsaturated lipids is most likely the result of an increased lateral pressure exerted on the protein in the interior of the bilayer (6, 27). Bilayers of short-chain PCs with two cis double bonds exhibit increasingly larger negative spontaneous curvatures as their chains are shortened (36). Intrinsic curvature stress is stored in these bilayers. This stress is relieved when OmpA, which because of girdles of aromatic residues near both interfaces is hourglass-shaped, is incorporated into such bilayers. The stored curvature stress energy thus stabilizes the folded structure of OmpA. The largest stabilizing effect is observed with C16:0C18:1PE, which also exhibits the largest negative spontaneous curvature.
of the shorter unsaturated lipids is most likely the result of an increased lateral pressure exerted on the protein in the interior of the bilayer (6, 27). Bilayers of short-chain PCs with two cis double bonds exhibit increasingly larger negative spontaneous curvatures as their chains are shortened (36). Intrinsic curvature stress is stored in these bilayers. This stress is relieved when OmpA, which because of girdles of aromatic residues near both interfaces is hourglass-shaped, is incorporated into such bilayers. The stored curvature stress energy thus stabilizes the folded structure of OmpA. The largest stabilizing effect is observed with C16:0C18:1PE, which also exhibits the largest negative spontaneous curvature.
This study also reveals an interesting aspect of the m value, which is highly correlated with the degree of hydrophobic mismatch, the acyl chain cis-unsaturation, and 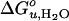 . Theories of the m value for water-soluble proteins agree that the parameter is proportional to the exposed hydrophobic surface area upon unfolding, ΔA, and the change in surface tension, dγ/d[denaturant] (25, 26)
. Theories of the m value for water-soluble proteins agree that the parameter is proportional to the exposed hydrophobic surface area upon unfolding, ΔA, and the change in surface tension, dγ/d[denaturant] (25, 26)
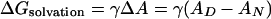 |
[4] |
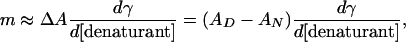 |
[5] |
where γ is the surface tension of the hydrophobic surface of a protein in water. Assuming that ΔA is little affected by the choice of lipids, we conclude that the observed m value changes are the result of lateral bilayer pressure changes, which in turn will change the γ of membrane proteins. Fig. 5B shows that m increases as the hydrophobic thickness and chain unsaturation of the bilayer increase. The former increases the local stress around the protein by lipid deformation (7, 35), and the latter increases intrinsic curvature stress and lateral pressure in the bilayer (6). This interpretation is consistent with and supports the interpretation of the previously discussed changes of 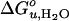 . As the saturated PC bilayers become very thin, the m values vanish and OmpA unfolds without dissociation from the surface of the bilayer (data not shown). In these cases, the two-state condition may no longer be valid and full sigmoidal curves are no longer observed (data not shown).
. As the saturated PC bilayers become very thin, the m values vanish and OmpA unfolds without dissociation from the surface of the bilayer (data not shown). In these cases, the two-state condition may no longer be valid and full sigmoidal curves are no longer observed (data not shown).
The absolute values of 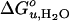 and m measured in SUVs may be shifted relative to those in LUVs because of additional curvature strain imposed by the smaller radius of SUVs compared to LUVs. However, the lipid-specific changes of these thermodynamic parameters should not be affected because they are measured relative to the reference C16:0C18:1PC/PG bilayer in SUVs and because the sizes of our vesicles measured by light scattering are independent of lipid composition (data not shown). Moreover, the effect of the overall macroscopic membrane curvature on even the absolute values of
and m measured in SUVs may be shifted relative to those in LUVs because of additional curvature strain imposed by the smaller radius of SUVs compared to LUVs. However, the lipid-specific changes of these thermodynamic parameters should not be affected because they are measured relative to the reference C16:0C18:1PC/PG bilayer in SUVs and because the sizes of our vesicles measured by light scattering are independent of lipid composition (data not shown). Moreover, the effect of the overall macroscopic membrane curvature on even the absolute values of 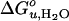 is probably small because the free energies of binding of hydrophobic 20- to 30-residue peptides to lipid bilayers are not much affected by macroscopic membrane curvature, even if enthalpies and entropies depend on curvature in a compensatory fashion (37).
is probably small because the free energies of binding of hydrophobic 20- to 30-residue peptides to lipid bilayers are not much affected by macroscopic membrane curvature, even if enthalpies and entropies depend on curvature in a compensatory fashion (37).
Fig. 6 schematically depicts how elastic bilayer properties modulate folding and unfolding of OmpA. When OmpA is under the elastic stress of bilayer forces, it unfolds completely and is fully excluded from the bilayer. In addition to the hydrophobic effect, intrinsic lipid shape-induced bilayer curvature stress and lipid deformation due to hydrophobic mismatch contribute significantly to 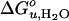 and m. However, OmpA incorporated in very thin lipid bilayers unfolds incompletely with smaller values of
and m. However, OmpA incorporated in very thin lipid bilayers unfolds incompletely with smaller values of 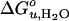 and m. It is likely that the protein assumes multiple fluctuating and partially membrane-associated conformations under these conditions. These insights into the stability of membrane proteins are likely general and also hold true for other membrane proteins, including helical ones, which because of their limited solubility in urea and guanidine hydrochloride are more difficult to study by these methods. However, other combinations of denaturants that may include strong detergents such as SDS might be suitable for similar studies on helical membrane proteins (27, 38). Our studies using the OmpA/urea model system suggest that nonspecific physical bilayer interactions and lipid packing are more important factors determining the stability of membrane proteins than specific chemical lipid interactions.
and m. It is likely that the protein assumes multiple fluctuating and partially membrane-associated conformations under these conditions. These insights into the stability of membrane proteins are likely general and also hold true for other membrane proteins, including helical ones, which because of their limited solubility in urea and guanidine hydrochloride are more difficult to study by these methods. However, other combinations of denaturants that may include strong detergents such as SDS might be suitable for similar studies on helical membrane proteins (27, 38). Our studies using the OmpA/urea model system suggest that nonspecific physical bilayer interactions and lipid packing are more important factors determining the stability of membrane proteins than specific chemical lipid interactions.
Fig. 6.

Cartoon depicting structures and bilayer forces acting on OmpA folding/unfolding under equilibrium conditions. When folding into most bilayers (left path), the process is two-state. The large black arrows indicate lateral bilayer pressure imparted on the lipid/protein interface in the hydrophobic core (red) of bilayers composed of lipids with negative intrinsic spontaneous curvature. Increasing this pressure increases the thermodynamic stability of the protein. The small black arrows indicate lipid deformation forces caused by hydrophobic mismatch between the protein and unstressed bilayers. These forces decrease the thermodynamic stability of the protein. When folding into thin bilayers (right path), the process is multistate, i.e., at least one equilibrium intermediate occurs. Water molecules penetrate more easily into the hydrophobic core (blue arrows) of more flexible and more dynamic thin bilayers, stabilizing equilibrium intermediates, decreasing the m- and 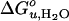 values until, in very thin bilayers composed of saturated lipids, complete unfolding (second step) can no longer be observed under any of our experimental conditions.
values until, in very thin bilayers composed of saturated lipids, complete unfolding (second step) can no longer be observed under any of our experimental conditions.
Acknowledgments
We thank S. Gruner and S. H. White for helpful comments on an early version of the manuscript. We also thank Mr. D. Rinehart for his expert technical assistance and V. Kiessling for providing software for data analysis. This work was supported by National Institutes of Health Grant GM51329.
Abbreviations: OmpA, outer membrane protein A; LUV, large unilamellar vesicles; NBD, [7-nitro-2–1,3-benzoxadiazol-4-yl]; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PG, phosphatidylglycerol; SUV, small unilamellar vesicles.
See Commentary on page 3995.
References
- 1.Martinac, B., Adler, J. & Kung, C. (1990) Nature 348, 261–263. [DOI] [PubMed] [Google Scholar]
- 2.Perozo, E., Cortes, D. M., Sompornpisut, P., Kloda, A. & Martinac, B. (2002) Nature 418, 942–948. [DOI] [PubMed] [Google Scholar]
- 3.Keller, S. L., Berzrukov, S. M., Gruner, S. M., Tate, M. W., Vodyanoy, I. & Parsegian, V. A. (1993) Biophys. J. 65, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown, M. F. (1994) Chem. Phys. Lipids 73, 159–180. [DOI] [PubMed] [Google Scholar]
- 5.Navarro, J., Toivio-Kinnucan, M. & Racker, E. (1984) Biochemistry 23, 130–135. [DOI] [PubMed] [Google Scholar]
- 6.Gruner, S. M. (1987) Proc. Natl. Acad. Sci. USA 82, 3665–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantor, S. C. (1999) Biophys. J. 76, 2625–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dill, K. A. (1990) Biochemistry 29, 7133–7155. [DOI] [PubMed] [Google Scholar]
- 9.White, S. H., Ladokhin, A. S., Jayasinghe, S. & Hristova, K. (2001) J. Biol. Chem. 276, 32395–32398. [DOI] [PubMed] [Google Scholar]
- 10.Abramson, J., Smirnova, I., Kasho, V., Verner, G., Kaback, H. R. & Iwata, S. (2003) Science 301, 610–615. [DOI] [PubMed] [Google Scholar]
- 11.Zhou, Y. & Bowie, J. U. (2000) J. Biol. Chem. 275, 6975–6979. [DOI] [PubMed] [Google Scholar]
- 12.Oxenoid, K., Sönnichsen, F. D. & Sanders C. R. (2002) Biochemistry 41, 12876–12882. [DOI] [PubMed] [Google Scholar]
- 13.Stowell, H. B. & Rees, D. C. (1995) Adv. Protein Chem. 46, 279–311. [DOI] [PubMed] [Google Scholar]
- 14.Haltia, T. & Freire, E. (1995) Biochim. Biophys. Acta 1228, 1–27. [DOI] [PubMed] [Google Scholar]
- 15.Pautsch, A. & Schulz, G. E. (1998) Nat. Struct. Biol. 5, 1013–1017. [DOI] [PubMed] [Google Scholar]
- 16.Arora, A., Ablidgaard, F., Bushweller, J. H. & Tamm, L. K. (2001) Nat. Struct. Biol. 8, 334–338. [DOI] [PubMed] [Google Scholar]
- 17.Kleinschmidt, J. H. & Tamm, L. K. (1996) Biochemistry 35, 12993–13000. [DOI] [PubMed] [Google Scholar]
- 18.Kleinschmidt, J. H. & Tamm, L. K. (2002) J. Mol. Biol. 324, 319–330. [DOI] [PubMed] [Google Scholar]
- 19.Mann, C. J., Royer, C. A. & Matthews, C. R. (1993) Protein Sci. 2, 1853–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arora, A., Rinehart, D., Szabo, G. & Tamm, L. K. (2000) J. Biol. Chem. 275, 1594–1600. [DOI] [PubMed] [Google Scholar]
- 21.Feng, Y., Yu, Z.-W. & Quinn, P. (2002) Chem. Phys. Lipids 114, 149–157. [DOI] [PubMed] [Google Scholar]
- 22.Kleinschmidt, J. H. & Tamm, L. K. (2002) Biophys. J. 83, 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flinkelstein, A. (1976) J. Gen. Physiol. 68, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kleinschmidt, J. H., den Blaauwen, T., Driessen, A. J. M. & Tamm, L. K. (1999) Biochemistry 38, 5006–5016. [DOI] [PubMed] [Google Scholar]
- 25.Shortle, D. (1995) Adv. Protein Chem. 46, 217–247. [DOI] [PubMed] [Google Scholar]
- 26.Myers, J. K., Pace, C. N. & Scholtz, J. M. (1995) Protein Sci. 4, 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curran, A. R., Templer, R. H. & Booth, P. J. (1999) Biochemistry 38, 9328–9336. [DOI] [PubMed] [Google Scholar]
- 28.Marsh, D. (1996) Biochim. Biophys. Acta 1286, 183–223. [DOI] [PubMed] [Google Scholar]
- 29.Szule, J. A., Fuller, N. & Rand, R. P. (2002) Biophys. J. 83, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis, B. & Engelman, D. M. (1983) J. Mol. Biol. 166, 211–217. [DOI] [PubMed] [Google Scholar]
- 31.Jayasinghe, S., Hristova, K. & White, S. H. (2001) J. Mol. Biol. 312, 927–934. [DOI] [PubMed] [Google Scholar]
- 32.Wimley, W. C. (2002) Protein Sci. 11, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards, F. M. (1985) Methods Enzymol. 115, 440–464. [DOI] [PubMed] [Google Scholar]
- 34.Tanford, C. (1980) The Hydrophobic Effect (Wiley, New York).
- 35.Nielsen, C., Goulian, M. & Andersen, O. S. (1998) Biophys. J. 74, 1966–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tate, M. W. & Gruner, S. M. (1987) Biochemistry 26, 231–236. [DOI] [PubMed] [Google Scholar]
- 37.Seelig, J. (2002) Curr. Topics Membr. 52, 31–56. [Google Scholar]
- 38.Lau, F. W. & Bowie, J. U. (1997) Biochemistry 36, 5884–5892. [DOI] [PubMed] [Google Scholar]