

Abstract
It has been proposed that hydroxyl radicals (·OH) generated in a perinuclear iron-dependent Fenton reaction are involved in O2-dependent gene expression. Thus, it was the aim of this study to localize the cellular compartment in which the Fenton reaction takes place and to determine whether scavenging of ·OH can modulate hypoxia-inducible factor 1 (HIF-1)-dependent gene expression. The Fenton reaction was localized by using the nonfluorescent dihydrorhodamine (DHR) 123 that is irreversibly oxidized to fluorescent rhodamine 123 while scavenging ·OH together with gene constructs allowing fluorescent labeling of mitochondria, endoplasmic reticulum (ER), Golgi apparatus, peroxisomes, or lysosomes. A 3D two-photon confocal laser scanning microscopy showed ·OH generation in distinct hot spots of perinuclear ER pockets. This ER-based Fenton reaction was strictly pO2-dependent. Further colocalization experiments showed that the O2-sensitive transcription factor HIF-1α was present at the ER under normoxia, whereas HIF-1α was present only in the nucleus under hypoxia. Inhibition of the Fenton reaction by the ·OH scavenger DHR attenuated HIF-prolyl hydroxylase activity and interaction with von Hippel–Lindau protein, leading to enhanced HIF-1α levels, HIF-1α transactivation, and activated expression of the HIF-1 target genes plasminogen activator inhibitor 1 and heme oxygenase 1. Further, ·OH scavenging appeared to enhance redox factor 1 (Ref-1) binding and, thus, recruitment of p300 to the transactivation domain C because mutation of the Ref-1 binding site cysteine 800 abolished DHR-induced transactivation. Thus, the localized Fenton reaction appears to impact the expression of hypoxia-regulated genes by means of HIF-1α stabilization and coactivator recruitment.
The heterogeneous pO2 distribution in tissues requires an O2-sensing system to adapt organ functions. Until now, contradictory evidence exists concerning the nature of the sensor. First, it was proposed that heme proteins (1) such as NADPH oxidases (2) or as exist within the mitochondrial electron-transport chain (3, 4) may act as O2 sensors by producing reactive oxygen species (ROS) as “second messengers.” Recently, it was proposed that a family of O2-using prolyl and asparagyl hydroxylases involved in the modification of HIFs may act as direct O2 sensors (5).
The role of ROS as O2 messengers has been supported by the finding that, similar to a typical response to hypoxia, treatment of healthy human volunteers with the antioxidant N-acetylcysteine enhanced the hypoxic ventilatory response and erythropoietin (EPO) concentration in blood (6). Thus, N-acetylcysteine or its biochemical derivatives, cysteine, and glutathione mimic hypoxia. In contrast, the ROS H2O2 mimicked normoxia in the expression of O2-dependent genes in cell cultures (7).
We have demonstrated (7, 8) that an H2O2-degrading perinuclear Fenton reaction [H2O2 + Fe2+ → Fe3+ + OH- + hydroxyl radical (·OH)] was involved in O2 signaling. However, it remained undetermined in which cellular compartment the Fenton reaction takes place and whether also transcription factors regulating the O2-dependent gene expression are located in this compartment.
A transcription factor who gained a central role in the O2-dependent modulation of gene expression is hypoxia-inducible factor 1 (HIF-1) (9, 10). HIF-1 is composed of the O2- and redox-sensitive α-subunit and the constitutively expressed HIF-1β (ARNT) subunit. Although other HIF isoforms appear to exist (5), HIF-1 is considered to be the major regulator of many physiologically important genes, including plasminogen activator inhibitor 1 (PAI-1), vascular endothelial growth factor (VEGF), and heme oxygenase 1 (HO-1) (11). Thus, it appears likely that the redox-sensitive HIF-1α may be a target within an O2-sensing system involving ROS and a Fenton reaction. To characterize the ROS and redox-sensitive part of the oxygensensing pathway further, it was the aim of this study to identify the compartment in which the Fenton reaction takes place and to investigate whether a compartment-specific inhibition of ·OH generation can interfere with the HIF-1-dependent gene expression.
Materials and Methods
All biochemicals and enzymes used were of analytical grade and obtained from commercial suppliers.
Cell Culture. HepG2 cells were kept in a normoxic atmosphere of 16% O2/79% N2/5% CO2 (vol) in MEM supplemented with 10% FCS for 24 h. At 24 h, medium was changed and culture was continued at normoxia (16% O2) or hypoxia (8% O2) (87% N2/5% CO2, vol).
Plasmid Constructs. The pGL3-EPO-hypoxia response element (HRE) luciferase plasmid, containing three EPO-HREs, was described (12). For pDsRed-endoplasmic reticulum (ER), DsRed from pDsRed-N1 (Clontech) was amplified by PCR and ligated into the NheI and BglII sites of pECFP-ER (Clontech). The pDsRed-Golgi and pDsRed-Mito constructs were constructed by replacing the BamHI and NotI enhanced cyan fluorescent protein (ECFP) fragment with pDsRed-N1 in pECFP-Golgi and pECFP-Mito. The plasmid pECFP-Peroxi was obtained from Clontech.
The pGL3PAI-766, containing the rat PAI-1 promoter from -766 to +33, as well as pGL3PAI-766M2, were described (13). The plasmids GFP-HIF1α (14), pG5E1B-LUC (15), pSG424 (19), Gal4-HIF1α-transactivation domain N (TADN), and HIF1α-TADC, as well as the Gal4-HIF1α-TADCM construct with an exchange of cysteine 800 into serine, were described (16, 17). The Gal4-HIF1α-TADNM construct, in which proline P564 was changed to alanine, was constructed by using the Quick-Change mutagenesis kit (Promega).
The plasmid pGEX-TADN was constructed by ligation of a 219-bp PCR fragment from rat HIF-1α, encompassing amino acids 531–604, into the BamHI and SalI sites of pGEX-5 × 1 (Amersham Biosciences), allowing generation of a GST fusion protein. The plasmids (pGEX-GST-TADN and pGEX-5 × 1) were transformed into Escherichia coli BL21(DE3), and protein expression was induced by using 0.1 mM isopropyl β-d-thiogalactoside for 4 h. After sonication and lysis (20 mM Tris·HCl,pH7.5/250 mM NaCl/2 mM MgCl2/1 mM DTT/10% glycerol), the clarified sonicates were incubated with glutathione Sepharose 4B (Amersham Biosciences) for 1 h at 4°C. After four washes in PBS, the GST-TADN and GST proteins were eluted in 50 mM Tris·HCl/10 mM reduced glutathione (pH 8.0). The integrity was assessed by SDS/PAGE, followed by Coomassie blue staining.
HIF-TADN Hydroxylation and von Hippel–Lindau (VHL) Pull-Down Assay. HepG2 cells homogenized at 4°C in 250 mM sucrose/50 mM Tris·HCl (pH 7.5) were centrifuged at 1,000 × g for 10 min, and the supernatant was centrifuged at 3,000 × g for 10 min. Again, supernatant was pelleted at 18,000 × g for 10 min and suspended in 40 mM Tris·HCl (pH 7.5). Extracts (300 μg/ml) were incubated at 37°C for 30 min in 40 mM Tris·HCl (pH 7.5)/0.5 mM DTT/50 μM ammonium ferrous sulfate/1 mM ascorbate/2 mg/ml BSA/0.4 mg/ml catalase/50,000 dpm of 5-[14C]2-oxoglutarate (Perkin–Elmer)/0.1 mM unlabeled 2-oxoglutarate with either 20 μg of GST protein or GST-TADN protein in the presence of various concentrations of dihydrorhodamine (DHR) or RH. Radioactivity associated to succinate was determined as described (18). The basal GST-dependent activity was subtracted from the GST-TADN-dependent activity.
For VHL pull-down assay, 35S-VHL was synthesized from pCMV-HA-VHL by using [35S]methionine and the TNT-coupled reticulocyte lysate system (Promega). The GST-TADN was incubated either without extract or with cell extracts, as described for the hydroxylation assay, either without or with 0.5 μM DHR or RH, respectively. The reaction products were incubated at 4°C in 200 μl of buffer (50 mM Tris·HCl, pH 8/120 mM NaCl/0.5% Nonidet P-40), supplemented with glutathione-Sepharose beads and 50,000 dpm of [35S]-VHL. After 2 h, beads were washed three times with cold buffer (20 mM Tris·HCl, pH 8/100 mM NaCl/1 mM EDTA/0.5% Nonidet P-40). Bound proteins were eluted in 10 mM reduced glutathione and analyzed by SDS/PAGE, followed by autoradiography.
RNA Preparation, Northern Blot Analysis, and Western Blot Analysis. Isolation of RNA and Northern blot analysis for PAI-1 and HO-1 were performed as described (19). Western blot analysis was carried out as described (20). In brief, media or lysates from cells were collected, and 100 μg of protein was loaded onto a 10% or 7.5% SDS-polyacrylamide gel and, after electrophoresis and blotting, probed with mouse mAb directed against PAI-1 (1:200 dilution; American Diagnostics, Pendleton, IN), HO-1 (1:500 dilution; Biomol, Plymouth Meeting, PA), or human HIF-1α (1:2000 dilution; BD Biosciences), or with a rabbit Ab directed against Golgi membrane (GM; 1:2,000 dilution; Bioscience, Göttingen, Germany). The enhanced chemiluminescence system (ECL; Amersham Biosciences) was used for detection.
Cell Transfection and Luciferase Assay. We transfected ≈4 × 105 cells per 60-mm dish, as described (20). In brief, 2.5 μg of pGl3-EPO-HRE LUC, pGL3PAI-766, and pGL3PAI-796M2 was transfected. After 5 h, medium was changed and cells were cultured under normoxia for 19 h. Then, medium was changed again and cells were stimulated with 6 μM DHR or rhodamine (RH) and cultured further for 24 h under normoxia or hypoxia. Transfection efficiency was controlled by cotransfection of 0.25 μg of Renilla Luciferase expression vector (pRLSV40; Promega).
To investigate HIF-1α transactivation, 2 μg of pG5-E1B-LUC was cotransfected with 500 ng of pSG424 or Gal4-TADN/TADC. To investigate HIF-1α nuclear translocation, 2.5 μg of GFP-HIF1α was transfected, medium was changed after 18 h, and cells were then cultured under normoxia for 30 h. Cells were then stimulated with either 6 μM DHR or 100 μM PDTC.
Measurement of ·OH Generation by Two-Photon Laser Confocal Microscopy (2P-CLSM). Intracellular ·OH generation by the Fenton reaction can be detected by irreversible conversion of nonfluorescent DHR into fluorescent RH 123 (21).
Cells were transfected with 5 μg of the pDsRed-ER, pDsRed-Mito, pDsRed-Golgi, pECFP-Golgi, pECFP-ER, pECFP-Mito, pDsRed-Peroxi, or pDsRed-Lyso gene constructs. Medium was changed after 19 h, and cells were cultured for 24 h. Cells were then transferred to a culture chamber on the microscope, enabling observation at 37°C under a variable gas atmosphere. Cells were treated with 30 μM DHR (Molecular Probes) for 5 min under normoxic or hypoxic conditions, and they were then washed without altering the pO2, as controlled by a polarographic pO2 catheter electrode (Licox, Kiel, Germany). The RH 123 fluorescence was recorded by using 2P-CLSM (8), and an 850-nm excitation wavelength was applied to colocalize RH 123 fluorescence (emission 530 nm) and ECFP fluorescence (emission 470 nm). We used an 875-nm excitation for RH 123 fluorescence and DsRed fluorescence (emission 580 nm). Fluorescence was registered by a photo multiplier, digitized, and visualized by the ez 2000 software (Version 2.1.4; Coord Automatisering, Hilversum, The Netherlands). Deconvolution, data reconstruction, and calculation of isosurfaces were performed on a Unix-based octane workstation (Silicon Graphics, Mountain View, CA), essentially as described (22).
Immunohistochemistry. HepG2 cells were placed on coverslips, transfected with pECFP-ER, and exposed to various pO2 distributions, as described above. Cells were fixed by ice-cold methanol/acetone (1:1) for 10 min, blocked with 3% BSA in PBS, and incubated with an anti-HIF-1α mAb (1:50 dilution; Transduction Laboratories, Lexington, KY) and a rabbit Living Colors Av peptide Ab (1:20 dilution; Clontech). Cells were washed in PBS before incubation with an Alexa Fluor 568-conjugated goat anti-mouse IgG secondary Ab (1:400 dilution; Molecular Probes) and a Cy2-conjugated goat anti-rabbit IgG secondary Ab (1:200 dilution; Dianova, Hamburg, Germany). Images of immunostained cells were captured by using 2P-CLSM, as described above, and images were visualized in false colors, as indicated.
Results
Localization of ·OH-Mediated RH Fluorescence at the ER. Because ·OH-mediated conversion of DHR to fluorescent RH 123 is localized in a perinuclear space (8), we aimed to identify this subcellular compartment. HepG2 cells transfected with pD-sRed-ER, pECFP-ER, pDsRed-Mito, pECFP-Mito, pECFP-Golgi, pDsRed-Peroxi, and pDsRed-Lyso were treated with 30 μM DHR for 5 min in a microscope tissue-culture chamber under normoxia. In a total of 20 experiments (pDsRed-ER, n = 3; pECFP-ER, n = 3; pDsRed-Mito, n = 2; p5CFP-Mito, n = 2; pDsRed-Peroxy, n = 2; pDsRed-Lyso, n = 2), RH 123 fluorescence and ECFP or DsRed fluorescence were recorded simultaneously by 2P-CLSM without any crosstalk because of the emission-filter settings. After deconvolution and reconstruction, DsRed or ECFP fluorescence overlapped with RH 123 fluorescence, indicating the site of ·OH generation.
The 3D reconstruction revealed that RH 123 fluorescence overlapped with DsRed or ECFP fluorescence when expressed in the ER (Fig. 1). No overlap with RH 123 fluorescence was observed when the ECFP or DsRed proteins were expressed in the mitochondria, Golgi apparatus, peroxisomes, or lysosomes (Fig. 1).
Fig. 1.
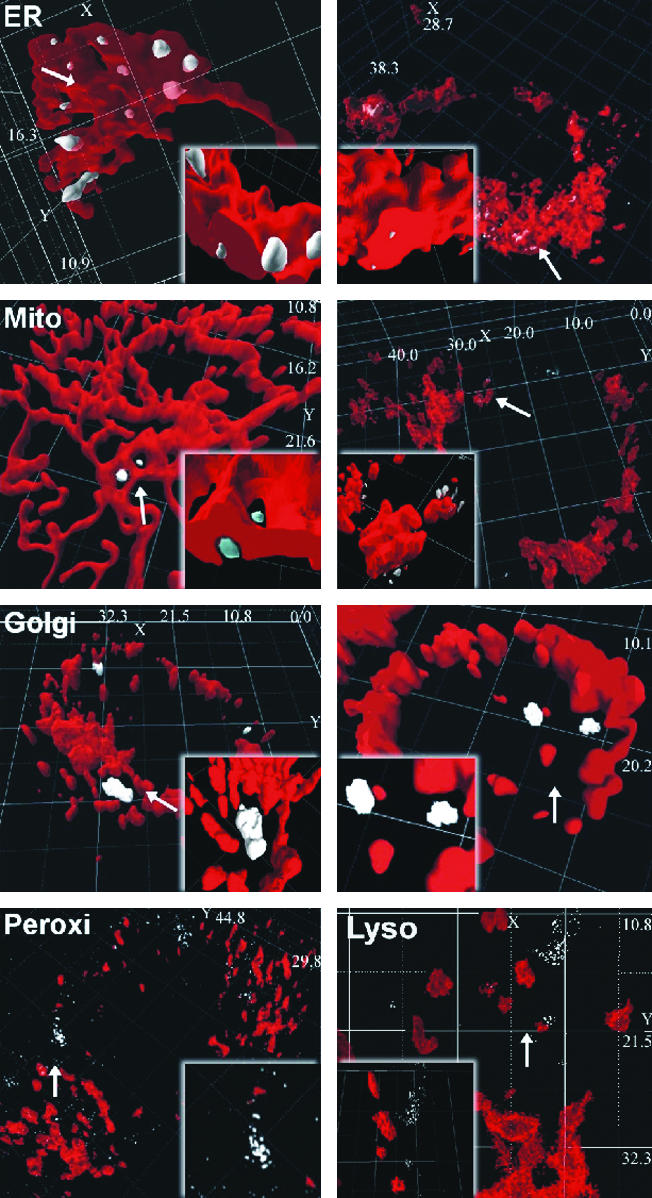
Detection of ·OH generation at the ER. HepG2 cells transfected with the pDsRed-ER (Right), pECFP-ER (Left), pDsRed-Mito (Right), pECFP-Mito (Left), pDsRed-Golgi (Right), pECFP-Golgi (Left), pDsRed-Lyso (Right), or pDsRed-Peroxi (Left) constructs were treated for 5 min with 30 μM DHR, converted into fluorescent RH 123 by ·OH that was generated by a Fenton reaction. Fluorescence was visualized by 2P-CLSM. The various subcellular compartments are shown in red, and the RH 123 fluorescence is shown in gray, indicating the local ·OH generation. Insets show images (×20 magnification) of the areas with ·OH generation (arrows). Dimensions of the x, y, and z axes are given in μm.
To investigate whether ·OH generation at ER is pO2-dependent, the pECFP-ER-transfected cells exposed for 1 h to hypoxia were treated with DHR. Under hypoxia, RH 123 fluorescence was abolished. By contrast, when steady-state normoxic conditions were reached within the microscope chamber (15 min after returning to normoxia), a clear overlap of ·OH generation and ECFP fluorescence in the ER was detected (Fig. 2A). The pO2-dependent ·OH generation was supported further by applying a photoreduction approach (23). The photoreduction effect is caused by blue-light illumination initiating a reduction of flavins and, concomitantly, an activation of flavin-containing oxidases. Subsequently, these oxidases reduce O2 to H2O2, which is then degraded to ·OH by the Fenton reaction. Thus, this light reaction, showing correct imaging of intracellular oxygen radical kinetics, is a control for intracellular ·OH production. Furthermore, it indicates that O2 absence is the major trigger for reduced intracellular ·OH production under hypoxia because the oxidases remain fully activated under blue light even under hypoxia. The challenge of pECFP-ER transfected cells with light of ≈450 nm for 5 s with a power of only 1.2 mW enhanced RH 123 fluorescence at the ER. However, this increase was seen predominantly under normoxia; under hypoxia, ·OH generation was minimal (Fig. 2A). Although photoreduction can be seen also in mitochondria because they contain FADH for generation of electrons, this reaction indicates the specificity of the O2-dependent ·OH generation at ER under normal conditions (Fig. 2A) because FADH reduction and ·OH generation in mitochondria occurred only on photoreduction.
Fig. 2.

Hypoxia-mediated inhibition of ·OH generation at the ER and translocation of HIF-1α. (A) HepG2 cells transfected with ECFP-ER or ECFP-Mito constructs were cultured under hypoxia for 60 min and treated for 5 min with 30 μM DHR before imaging. Cells were then exposed to normoxia and imaged again by 2P-CLSM. Insets show ·OH generation mediated by photoreduction. Cells were challenged with 450-nm light for 5 s under hypoxia or normoxia and imaged. RH 123 fluorescence indicating ·OH generation is shown in white, and fluorescence, indicating subcellular compartments, is shown in red. (B) For detection of endogenous HIF-1α in pECFP-ER transfected HepG2 cells, cells were exposed to normoxia (60 min) or hypoxia (60 min) and immunostained with an anti-HIF-1α Ab and a Living Colors Av peptide Ab. HIF-1α (yellow-orange) and ER (red) are shown. Dimensions of the x, y, and z axes are given in μm.
Oxygen-Dependent ·OH Generation and Colocalization of HIF-1α with the ER Under Normoxia. To investigate whether ·OH generation at ER may target HIF-1α, we examined whether HIF-1α can be found at the ER. For this examination, cells transfected with pECFP-ER were incubated under normoxia (60 min) or hypoxia (60 min) and immunostained with an anti-HIF-1α Ab. Under normoxia, the fluorescence indicating ER overlapped with the fluorescence indicating the presence of HIF-1α. Under hypoxia, in contrast, the ER fluorescence and the HIF-1α fluorescence were dissociated with all of the HIF-1α fluorescence within the nucleus (Fig. 2B).
Induction of HIF-1-Dependent Genes by DHR. To investigate whether scavenging of ·OH by DHR at ER interferes with the O2 signal modulating HIF-1-dependent genes such as PAI-1 and HO-1, cells were treated with DHR under normoxia and hypoxia, and PAI-1 and HO-1 expression were measured.
Hypoxia induced PAI-1 and HO-1 mRNA expression. Similarly, treatment with DHR induced PAI-1 and HO-1 mRNA under normoxia. Treatment with DHR under hypoxia did not further enhance mRNA expression of both genes. The DHR-induced mRNA increase was followed by an increase in PAI-1 and HO-1 proteins (Fig. 3).
Fig. 3.

Induction of PAI-1 and HO-1 expression by DHR. Cells were treated with 6 μM DHR and cultured under normoxia (16% O2) or hypoxia (8% O2) for 3 h. (A) Left shows PAI-1 and HO-1 mRNA, as measured by Northern blotting. The RNA level under normoxia was set at 100%. Right shows the PAI-1 and HO-1 protein levels, as measured by Western blotting. The protein level under normoxia was set at 100%. Values are given as mean ± SEM of three independent experiments. Student's t test for paired values was performed to determine significant difference. *, 16% O2 vs. 8% O2. **, 16% O2 vs. 16% O2 + DHR; P ≤ 0.05. (B) Representative Northern blot and Western blot analyses. For Northern blot analysis, 20 μg of RNA was hybridized to PAI-1, HO-1, and β-actin antisense RNA probes. Western blot analyses were performed for 100 μg of protein from the medium or the whole-cell extract with an Ab directed against PAI-1, HO-1, and GM.
HIF-1 activates gene expression by binding to HREs. In cells transfected with pGL3PAI-766, LUC activity was induced 2-fold under hypoxia (Fig. 4). The treatment with DHR increased LUC activity 3-fold under normoxia and 3.5-fold under hypoxia (Fig. 4). Mutation of the HRE in pGL3PAI-766M2 abolished both hypoxia- and DHR-mediated induction of LUC activity (Fig. 4). Furthermore, in cells transfected with pGL3-EPO-HRE, hypoxia as well as DHR induced LUC activity 2-fold. DHR treatment under hypoxia induced LUC activity 4-fold. Treatment with RH 123, the final product of DHR conversion, had no effect on LUC activity, indicating the specificity of the DHR effect (Fig. 4).
Fig. 4.

Mimicry of hypoxia by DHR in cells transfected with HIF-1-dependent PAI-1 promoter and EPO-HRE luciferase gene constructs. Cells were transfected with the wild-type rat PAI-1 promoter LUC construct (pGL3PAI766), the PAI-1 promoter construct mutated at the HRE-2 site (pGL3PAI766-M2), or a luciferase construct containing three EPO-HREs in front of the SV40 promoter (pGL3-EPO-HRE). The cells were treated with 6 μM DHR or RH and cultured for 24 h under normoxia (16% O2) or hypoxia (8% O2). In each experiment, the LUC activity of pGL3-EPO-HRE, pGL3PAI766, or pGL3PAI766-M2 cells at 16% O2 was set at 100%, respectively. In pGL3PAI766-M2, the wild-type sequence is shown on the upper strand, and mutated bases are given in lowercase letters. Values are given as mean ± SEM of three independent experiments. Student's t test for paired values was performed to determine significant difference. *, 16% O2 vs. 8% O2. **, 16% O2 vs. 16% O2 + DHR, or 16% O2 vs. 8% O2 + DHR; P ≤ 0.05.
Induction of HIF-1α Protein and Nuclear Translocation of HIF-1α by DHR. It was next examined whether the ·OH scavenger DHR could induce HIF-1α protein levels. Hypoxia induced HIF-1α protein levels in HepG2 cells. Treatment with DHR under normoxia also increased HIF-1α, whereas RH 123 had no effect on HIF-1α protein levels (Fig. 5).
Fig. 5.

Induction of HIF-1α protein levels by DHR. Cells were treated with either 6 μM DHR or 6 μM RH and incubated for 4 h under normoxia (16% O2) or hypoxia (8% O2). (A) The HIF-1α protein level under normoxia was set at 100%. Values are given as mean ± SEM of three independent experiments. Student's t test for paired values was performed to determine significant difference. *, HIF-1α 16% O2 vs. HIF-1α 8% O2. **, HIF-1α 16% O2 vs. HIF-1α 16%O2 + DHR; P ≤ 0.05. (B) Representative Western blot. We subjected 100 μg of protein to Western blot analysis with an Ab directed against HIF-1α or GM.
To investigate whether the mimicry of hypoxia by DHR influences HIF-1α nuclear translocation, cells were transfected with an expression vector for GFP-tagged HIF-1α. Nuclear accumulation of GFP-HIF-1α was observed after treatment with either hypoxia or DHR. In addition, PDTC, which can act as radical scavenger, also induced nuclear accumulation of HIF-1α (Fig. 6).
Fig. 6.

Induction of HIF-1α nuclear translocation by DHR. Cells were transfected with GFP-HIF-1α and cultured under normoxia (16% O2). After 48 h, cells were cultured under hypoxia (4 h) or treated with either 6 μM DHR or 100 μM PDTC under normoxia. (A) Green fluorescence indicates the accumulation of GFP-HIF-1α. (B) Microscopic images of the same fields as shown in A; the 4′,6-diamidino-2-phenylindole (DAPI) fluorescence indicates the nucleus.
Induction of HIF-1α Transactivation by DHR. There are two TADs present in HIF-1α, TADN (N-terminal; amino acids 531–575) and TADC (C-terminal; amino acids 786–826). To investigate whether DHR enhances HIF-1α transactivation, cells were cotransfected with pG5-E1B-LUC containing five Gal4-responsive elements and constructs expressing fusion proteins of the Gal4-DNA binding domain (Gal4) and either TADN or TADC.
After transfection of pG5-E1B-LUC with empty Gal4 expression vector neither hypoxia, DHR, nor RH 123 enhanced LUC activity. Cotransfection of pG5-E1B-LUC and Gal4-HIF1α-TADN enhanced LUC activity 200-fold (Fig. 7A). Hypoxia increased LUC activity in Gal4-HIF1α-TADN cotransfected cells. Exposure to DHR mimicked hypoxia, enhancing LUC activity 350-fold under normoxia, whereas RH 123 had no effect (Fig. 7A).
Fig. 7.

Induction of HIF-1α transactivation by DHR by inhibition of prolyl hydroxylase activity. (A) Cells were cotransfected with a luciferase reporter pG5-E1B-LUC and various fusion gene constructs in which the Gal4 DNA binding domain was fused to either HIF-1α TADN or TADC (Left). Mutations in constructs are shown in italics. After 24 h, transfected cells were treated with either 6 μM DHR or 6 μM RH and then cultured for 24 h under normoxia (16% O2) or hypoxia (8% O2). Values are given as mean ± SEM of four independent experiments. Student's t test for paired values was performed to determine significant difference. *, 16% O2 (Control) vs. 8% O2. **, Control vs. DHR; P ≤ 0.05. (B) VHL pull-down assay. The GST-TADN-HIF-1α protein was incubated either without extract (-E) or with extract supplemented with cofactors (+E) (see Materials and Methods) in the presence of either DHR or RH. Glutathione-Sepharose beads and [35S]VHL were added, and bound VHL was recovered, subjected to SDS/PAGE, and visualized. The input remains from directly loaded [35S]VHL. The two bands represent the 213- and 160-aa VHL products. (C) Dose-dependent inhibition of HIF prolyl hydroxylase activity by DHR. The GST-TADN-HIF-1α protein or the GST protein were incubated with HepG2 cell extract, cofactors, and 5-[14C]2-oxoglutarate in the presence of various DHR or RH concentrations. Radioactivity associated to succinate was determined. In each experiment, the basal HIF-TADN-dependent activity, which was set at 100%, was determined by subtracting the GST-associated activity. Values are given as mean ± SEM from three independent culture experiments, with each point measured in duplicate.
Cotransfection of pG5-E1B-LUC and Gal4-HIF1α-TADNM encoding a fusion protein with a hydroxylation-resistant mutation (P564A), which thus inhibits binding of VHL and subsequent degradation, led to an enhancement of LUC activity and a loss in the induction by hypoxia as well as by DHR (Fig. 7A). This result suggested that scavenging of ·OH by DHR may inhibit HIF-1α–VHL interaction because of reduction of prolyl hydroxylase activity by Fe redox chemistry. To investigate this possibility, VHL pull-down assays were performed. Binding of radioactive VHL to GST-TADN was reduced when extracts from DHR-treated cells were used to hydroxylate the GST-TADN protein. By contrast, use of cell extracts from RH 123-treated cells did not disturb HIF-1α–VHL interaction (Fig. 7B).
Furthermore, the total prolyl hydroxylase activity present in the cell extracts was measured by the formation of succinate from 2-oxoglutarate. It was found that addition of DHR to the assay had an inhibitory effect on prolyl hydroxylase activity, whereas RH 123 did not influence prolyl hydroxylase activity (Fig. 7C).
Moreover, when Gal4-HIF1α-TADC was cotransfected with pG5-E1B-LUC, the transactivation was lower than transactivation with the TADN. Again, however, hypoxia and DHR enhanced LUC activity, whereas RH 123 had no effect (Fig. 7A). The cotransfection of Gal4-HIF1α-TADC, in which the critical site for redox modification cysteine 800 was rendered nonfunctional, displayed a loss in induction of LUC activity by hypoxia as well as by DHR. RH 123, as in the other experiments, had no effect (Fig. 7A).
Discussion
In this study, the usage of gene constructs allowing the cell compartment-specific expression of the fluorescent proteins ECFP or DsRed together with the ·OH scavenger DHR enabled the in vivo localization of an H2O2-using Fenton reaction within the ER. Scavenging of ·OH generation at the ER by DHR interfered with O2 signaling, leading to an enhanced activation of HIF-1, HIF-1-dependent PAI-1, and HO-1 gene expression.
ROS as Messengers of the O2 Signal: Localization and Kinetics. The present study localized the Fenton reaction at the ER but not at mitochondria or other intracellular compartments. This finding is in line with a study showing a perinuclear Fenton reaction, as well as iron deposits, close to the nucleus in intimate contact with ER (16). To localize the Fenton reaction, we used 2P-CLSM, which is nonphototoxic, because of infrared light (23). Furthermore, wavelength tunability enabled optimal fluorescence excitation without crosstalk of fluorescence, and maintenance of physiological conditions in our microscope culture minimized cell stress. The finding that DHR was fully convertible to RH 123 (which is irreversible) only under normoxia in combination with 5-s blue-light illumination renders short-term blue-light illumination an ideal proof of intracellular ROS turnover (Fig. 2 A). Under these conditions, hypoxia was accompanied by a decrease in ROS levels, in agreement with earlier studies (24). However, other studies (3, 25) have described enhanced ROS under hypoxia and showed reversibility of normally irreversible ROS-induced dye oxidation, suggesting that reversibility of the signal was because of fluorescence-intensity changes of the oxidized dye. Thus, the present study underlines the importance of ROS as possible second messengers for O2 signaling.
Redox State and Hypoxia-Regulated Gene Expression. Our study demonstrated that, under normal conditions, ·OH generation takes place at the ER but not within mitochondria. This finding is supported by the fact that, under physiological conditions, mitochondria work at a membrane potential of about -120 mV. Thus, ROS generation in mitochondria can be measured only when this potential decreases below -120 mV, which occurs under stress-induced increases of calcium ions or decreases of the ATP/ADP ratio (26). Thus, this study also suggests that an operating respiratory chain is not involved in ROS generation (26). Further, the relevance of the mitochondrial respiratory chain as oxygen sensor was questioned in experiments with various cell lines defective in components of the electron-transport chain. These studies showed decreased H2O2 levels under hypoxia and activation of HIF-1α with no difference in wild-type cells (27–29).
The H2O2 levels are controlled mainly by glutathione peroxidase in cytosol and mitochondria or by catalase in peroxisomes. Because glutathione peroxidase (KM =100 μM) and catalase (KM = 100 mM) (30) require relatively high H2O2 concentrations, it seems conceivable that lower concentrations may be converted nonenzymatically in an ER-localized Fenton reaction. The resulting ·OH can then directly or indirectly modify transcription factors, such as HIF-1α (8). This proposal is in line with this study and investigations showing that H2O2 destabilized HIF-1α and HIF-2α (EPAS-1) (31, 32). Moreover, our study shows a compartmentalization of the redox-dependent regulation of HIF-1α because ·OH and ER fluorescence coincide with HIF-1α under normoxia (Fig. 2B). Furthermore, scavenging of ·OH by DHR at ER promoted HIF-1α nuclear translocation as well as HIF-1 activity, which subsequently enhanced PAI-1 and HO-1 gene expression (Figs. 3 and 4). Thereby, it remains uncertain why DHR and hypoxia were not additive in PAI-1 promoter regulation, but this phenomenon may depend on a specific PAI-1 promoter context.
The localization of a substantial amount of HIF-1α to ER under normoxia does not seem to be caused by an ER retention sequence but by the interaction of HIF-1α with VHL, which is retained at the ER by a 64-aa region (33). Because interaction with VHL is a prerequisite for HIF-1α degradation, it might be true that HIF-1α, which did not colocalize with ER under normoxia, represents a part already associated with proteasomes.
The scavenging of ·OH by DHR acted mainly on TADN and TADC (Fig. 7A). These domains appear to be affected mainly by a new family of prolyl (34–37) and asparagyl (38) hydroxylases. Especially for HIF-1α, hydroxylation of Pro-564 under normoxia would enable VHL binding, which then targets HIF-1α for proteasomal degradation (17). However, the activity of prolyl hydroxylase depends on Fe2+, thus pointing to the modulation of its activity by a redox cycle. This proposal was supported by the present study because scavenging of ·OH that was formed during an Fe2+-dependent Fenton reaction inhibited HIF-1α prolyl hydroxylase activity and binding of VHL to TADN (Fig. 7 B and C).
The HIF-1α-TADC appeared to be critically regulated by recruitment of coactivators, such as CBP/p300 and SRC-1 (16, 39). Thereby, interaction of CBP/p300 and SRC-1 with TADC depended on the redox state of cysteine 800, which can be modified by Ref-1 (16, 31, 39). Because mutation of C-800 abolished DHR-mediated TADC activation, DHR seems to affect the redox status of Cys-800, and thus the recruitment of CBP/p300, directly. This mechanism may act together with the factor-inhibiting HIF (FIH-1), which has been found to hydroxylate Asn-803 of HIF-1α-TADC under normoxia, thus inhibiting binding of CBP/p300 (40). Similarly, the modification of certain cysteine residues may also account for redox modulation of other transcription factors, such as c-Jun or NF-κB (1).
In summary, the present study has shown that generation of ·OH in a Fenton reaction at the ER contributes to HIF-1α regulation by either modulation of prolyl hydroxylase activity or interference with redox-sensitive residues, such as Cys-800, within the TADC. Thus, under normal physiological conditions, the changes of the redox status within a cell may contribute to an efficient and fast-responding O2-sensing system.
Acknowledgments
We thank Dr. Patrick Maxwell for the kind gift of the pCMV-HA-VHL construct. This study was supported by Deutsche Forschungsgemeinschaft Grants SFB 402, Teilprojekt A1, and GRK 335 (to T.K.); Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie Grant 13N7447/5 (to H.A.); and grants from Silicon Graphics, Nikon, and Newport (to H.A.).
Abbreviations: ·OH, hydroxyl radical; HIF-1, hypoxia-inducible factor 1; DHR, dihydrorho-damine; ER, endoplasmic reticulum; VHL, von Hippel–Lindau; TADx; transactivation domain x; ROS, reactive oxygen species; EPO, erythropoietin; HRE, hypoxia response element; ECFP, enhanced cyan fluorescent protein; RH, rhodamine; GM, Golgi membrane; 2P-CLSM, two-photon laser confocal microscopy.
References
- 1.Bunn, H. F. & Poyton, R. O. (1996) Physiol. Rev. 76, 839-885. [DOI] [PubMed] [Google Scholar]
- 2.Gorlach, A., Holtermann, G., Jelkmann, W., Hancock, J. T., Jones, S. A., Jones, O. T. & Acker, H. (1993) Biochem. J. 290, 771-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandel, N. S., McClintock, D. S., Feliciano, C. E., Wood, T. M., Melendez, J. A., Rodriguez, A. M. & Schumacker, P. T. (2000) J. Biol. Chem. 275, 25130-25138. [DOI] [PubMed] [Google Scholar]
- 4.Agani, F. H., Pichiule, P., Chavez, J. C. & LaManna, J. C. (2000) J. Biol. Chem. 275, 35863-35867. [DOI] [PubMed] [Google Scholar]
- 5.Wenger, R. H. (2002) FASEB J 16, 1151-1162. [DOI] [PubMed] [Google Scholar]
- 6.Hildebrandt, W., Alexander, S., Bartsch, P. & Droge, W. (2002) Blood 99, 1552-1555. [DOI] [PubMed] [Google Scholar]
- 7.Kietzmann, T., Fandrey, J. & Acker, H. (2000) News Physiol. Sci. 15, 202-208. [DOI] [PubMed] [Google Scholar]
- 8.Kietzmann, T., Porwol, T., Zierold, K., Jungermann, K. & Acker, H. (1998) Biochem. J. 335, 425-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenza, G. L. (2001) Pediatr. Res. 49, 614-617. [DOI] [PubMed] [Google Scholar]
- 10.Wenger, R. H. & Gassmann, M. (1997) Biol. Chem. 378, 609-616. [PubMed] [Google Scholar]
- 11.Semenza, G. L. (1998) Curr. Opin. Genet. Dev. 8, 588-594. [DOI] [PubMed] [Google Scholar]
- 12.Kietzmann, T., Cornesse, Y., Brechtel, K., Modaressi, S. & Jungermann, K. (2001) Biochem. J. 354, 531-537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kietzmann, T., Roth, U. & Jungermann, K. (1999) Blood 94, 4177-4185. [PubMed] [Google Scholar]
- 14.Kallio, P. J., Okamoto, K., O'Brien, S., Carrero, P., Makino, Y., Tanaka, H. & Poellinger, L. (1998) EMBO J. 17, 6573-6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruger, M., Schwaninger, M., Blume, R., Oetjen, E. & Knepel, W. (1997) Naunyn Schmiedeberg's Arch. Pharmacol. 356, 433-440. [DOI] [PubMed] [Google Scholar]
- 16.Carrero, P., Okamoto, K., Coumailleau, P., O'Brien, S., Tanaka, H. & Poellinger, L. (2000) Mol. Cell. Biol. 20, 402-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanimoto, K., Makino, Y., Pereira, T. & Poellinger, L. (2000) EMBO J. 19, 4298-4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baader, E., Tschank, G., Baringhaus, K. H., Burghard, H. & Gunzler, V. (1994) Biochem. J. 300, 525-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kietzmann, T., Roth, U., Freimann, S. & Jungermann, K. (1997) Biochem. J. 321, 17-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Immenschuh, S., Hinke, V., Ohlmann, A., Gifhorn-Katz, S., Katz, N., Jungermann, K. & Kietzmann, T. (1998) Biochem. J. 334, 141-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehleben, W., Porwol, T., Fandrey, J., Kummer, W. & Acker, H. (1997) Kidney Int. 51, 483-491. [DOI] [PubMed] [Google Scholar]
- 22.Strohmaier, A. R., Porwol, T., Acker, H. & Spiess, E. (2000) Cells Tissues Organs 167, 1-8. [DOI] [PubMed] [Google Scholar]
- 23.Hockberger, P. E., Skimina, T. A., Centonze, V. E., Lavin, C., Chu, S., Dadras, S., Reddy, J. K. & White, J. G. (1999) Proc. Natl. Acad. Sci. USA 96, 6255-6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fandrey, J. & Genius, J. (2000) Adv. Exp. Med. Biol. 475, 153-159. [DOI] [PubMed] [Google Scholar]
- 25.Chandel, N. S., Maltepe, E., Goldwasser, E., Mathieu, C. E., Simon, M. C. & Schumacker, P. T. (1998) Proc. Natl. Acad. Sci. USA 95, 11715-11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, I., Bender, E., Arnold, S. & Kadenbach, B. (2001) Biol. Chem. 382, 1629-1636. [DOI] [PubMed] [Google Scholar]
- 27.Srinivas, V., Leshchinsky, I., Sang, N., King, M. P., Minchenko, A. & Caro, J. (2001) J. Biol. Chem. 276, 21995-21998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaux, E. C., Metzen, E., Yeates, K. M. & Ratcliffe, P. J. (2001) Blood 98, 296-302. [DOI] [PubMed] [Google Scholar]
- 29.Searle, G. J., Hartness, M. E., Hoareau, R., Peers, C. & Kemp, P. J. (2002) Biochem. Biophys. Res. Commun. 291, 332-337. [DOI] [PubMed] [Google Scholar]
- 30.Chance, B., Sies, H. & Boveris, A. (1979) Physiol. Rev. 59, 527-605. [DOI] [PubMed] [Google Scholar]
- 31.Huang, L. E., Arany, Z., Livingston, D. M. & Bunn, H. F. (1996) J. Biol. Chem. 271, 32253-32259. [DOI] [PubMed] [Google Scholar]
- 32.Wiesener, M. S., Turley, H., Allen, W. E., Willam, C., Eckardt, K. U., Talks, K. L., Wood, S. M., Gatter, K. C., Harris, A. L., Pugh, C. W. et al. (1998) Blood 92, 2260-2268. [PubMed] [Google Scholar]
- 33.Schoenfeld, A. R., Davidowitz, E. J. & Burk, R. D. (2001) Int. J. Cancer 91, 457-467. [DOI] [PubMed] [Google Scholar]
- 34.Jaakkola, P., Mole, D. R., Tian, Y. M., Wilson, M. I., Gielbert, J., Gaskell, S. J., Kriegsheim, A., Hebestreit, H. F., Mukherji, M., Schofield, C. J., et al. (2001) Science 292, 468-472. [DOI] [PubMed] [Google Scholar]
- 35.Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J. M., Lane, W. S. & Kaelin. (2001) Science 292, 464-468. [DOI] [PubMed] [Google Scholar]
- 36.Epstein, A. R., Gleadle, J. M., McNeill, L. A., Hewitson, K. S., O'Rourke, J., Mole, D. R., Mukherji, M., Metzen, E., Wilson, M. I., Dhanda, A., et al. (2001) Cell 107, 43-54. [DOI] [PubMed] [Google Scholar]
- 37.Oehme, F., Ellinghaus, P., Kolkhof, P., Smith, T., Ramakrishnan, S., Hutter, J., Schramm, M. & Flamme, I. (2002) Biochem. Biophys. Res. Commun. 296, 343. [DOI] [PubMed] [Google Scholar]
- 38.Lando, D., Peet, DJ., Whelan, DA., Gorman, J. & Whitelaw, M. L. (2002) Science 295, 858-861. [DOI] [PubMed] [Google Scholar]
- 39.Ema, M., Hirota, K., Mimura, J., Abe, H., Yodoi, J., Sogawa, K., Poellinger, L. & Fujii-Kuriyama, Y. (1999) EMBO J. 18, 1905-1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lando, D., Peet, D. J., Gorman, J. J., Whelan, D. A., Whitelaw, M. L. & Bruick, R. K. (2002) Genes Dev. 16, 1466-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]