

Abstract
Adipocyte precursor cells give raise to two major cell populations with different physiological roles: white and brown adipocytes. Here we demonstrate that the retinoblastoma protein (pRB) regulates white vs. brown adipocyte differentiation. Functional inactivation of pRB in wild-type mouse embryo fibroblasts (MEFs) and white preadipocytes by expression of simian virus 40 large T antigen results in the expression of the brown fat-specific uncoupling protein 1 (UCP-1) in the adipose state. Retinoblastoma gene-deficient (Rb–/–) MEFs and stem cells, but not the corresponding wild-type cells, differentiate into adipocytes with a gene expression pattern and mitochondria content resembling brown adipose tissue. pRB-deficient MEFs exhibit an increased expression of the Forkhead transcription factor Foxc2 and its target gene cAMP-dependent protein kinase regulatory subunit RIα, resulting in increased cAMP sensitivity. Suppression of cAMP-dependent protein kinase activity in Rb–/–MEFs blocked the brown adipocyte-like gene expression pattern without affecting differentiation per se. Immunohistochemical studies revealed that pRB is present in the nuclei of white but not brown adipocyte precursor cells at a developmental stage where both cell types begin to accumulate lipid and brown adipocytes express UCP-1. Furthermore, pRB rapidly undergoes phosphorylation upon cold-induced neodifferentiation and up-regulation of UCP-1 expression in brown adipose tissue. Finally, down-regulation of pRB expression accompanies transdifferentiation of white into brown adipocytes in response to β3-adrenergic receptor agonist treatment. We propose that pRB acts as a molecular switch determining white vs. brown adipogenesis, suggesting a previously uncharacterized function of this key cell cycle regulator in adipocyte lineage commitment and differentiation.
Adipose tissue comes in two colors, white and brown. White adipose tissue (WAT) stores energy, whereas brown adipose tissue (BAT) expressing the uncoupling protein 1 (UCP-1) has the ability to dissipate energy through adaptive thermogenesis (1, 2). Genetic ablation of BAT in mice causes obesity without hyperphagia, demonstrating the important energy-dissipating role of this tissue (3). Little is known about regulatory circuits controlling white vs. brown adipocyte differentiation, and the key regulators, peroxisome proliferator-activated receptor γ (PPARγ) and CCAAT/enhancer-binding proteins (C/EBPs), seem to play similar roles in white and brown adipose conversion (2). A number of coactivators have been implicated in the control of white vs. brown adipocyte differentiation (4, 5), with PPARγ coactivator 1α (PGC-1α) being a prime candidate regulator of brown adipogenesis. Interestingly, fat-specific overexpression of the Forkhead transcription factor Foxc2 in mice led to a lean phenotype with conversion of white fat depots toward a brown fat-like phenotype (6).
The retinoblastoma protein (pRB), encoded by the retinoblastoma gene (Rb), is the founding member of a protein family designated pocket proteins, which also includes p107 and p130. pRB is a central regulator of the mammalian cell cycle (7), and in addition, numerous studies have addressed the role of pRB in cellular differentiation (8). Mouse embryo fibroblasts (MEFs) lacking pRB are unable to undergo adipose conversion in response to treatment with standard adipogenic inducers (9). The defective differentiation of Rb–/– MEFs, however, can be bypassed by administration of a PPARγ ligand or by partial inhibition of extracellular signal-regulated kinase 1/2 activity (10, 11).
In this study, we address the potential role of pRB as a regulator of white vs. brown adipocyte differentiation using a combination of in vitro and in vivo approaches. We present evidence that pRB acts as a molecular switch determining whether adipocyte differentiation proceeds toward the white or the brown lineage.
Materials and Methods
Animals, Immunohistochemistry, and Morphometry. C57BL/6 mice and Sprague–Dawley rats (12) were use for animal studies. All experiments were performed in compliance with Swedish and Italian institutional guidelines. Experimental details are given in Supporting Text, which is published as supporting information on the PNAS web site.
Cell Culture and Differentiation. MEFs were cultured as described (10). 3T3-L1 and HIB-1B cells were grown in DMEM containing either 10% bovine serum (before differentiation) or 10% FBS (during differentiation). Packaging of retrovirus and transductions were performed as described (13). Wild-type MEFs and 3T3-L1 cells were transduced and selected with puromycin. Selected clones were pooled and replated for differentiation. Rb–/– embryonic stem (ES) cells have been described (14), and the generation of p107–/–p130–/– ES cells will be described elsewhere (J.-H. Dannenberg and H.t.R., unpublished work). Differentiation of ES cells was performed as described (15) except that rosiglitazone (1 μM) and T3 (10 nM) (Sigma) were added from day 17 to day 27. The cells were harvested for RNA isolation on day 27. Oil Red O stainings were performed as described (10). Details of the differentiation protocols and the retroviral vectors are given in Supporting Text.
Transient Transfections. MEFs were transfected in 12-well plates by using the FuGENE 6 Transfection Reagent (Roche Applied Science). Plasmids used were FH-Luc (containing six consensus Forkhead-binding sites) (kindly provided by René Medema) (200 ng per well), pCMV-Renilla (2 ng per well) and pBluescript (to a total of 400 ng per well). Firefly and Renilla luciferase activities were measured by using the Dual-Luciferase Reporter Assay System (Promega). In initial control experiments, the FH-Luc reporter plasmid was effectively activated by cotransfection of a FOXC2 expression vector.
Normal and Multiplex RT-PCR. RT-PCR was performed as described (10). Primer sets specific for the indicated mRNAs were designed, and the number of cycles (between 15 and 22) used for the individual primer sets was chosen based on mRNA abundance. Primer sequences are available on request. TATA-binding protein (TBP) or β-actin primer sets were used as internal standards in all PCR assays. For the RT-PCR in Fig. 1F, 30 cycles of cold PCR were applied.
Fig. 1.

Sequestering of pocket proteins by SV40 TAg in wild-type MEFs and 3T3-L1 cells results in expression of UCP-1 in the adipose state. For A–D, cells were transduced with either control virus or virus encoding TAg or TAg-K1. Puromycin-selected cells were induced to differentiate in the presence of rosiglitazone. (A) Western blot analysis of the expression of TAg. Equal levels of TAg and TAg-K1 were expressed in MEFs as well as in 3T3-L1 cells. (B) Morphological differentiation. Cells were stained with Oil Red O to visualize lipid accumulation. Shown are whole stained dishes. (C) Expression of UCP-1 in day 10 adipocytes analyzed by multiplex RT-PCR. The TBP primer set was included as an internal reference (not shown). (D) Expression of UCP-1 in day 10 adipocytes analyzed by Western blotting. One hundred micrograms of whole-cell protein was loaded in each lane. (E) Quantitative comparison of UCP-1 expression by real-time PCR. All cells were differentiated in the presence of rosiglitazone and stimulated from day 8 to day 10 with isoproterenol and 9-cis-retinoic acid. The expression of UCP-1 in the individual samples was normalized to the expression of TBP, and the normalized expression in HIB-1B cells was set as 1. Error bars represent the SD of each sample analyzed in triplicate. (F) Wild-type, Rb–/–, and p107–/–p130–/– ES cells were induced to differentiate in the presence of rosiglitazone and T3, and the expression of UCP-1, PGC-1α, PPARγ, C/EBPα, and aP2 was analyzed by RT-PCR. The TBP primer set was used as a control for equal cDNA input.
Real-Time Quantitative PCR. Real-time PCR assays were run on an ABI Prism 7700 real-time PCR machine (PE Applied Biosystems). Reactions were performed according to the manufacturer's instructions by using SYBR green PCR Master Mix. Primer sequences are available on request. The expression of selected genes was in all cases normalized to the expression of TBP.
Western Blotting. Preparation of whole-cell extracts, SDS/PAGE gel electrophoresis, and Western blotting were performed as described (10). Antibodies are described in Supporting Text.
Staining of Mitochondria and Electron Microscopy. MitoTracker green stainings were performed as described by the manufacturer (Molecular Probes). Electron microscopy was performed as described (16).
Results
Inactivation of pRB in White Preadipocytes by Simian Virus 40 (SV40 Large T Antigen (TAg) Leads to Brown Adipocyte-Specific Gene Expression in the Adipose State. Forced expression of TAg is widely used for the establishment of brown preadipocyte cell lines, including the HIB-1B cell line (17–20). In contrast, SV40 TAg has been shown to inhibit the differentiation of white preadipocytes (21, 22). These observations indicate that TAg differentially modulates white and brown adipocyte differentiation, inhibiting the former and facilitating the latter. TAg interacts physically and functionally with numerous cellular proteins, including the pocket proteins, p53, p300, and cAMP response element-binding protein (CREB)-binding protein (23).
To investigate whether functional inactivation of pocket proteins is causally related to differentiation into brown adipocytes, we expressed wild-type and mutant SV40 TAg in cells that normally differentiate into white adipocytes (Fig. 1 A). Expression of TAg in wild-type MEFs and 3T3-L1 white preadipocytes did not inhibit adipose conversion induced by the PPARγ ligand rosiglitazone (Fig. 1B and data not shown). Brown fat cells respond to β-adrenergic stimulation by increasing the expression of UCP-1, and this increase is further augmented by simultaneous treatment with 9-cis-retinoic acid (1, 2, 24). Treatment of fully differentiated adipocytes with isoproterenol and 9-cis-retinoic acid for 48 h before harvesting induced significant expression of UCP-1 mRNA and protein in TAg-expressing adipocytes derived from both wild-type MEFs and 3T3-L1 cells but not in control cells (Fig. 1 C and D). Similar results were observed in the 3T3-F442A cell line (data not shown). To test whether the up-regulation of UCP-1 expression was linked to functional inactivation of pocket proteins or to alternative activities of TAg, we expressed a mutant of TAg specifically defective in binding to pRB, p107, and p130 (designated TAg-K1) (Fig. 1 A). UCP-1 was not induced in adipocytes expressing TAg-K1, demonstrating that the sequestering of one or more pocket proteins by TAg allowed induction of UCP-1 in the adipose state (Fig. 1 C and D). Real-time quantitative PCR analyses of MEF-TAg, 3T3-L1-TAg, and HIB-1B adipocytes showed that the expression of UCP-1 was ≈6-fold higher in MEF-TAg and 3T3-L1-TAg compared with HIB-1B fat cells (Fig. 1E).
Rb–/– Mouse Embryonic Stem Cells Differentiate into UCP-1-Expressing Adipocytes. To examine whether TAg facilitated differentiation into brown adipocytes by functional inactivation of pRB or p107 and p130, we took advantage of ES cells lacking pRB or lacking both p107 and p130. By directed differentiation into adipocytes (15), we observed that UCP-1 was expressed exclusively in Rb–/– cells, and that PGC-1α was expressed at higher levels in Rb–/– compared with wild-type and p107–/–p130–/– cells (Fig. 1F). In contrast, standard adipocyte markers were induced to approximately the same level (Fig. 1F). Thus pRB, but not p107 and/or p130, appears to be an important target of TAg in determining UCP-1 expression.
Rb–/– MEFs Differentiate to Fat Cells with Characteristics of Brown Adipocytes. Differentiation of wild-type and Rb–/– MEFs progresses with approximately the same pace and efficiency in the presence of rosiglitazone (Fig. 2A and data not shown). UCP-1 mRNA, however, was induced only in differentiating Rb–/– MEFs, and the level of UCP-1 mRNA in these cells was ≈25% of that in BAT (Fig. 2B). Similarly, UCP-1 protein expression was induced exclusively in pRB-deficient cells (Fig. 2C). It is noteworthy that the level of UCP-1 protein in Rb–/– adipocytes was nearly as high as in BAT and much higher than in HIB-1B adipocytes (Fig. 2D). The differential expression of UCP-1 was confirmed in two wild-type and two Rb–/– MEF preparations (data not shown). PPARα expression was induced to 4-fold higher levels in Rb–/– compared with Rb+/+ adipocytes (Fig. 2B), consistent with the differential expression in WAT and BAT (25). PGC-1α and -1β (26) were strongly induced during adipose conversion of pRB-deficient cells but were barely expressed and weakly induced in wild-type cells (Fig. 2B). We identified a third nuclear receptor cofactor with a dramatically higher expression level in BAT than in WAT, the nuclear receptor-binding factor 1 (NRBF-1) (27). NRBF-1 was strongly induced during the differentiation of Rb–/– MEFs but not during conversion of wild-type MEFs (Fig. 2B).
Fig. 2.

The gene expression pattern in differentiated Rb–/– MEFs resembles that of brown fat. Rb+/+ and Rb–/– MEFs were induced to differentiate, and RNA and protein extracts were prepared at the indicated days. The expression of selected genes was analyzed by multiplex RT-PCR or Western blotting. RNA from WAT and BAT was included as references for the multiplex RT-PCR. (A) Analyses of adipocyte marker gene expression during differentiation of wild-type and Rb–/– MEFs. (B) Analyses of BAT-related gene expression by multiplex RT-PCR in differentiating MEFs. (C) Western blot analysis of gene expression in differentiating MEFs. Antibodies used were against UCP-1, serine-133 phosphorylated CREB (P-CREB), total CREB (control for equal loading), and CDK4 (control for equal loading). One hundred micrograms of whole-cell protein were loaded in each lane. (D) Comparison of UCP-1 protein levels in Rb+/+ adipocytes (day 10, not treated), HIB-1B adipocytes (day 10, stimulated with isoproterenol and 9-cis-retinoic acid as described in Fig. 1E), Rb–/– adipocytes (day 10, not stimulated), and BAT from a mouse kept at room temperature. The amounts of whole-cell protein loaded in each lane are indicated. Antibodies against total CREB and CDK4 were used as loading controls.
A major difference between WAT and BAT is mitochondrial activity and numbers, both of which are higher in BAT (28). We measured the levels of mRNAs encoding two enzymes of the respiratory chain, ATP synthase β and cytochrome c oxidase II (COX II). Both mRNAs were significantly induced during differentiation of Rb–/– cells, but little change was observed in wild-type cells (Fig. 2B). Expression of the mitochondrial transcription factor A, an important regulator of mitochondrial biogenesis (29, 30), was barely induced in wild-type cells, whereas it was induced 5-fold in Rb–/– cells during differentiation (Fig. 2B). Accordingly, staining of fully differentiated Rb+/+ and Rb–/– adipocytes with MitoTracker Green revealed a stronger staining of pRB-deficient cells (Fig. 3A). Consistently, electron microscopy revealed that Rb–/– adipocytes contain more mitochondria than wild-type adipocytes (Fig. 3B). The difference in mitochondrial numbers and mitochondria-related gene expression as a function of pRB status was specific for the adipose state, because no significant difference was observed between wild-type and Rb–/– MEFs at day 0 (Fig. 2B and data not shown).
Fig. 3.

High content of mitochondria in Rb–/– adipocytes. Wild-type and Rb–/– adipocytes were analyzed for mitochondrial abundance by MitoTracker Green staining and electron microscopy. (A) Representative area of live wild-type and Rb–/– adipocytes stained with MitoTracker Green. The same panel of cells was captured by both light and fluorescence microscopy. (B) Electron microscopic analysis of mitochondrial numbers in wild-type and Rb–/– adipocytes. Two representative cells of each genotype are shown.
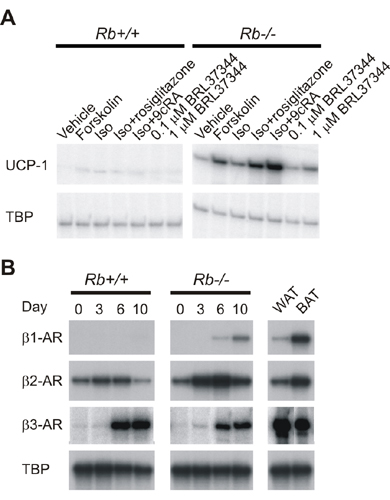
UCP-1 expression was induced in Rb–/– adipocytes upon treatment with β-adrenergic agonists, and this induction was potentiated by simultaneous treatment with 9-cis-retinoic acid (Fig. 7A, which is published as supporting information on the PNAS web site). The expression of UCP-1 mRNA was 3- to 4-fold higher in Rb–/– MEF-derived adipocytes than in the TAg-expressing cells described above after treatment with isoproterenol and 9-cis-retinoic (Fig. 1E). Because this in part could be because of a differential expression of β-adrenergic receptors (β-ARs), we measured the expression of these during differentiation of wild-type and Rb–/– MEFs. Whereas we found higher levels of β1-AR and β2-AR in Rb–/– adipocytes, β3-AR was induced to approximately the same level in wild-type and pRB-deficient cells (Fig. 7B).
Differentiation of Rb–/– Fibroblasts into Brown Adipocytes Is Coupled to Increased cAMP Sensitivity. Mice overexpressing Foxc2 in fat show a conversion of white into brown fat-like depots (6). One likely explanation for this is enhanced sensitivity to cAMP signaling because of an increased expression of the sensitizing cAMP-dependent protein kinase (PKA) subunit RIα in fat (6). We hypothesized that a similar mechanism might be involved in determining the path of adipose conversion followed by wild-type and Rb–/– cells. The expression of Foxc2 declined after day 3 of the differentiation in both wild-type and Rb–/– MEFs (Fig. 4A). At all 4 days tested, however, the expression of Foxc2 mRNA was 2- to 3-fold higher in Rb–/– compared with wild-type MEFs (Fig. 4A). Accordingly, a Forkhead-responsive luciferase reporter plasmid had a 3-fold higher activity in Rb–/– MEFs compared to wild-type MEFs (Fig. 4B). The expression of RIα was ≈3- and 1.5-fold higher in Rb–/– relative to wild-type cells at days 0 and 10, respectively (Fig. 4C). We measured the levels of serine-133 phosphorylated CREB (designated P-CREB) as a measure of cAMP-dependent activation of PKA. The level of P-CREB was highest at day 0 in both Rb+/+ and Rb–/– cells, but the level was significantly higher in the latter (Fig. 2C). The increased level of P-CREB in the pRB-deficient relative to wild-type cells was also prominent at days 1 and 2. To determine whether increased PKA activation in the early stages of differentiation of Rb–/– MEFs contributed to their brown adipogenic fate, we tested the effect of the PKA inhibitor H-89. As shown in Fig. 4D, addition of H-89 essentially blocked induction of UCP-1 protein expression in pRB-deficient cells. Real-time PCR demonstrated a 15-fold decrease in UCP-1 mRNA levels and a 2-fold decrease in PGC-1α and mitochondrial transcription factor A mRNA levels in response to H-89 treatment (Fig. 8, which is published as supporting information on the PNAS web site). The strong suppression of UCP-1 expression required treatment with H-89 before day 2 of the differentiation program (data not shown), consistent with the differences in P-CREB levels in Rb+/+ and Rb–/– cells at these days (Fig. 2C). Importantly, H-89 did not inhibit the adipose conversion of the Rb–/– cells, as determined by Oil Red O staining (data not shown) and expression of PPARγ2 and aP2 mRNAs (Fig. 8, which is published as supporting information on the PNAS web site). Therefore, increased cAMP sensitivity during the early stages of differentiation of pRB-deficient cells contributes to their brown-like adipogenic fate.
Fig. 4.

Increased expression of Foxc2 and RIα throughout adipose conversion of Rb–/– MEFs. (A) The expression of Foxc2 at the indicated days during differentiation was measured by multiplex RT-PCR. A TBP primer set was included as an internal standard. (B) Activity of a Forkhead-responsive firefly luciferase reporter plasmid in Rb+/+ and Rb–/– MEFs. Cells transfected with FH-Luc and pCMV-Renilla were harvested after 24 h, and the activity of firefly luciferase was normalized to that of Renilla luciferase. Transfections were performed in triplicate and repeated twice. The activity in Rb+/+ MEFs was in each experiment set as 1. Shown is the mean activity in the two experiments (±range). (C) The expression of RIα was analyzed on days 0 and 10 by real-time PCR as described in Materials and Methods and normalized to the expression of TBP. The normalized expression in day 0 Rb+/+ MEFs was set as 1. Bars represent the mean value obtained in two independent differentiations, and error bars represent the range measured in these. (D) The expression of UCP-1 in day 10 Rb–/– adipocytes treated with either H-89 or vehicle during their differentiation was analyzed by Western blotting. Shown are the results of two independent experiments. Whole cell protein from wild-type adipocytes (day 10) is shown for comparison. We loaded 50 μg of protein in each lane, and antibody against CDK4 was used as a loading control.
The Absence of Nuclear pRB Characterizes Early Stages of Brown Adipocyte Differentiation, and Down-Regulation of pRB Expression Accompanies Transdifferentiation of White into Brown Adipocytes. The ability of pRB to determine white vs. brown adipocyte differentiation in vitro suggested that pRB expression or activity might be differentially regulated during the development of WAT and BAT. To investigate this, we used immunohistochemical analyses to study expression and cellular localization of pRB during development of interscapular BAT and epididymal WAT. We used the last period of fetal growth for the interscapular depot (BAT anlage) and the early postnatal period for the epididymal depot. The BAT anlage at this stage is mainly occupied by UCP-1-positive brown adipocyte precursors with small lipid droplets and numerous mitochondria (31, 32). It is noteworthy that no pRB immunoreactivity was observed in these brown adipocyte precursors (Fig. 5A), whereas distinct nuclear staining was observed in control tissues from the same fetal specimen (intestinal villi, skin, and testis, Fig. 5A Inset, and data not shown). In contrast, pRB was present in the nuclei of mature brown fat cells from 10-day-old mice (Fig. 5B). Epididymal fat pads from 6- to 10-day-old mice are mainly composed of poorly differentiated adipoblasts without lipid droplets and white adipocyte precursors with lipid droplets (31). These white precursors are quite different from brown precursors in their morphology and do not express UCP-1 (33). We found nuclear pRB immunoreactivity in the lipid droplet-containing white adipocyte precursors in all specimens from 6- to 10-day-old mice (Fig. 5 C and D and data not shown). No pRB immunoreactivity was observed in adipoblasts.
Fig. 5.

Differential expression of pRB during development of WAT and BAT and during transdifferentiation of white into brown adipocytes. (A) BAT anlage of an embryonic day 19 mouse fetus. No nuclear pRB immunoreactivity is observed. (Inset) Internal positive control of the same fetus showing pRB-positive nuclei of apical cells of intestinal villi (arrowheads). (B) BAT of a 10-day-old mouse. Nuclei of well differentiated brown adipocytes are pRB-positive (*), and nuclei of endothelial cells are negative (arrowheads). (C) Epididymal fat pad of a 9-day-old mouse. f, fat pad; ep, epididymus; t, testis. (D) Epididymal WAT of a 9-day-old mouse (enlargement of the squared area in C). Adipocyte precursors with lipids droplets (*) show nuclear staining. Endothelial cells (e) and adipoblasts (a) are pRB-negative. (E) Retroperitoneal WAT of a 20-week-old rat treated with the β3-adrenergic agonist CL-316243 for 7 days. Most of the transdifferentiating multilocular adipocytes exhibit pRB negative nuclei (arrows). A unilocular adipocyte with positive nucleus is visible (arrowhead). (F) Same tissue as in E. A unilocular adipocyte with positive nucleus is visible (arrowhead). (Bars: A and B = 30 mM; A Inset = 60 mM; D and F = 10 mM; C = 200 mM; E = 15 mM.)
The retroperitoneal fat pad of 20-week-old rats contains mature white unilocular adipocytes (12). After 7 days of treatment with the β3-adrenergic receptor agonist CL-316243, 17% of the fat cells in the fat pad resembled immature brown adipocytes with a multilocular appearance, some of which stain positive for UCP-1 (12). These multilocular cells have been shown to stain negative for BrdUrd, thereby ruling out that they are the result of proliferation of brown adipocyte precursor cells present in the fat pad (12). Together with the overall decline in unilocular white adipocyte numbers in response to CL-316243, these results suggest that the multilocular adipocytes arise as the result of transdifferentiation of white fat cells. In nontreated retroperitoneal fat, pRB immunoreactivity was found only in the nuclei of adipocytes, and morphometry showed that 68 ± 2.6% (mean ± SD) of these stained positive. After 7 days of treatment with CL-316243, the percentage of pRB-positive nuclei had declined to 37 ± 2.9% (P < 0.0005). These transdifferentiating multilocular adipocytes (87 ± 2.3%) stained negative for pRB (Fig. 5E), whereas adipocytes with typical unilocular morphology exhibited distinct nuclear pRB immunoreactivity (Fig. 5 E and F). In summary, our data show that pRB is absent from brown adipocyte precursors but present in white adipocyte precursors and mature white and brown adipocytes. Moreover, expression of pRB is strongly down-regulated in a rat model of white into brown adipocyte transdifferentiation.
pRB Is Inactivated by Phosphorylation in BAT upon Exposure to Cold. Transfer of mice to cold induces a rapid induction of UCP-1 expression and neodifferentiation in interscapular BAT. We performed a time-course analysis of pRB phosphorylation and UCP-1 induction in mice transferred from 30°C to 4°C. UCP-1 was strongly induced in BAT, reaching a maximum after 2 days, whereas no expression was observed in epididymal WAT (Fig. 6). pRB phosphorylation in BAT was induced within 1 day, whereas no change was observed in epididymal WAT (Fig. 6). Obviously, enhanced pRB phosphorylation is expected in cold-induced neodifferentiation in BAT, because it involves DNA replication and proliferation of cells in the tissue (34). However, it is noteworthy that the kinetics of cold-induced pRB phosphorylation seems to mirror induction of UCP-1 expression more closely than thymidine incorporation and DNA accumulation, which were previously shown to reach maxima at much later time points (34).
Fig. 6.

Phosphorylation of pRB is rapidly induced in interscapular BAT but not in epididymal WAT upon cold exposure. C57BL/6 mice were preacclimated to 30°C for 2 weeks and then transferred to 4°C. Interscapular BAT and epididymal WAT were isolated at the indicated days after cold exposure and prepared for SDS/PAGE. Levels of UCP-1 expression, pRB phosphorylation, and total pRB were determined by Western blotting. P-pRB indicates Ser 780-phosphorylated pRB. An antibody against actin was used as a loading control.
Discussion
Studies have shown that pRB is required for spontaneous white adipogenesis and for white adipose conversion in response to a standard adipogenic treatment (9, 10), and it was reported that SV40 TAg inhibits white adipocyte differentiation in part by inactivation of pocket proteins (21, 22). Interestingly, expression of TAg is routinely used to generate brown preadipocyte cell lines. Even more intriguing is the observation that transgenic mice expressing SV40 TAg from a fat-specific promoter exhibit a partial conversion of WAT into BAT (20). These findings suggest that SV40 TAg has opposing effects on white and brown adipogenesis, inhibiting the former but facilitating the latter.
We have shown that the administration of a synthetic PPARγ ligand rescues differentiation of pRB-deficient cells (10). Here we show that the same is true for TAg-expressing white preadipocytes. Surprisingly, we found that cells with functional inactivation of pRB by TAg or genetic ablation of the Rb gene in the adipose state displayed a gene expression pattern closely resembling that of BAT, including expression of UCP-1. In particular, Rb–/– MEFs differentiated into adipocytes expressed UCP-1 protein at levels comparable to that of BAT. Moreover, Rb–/– adipocytes accumulated many more mitochondria than wild-type adipocytes. To our knowledge, pRB is the first example of a protein having opposing roles in white and brown adipogenesis, being required for the differentiation of white adipocytes in response to no or standard inducers (9, 10), but as shown here inhibiting the formation of brown fat cells in response to stronger inducers. The observation that transgenic mice expressing SV40 TAg in fat show a conversion of white into brown fat (20) indicates that impairing the activity of pRB promotes brown adipogenesis also in vivo. In line with these observations, our immunohistochemical analyses of pRB expression indicate that pRB is present in the nuclei in white but not in brown adipocyte precursors during development. Moreover, we demonstrate that pRB rapidly becomes inactivated by phosphorylation in BAT of mice exposed to cold, a condition that activates UCP-1 expression, but also promotes a significant neodifferentiation of BAT. Finally, transdifferentiation of retroperitoneal white adipocytes into brown-like multilocular adipocytes induced by β3-adrenergic stimulation is accompanied by a marked decline in pRB expression.
How does pRB inhibit brown adipogenesis? We show that Rb–/– fibroblasts have an increased expression of Foxc2 and its target gene RIα, indicative of an enhanced sensitivity of cAMP-mediated PKA activation. Suppression of PKA activity by H-89 during differentiation of Rb–/– MEFs largely blocks the induction of UCP-1 expression. Therefore, we propose a model for the brown adipocyte differentiation of Rb–/– cells in which higher expression of Foxc2 early during the differentiation leads to a higher expression of RIα, which sensitizes the cells to cAMP (as reflected by P-CREB levels). CREB activity in turn assists in the activation of PGC-1α expression (35), which then coactivates the brown adipose conversion to completion, including UCP-1 expression, mitochondrial gene expression, and biogenesis. However, we cannot rule out the involvement of pRB in other mechanisms inhibiting brown adipogenesis. pRB may interfere with the function of the thyroid hormone receptors (36), which play important roles in brown adipocyte differentiation and function (1). Moreover, it is possible that pRB directly represses the expression of PGC-1α and/or -1β by an as-yet-unknown mechanism, thereby indirectly suppressing brown adipose conversion.
Although the detailed mechanisms by which pRB status potentially governs commitment to the white or the brown adipocyte lineage in vivo remain to be elucidated, this paper describes how pRB functions as a molecular switch that determines whether adipocyte precursor cells in vitro follow a path leading to differentiation into white or brown adipocytes. Obviously, the activity of pRB can be modulated in many ways at the cellular level, e.g., by being expressed at low levels (or not being expressed at all), inactivated by hyperphosphorylation, or sequestered by a cellular protein in a manner similar to the situation in our SV40 TAg experiments. Using various mouse and rat models of brown adipocyte differentiation in vivo, we show examples of the first two mechanisms. Therefore, these findings are compatible with our model that a stage-specific absence or inactivation of pRB provides a window permissible for brown adipocyte differentiation and UCP-1 expression. A more detailed understanding of events acting upstream and downstream of pRB will help further elucidating mechanisms differentially controlling white and brown adipose conversion of precursor cells. In time, designed interference with the balance between white and brown adipocyte differentiation might be relevant in the treatment of obesity and obesity-associated disorders.
Supplementary Material
Acknowledgments
We thank Drs. James A. DeCaprio (Dana–Farber Cancer Institute, Boston) and René Medema (The Netherlands Cancer Institute, Amsterdam) for the kind gifts of plasmids. We thank Novo Nordisk for the gifts of rosiglitazone and insulin. This work was conducted within the Center for Experimental BioInformatics and supported by the Danish Biotechnology Program, the Danish Cancer Society, the Danish Natural Science Research Council, the Novo Nordisk Foundation, The Swedish Science Research Council, and the European Community (Contract QLK1-CT-2001-00183). J.B.H. thanks the Federation of European Biochemical Societies for financial support.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: WAT, white adipose tissue; CREB, cAMP response element-binding protein; BAT, brown adipose tissue; PPAR, peroxisome proliferator-activated receptor; PGC-1, PPARγ coactivator 1; PKA, cAMP-dependent protein kinase; C/EBP, CCAAT/enhancer-binding protein; pRB, retinoblastoma protein; Rb, retinoblastoma gene; MEF, mouse embryo fibroblast; ES cells, embryonic stem cells; SV40, simian virus 40; TAg, large T antigen; TBP, TATA-binding protein; P-CREB, phosphorylated CREB; UCP-1, uncoupling protein 1.
References
- 1.Lowell, B. B. & Spiegelman, B. M. (2000) Nature 404, 652–660. [DOI] [PubMed] [Google Scholar]
- 2.Rosen, E. D., Walkey, C. J., Puigserver, P. & Spiegelman, B. M. (2000) Genes Dev. 14, 1293–1307. [PubMed] [Google Scholar]
- 3.Lowell, B. B., S-Susulic, V., Hamann, A., Lawitts, J. A., Himms-Hagen, J., Boyer, B. B., Kozak, L. P. & Flier, J. S. (1993) Nature 366, 740–742. [DOI] [PubMed] [Google Scholar]
- 4.Picard, F., Géhin, M., Annicotte, J.-S., Rocchi, S., Champy, M.-F., O'Malley, B. W., Chambon, P. & Auwerx, J. (2002) Cell 111, 931–941. [DOI] [PubMed] [Google Scholar]
- 5.Puigserver, P., Wu, Z., Park, C. W., Graves, R., Wright, M. & Spiegelman, B. M. (1998) Cell 92, 829–839. [DOI] [PubMed] [Google Scholar]
- 6.Cederberg, A., Grønning, L. M., Ahrén, B., Taskén, K., Carlsson, P. & Enerbäck, S. (2001) Cell 106, 563–573. [DOI] [PubMed] [Google Scholar]
- 7.Harbour, J. W. & Dean, D. C. (2000) Genes Dev. 14, 2393–2409. [DOI] [PubMed] [Google Scholar]
- 8.Lipinski, M. M. & Jacks, T. (2000) Oncogene 18, 7873–7882. [DOI] [PubMed] [Google Scholar]
- 9.Chen, P.-L., Riley, D. J., Chen, Y. & Lee, W.-H. (1996) Genes Dev. 10, 2794–2804. [DOI] [PubMed] [Google Scholar]
- 10.Hansen, J. B., Petersen, R. K., Larsen, B. M., Bartkova, J., Alsner, J. & Kristiansen, K. (1999) J. Biol. Chem. 274, 2386–2393. [DOI] [PubMed] [Google Scholar]
- 11.Hansen, J. B., Petersen, R. K., Jørgensen, C. & Kristiansen, K. (2002) J. Biol. Chem. 277, 26335–26339. [DOI] [PubMed] [Google Scholar]
- 12.Himms-Hagen, J., Melnyk, A., Zingaretti, M. C., Ceresi, E., Barbatelli, G. & Cinti, S. (2000) Am. J. Physiol. 279, C670–C681. [DOI] [PubMed] [Google Scholar]
- 13.Hansen, J. B., Zhang. H., Rasmussen, T. H., Petersen, R. K., Flindt, E. N. & Kristiansen, K. (2001) J. Biol. Chem. 276, 3175–3182. [DOI] [PubMed] [Google Scholar]
- 14.Dannenberg, J.-H., van Rossum, A., Schuijff, L. & te Riele, H. (2000) Genes Dev. 14, 3051–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosen, E. D., Sarraf, P., Troy, A. E., Bradwin, G., Moore, K., Milstone, D. S., Spiegelman, B. M. & Mortensen, R. M. (1999) Mol. Cell 4, 611–617. [DOI] [PubMed] [Google Scholar]
- 16.Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., Troy, A., Cinti, S., Lowell, B. B., Scarpulla, R. C. & Spiegelman, B. M. (1999) Cell 98, 115–124. [DOI] [PubMed] [Google Scholar]
- 17.Benito, M., Porras, A. & Santos, E. (1993) Exp. Cell Res. 209, 248–254. [DOI] [PubMed] [Google Scholar]
- 18.Kozak, U. C., Held, W., Kreutter, D. & Kozak, L. P. (1992) Mol. Endocrinol. 6, 763–772. [DOI] [PubMed] [Google Scholar]
- 19.Rohlfs, E. M., Daniels, K. W., Premont, R. T., Kozak, L. P. & Collins, S. (1995) J. Biol. Chem. 270, 10723–10732. [DOI] [PubMed] [Google Scholar]
- 20.Ross, S. R., Choy, L., Graves, R. A., Fox, N., Solevjeva, V., Klaus, S., Ricquier, D. & Spiegelman, B. M. (1992) Proc. Natl. Acad. Sci. USA 89, 7561–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherington, V., Morgan, B., Spiegelman, B. M. & Roberts, T. M. (1986) Proc. Natl. Acad. Sci. USA 83, 4307–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Higgins, C., Chatterjee, S. & Cherington, V. (1996) J. Virol. 70, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali, S. H. & DeCaprio, J. A. (2001) Semin. Cancer Biol. 11, 15–23. [DOI] [PubMed] [Google Scholar]
- 24.Puigserver, P., Vazquez, F., Bonet, M. L., Pico, C. & Palou, A. (1996) Biochem. J. 317, 827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Issemann, I. & Green, S. (1990) Nature 347, 645–650. [DOI] [PubMed] [Google Scholar]
- 26.Lin, J., Puigserver, P., Donovan, J., Tarr, P. & Spiegelman, B. M. (2002) J. Biol. Chem. 277, 1645–1648. [DOI] [PubMed] [Google Scholar]
- 27.Masuda, N., Yasumo, H., Furusawa, T., Tsukamoto, T., Sadano, H. & Osumi, T. (1998) Gene 221, 225–233. [DOI] [PubMed] [Google Scholar]
- 28.Ricquier, D. & Bouillaud, F. (2000) J. Physiol. 529, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsson, N.-G., Wang, J., Wilhelmsson, H., Oldfors, A., Rustin, P., Lewandoski, M., Barsh, G. S. & Clayton, D. A. (1998) Nat. Genet. 18, 231–236. [DOI] [PubMed] [Google Scholar]
- 30.Scarpulla, R. C. (2002) Biochim. Biophys. Acta 1576, 1–14. [DOI] [PubMed] [Google Scholar]
- 31.Cinti, S. (2002) J. Endocrinol. Invest. 25, 823–835. [DOI] [PubMed] [Google Scholar]
- 32.Houstek, J., Kopecky, J., Rychter, Z. & Soukup, T. (1988) Biochim. Biophys. Acta 935, 19–25. [DOI] [PubMed] [Google Scholar]
- 33.Moulin, K., Truel, N., André, M., Arnauld, E., Nibbelink, M., Cousin, B., Dani, C., Pénicaud, L. & Casteilla, L. (2001) Biochem. J. 356, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rehnmark, S. & Nedergaard, J. (1989) Exp. Cell Res. 180, 574–579. [DOI] [PubMed] [Google Scholar]
- 35.Herzig, S., Long, F., Jhala, U. S., Hedrick, S., Quinn, R., Bauer, A., Rudolph, R., Schutz, G., Yoon, C., Puigserver, P., Spiegelman, B. & Montminy, M. (2001) Nature 413, 179–183. [DOI] [PubMed] [Google Scholar]
- 36.Chang, K.-H., Chen, Y., Chen, T.-T., Chou, W.-H., Chen, P.-L., Ma, Y.-Y., Yang-Feng, T. L., Leng, X., Tsai, M.-J., O'Malley, B. W. et al.. (1997) Proc. Natl. Acad. Sci. USA 94, 9040–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}