

Abstract
Background
This study aimed to evaluate the efficacy of mesenchymal stem cell (MSC) transplantation to mitigate abnormalities in cardiac-specific and systemic metabolism mediated by a combination of a myocardial infarction and diet-induced insulin resistance.
Methods
C57BL/6 mice were high-fat fed for eight weeks prior to induction of a myocardial infarction via chronic ligation of the left anterior descending coronary artery. MSCs were administered directly after myocardial infarction induction through a single intramyocardial injection. Echocardiography was performed prior to the myocardial infarction as well as seven and 28 days post-myocardial infarction. Hyperinsulinemic-euglycemic clamps coupled with 2-[14C]deoxyglucose were employed 36 days post-myocardial infarction (13 weeks of high-fat feeding) to assess systemic insulin sensitivity and insulin-mediated, tissue-specific glucose uptake in the conscious, unrestrained mouse. High-resolution respirometry was utilized to evaluate cardiac mitochondrial function in saponin-permeabilized cardiac fibers.
Results
MSC administration minimized the decline in ejection fraction following the myocardial infarction. The greater systolic function in MSC-treated mice was associated with increased in vivo cardiac glucose uptake and enhanced mitochondrial oxidative phosphorylation efficiency. MSC therapy promoted reductions in fasting arterial glucose and fatty acid concentrations. Additionally, glucose uptake in peripheral tissues including skeletal muscle and adipose tissue was elevated in MSC-treated mice. Enhanced glucose uptake in these tissues was associated with improved insulin signalling as assessed by Akt phosphorylation and prevention of a decline in GLUT4 often associated with high-fat feeding.
Conclusions
These studies provide insight into the utility of MSC transplantation as a metabolic therapy that extends beyond the heart exerting beneficial systemic effects on insulin action.
Keywords: Diabetes, Glucose uptake, Isotopic tracer, Mitochondria, Myocardial infarction
Background
The regional loss of cardiac myocytes following a myocardial infarction (MI) compromises the ability of the heart to pump blood to peripheral sites and may initiate compensatory mechanisms in an attempt to preserve cardiac function [1]. Researchers in regenerative medicine have become excited by the possibility of repairing and/or replacing pathological tissue via administration of exogenous cells [2-4]. Experimental study and clinical trials have identified cell-based therapies to consistently improve systolic function [5-10]. However, cell transplantation has been less than adequate at replacing the lost cells; which may be, in part, due to poor cell persistence following administration in the infarcted heart [11,12].
In the presence of poor cell survival and engraftment, the mechanisms promoting beneficial results reported following stem cell transplantation have been subject to an increasing number of studies that suggest cell-mediated paracrine effects are likely the major mediator for the improvement in cardiac function and the attenuation or slowing of the maladaptive processes [10]. The release of paracrine factors have a potential regulatory role in various processes that influence cardiac function including anti-apoptotic signalling and neovascularization as well as the modulation of inflammation, fibrosis, cardiac contractility, host stem cell activation and metabolism [10,13].
In terms of cardiac metabolism, animal studies indicate the ability of stem cell transplantation, specifically mesenchymal stem cell (MSC) transplantation, to lessen aberrations in glucose utilization, mitochondrial function and high-energy phosphate provision in the infarcted heart [14-18]. Since diabetes and heart disease often co-aggregate [19-21], the absence of metabolic dysfunction associated with type 2 diabetes in previous studies may not provide a complete model in assessing the utility of cell transplantation for the infarcted heart. Of particular interest is insulin resistance, as it enhances the risk of experiencing cardiovascular disease and a MI [22,23]. Moreover, individuals with insulin resistance and type 2 diabetes exhibit a greater probability of developing heart failure and higher mortality following a MI [22,24,25].
From a mechanistic perspective, insulin resistance may enhance cardiac pathology following a MI by inhibiting changes in metabolic characteristics that are initially suggested to be adaptive [26-30]. The cardiac metabolic phenotype shifts away from its predominant reliance on fatty acid oxidation towards increased glucose utilization early post-MI [26-30]. Glucose oxidation is more oxygen efficient and may provide a more effective means of ATP provision [31]. An alternative means of providing energy for cardiac contraction in an economical fashion involves the down-regulation of mitochondrial uncoupling proteins (UCPs) in the failing heart [31,32]. UCPs create an environment where oxygen consumption does not contribute to ATP synthesis, which is achieved through the uncoupling of the electrochemical gradient and ADP phosphorylation [33]. These adaptations in cardiac metabolic pathways may assist in meeting the energetic demand of contraction. However, this metabolic flexibility demonstrated by the heart is impeded in insulin resistance [34]. Insulin resistance inhibits glucose uptake creating a situation of severe detriment given both fatty acid and glucose metabolism are impaired and energy provision is compromised in the insulin resistant, infarcted heart [34]. Further insult occurs from hyperglycemia and hyperlipidemia [35,36]. For example, elevated circulating fatty acids promote an increase in cardiac UCP3 levels [37]. The increase in UCP3 may stimulate higher mitochondrial uncoupling and further energy demand/supply disparities.
Given the deleterious impact of insulin resistance on the infarcted heart, this study aimed to identify whether the efficacy of MSC administration to minimize alterations in metabolic processes that assist in ATP provision following a MI is maintained in a murine model of diet-induced insulin resistance/MI.
Methods
See Figure 1 for a schematic of experimental outline.
Figure 1.
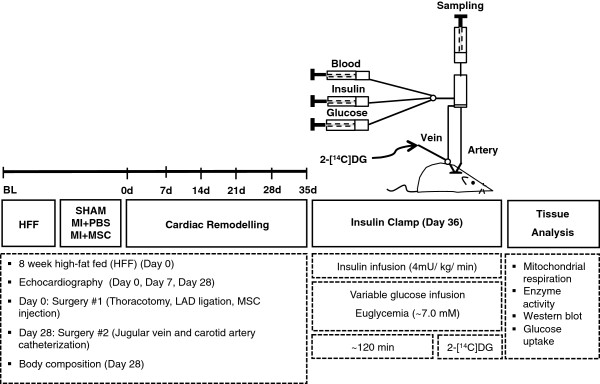
Schematic representation of experimental procedures and timeline. Echocardiography on conscious mice was performed prior to, seven and 28 days following ligation of the left anterior descending coronary artery (LAD). Arterial and jugular catheterization was performed 28 days following the LAD ligation for the sampling and infusion protocols of the hyperinsulinemic-euglycemic (insulin) clamp. Insulin clamps were performed following seven days of recovery from the catheterization surgeries (36 days post-LAD ligation/13 weeks of high-fat feeding) to assess insulin action in the conscious, unrestrained mouse. Isotopic tracer 2-[14C]deoxyglucose (2-[14C]DG) administration during the insulin clamps allowed for whole body and tissue-specific substrate uptake to be assessed in vivo. Additional experiments included evaluation of mitochondrial respiration in permeabilized cardiac tissue and key molecular regulators of metabolism by immunoblotting. MI + PBS, myocardial infarction + phospho-buffered saline; MI + MSC, myocardial infarction + mesenchymal stem cells.
Animal characteristics and myocardial infarction
Procedures were approved by the Vanderbilt University Animal Care and Use Committee and performed according to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85–23, revised 1996, A3227-01). Thirteen week-old, eight week high-fat fed (HFF) (F3282, 60% calories from fat, Bio-Serv, Frenchtown, NJ, USA), C57BL/6J, male mice (Jackson Laboratories, Bar Harbour, ME, USA) were randomly separated into three groups: SHAM, MI + phosphate buffered saline (MI + PBS) and MI + mesenchymal stem cells (MI + MSC). A left anterior descending coronary artery (LAD) chronic ligation induced a MI as described [18]. Following the ligation, 25 μl of PBS (pH 7.2) or 2.5 × 105 MSCs in 25 μl of PBS (pH 7.2) were injected into the blanching peri-infarct area of the MI + PBS or MI + MSC mice, respectively.
Mesenchymal stem cells
MSCs reputedly display immunoprivileged characteristics that make the use of this cell type attractive for cell transplantation purposes [38-40]. These unique characteristics provide promise for use of MSCs in not only autologous transplantation but allogenic and xenogenic transplantation. Given this, human MSCs were purchased from the Texas A&M Health Science Center College of Medicine Institute for Regenerative (Temple, TX, USA) that provides standardized MSC preparations through support from the NIH/NCRR (P40 RR 17447–06). The MSCs have been described to consistently exhibit trilineage potential and be CD90+, CD105+, CD49c+, CD49f+, CD166+, CD105+, CD29+, CD44+, CD45-, CD34-, CD117- and CD36-[41]. MSCs were expanded in alpha minimum essential medium (αMEM) with L-glutamine (Life Technologies, Burlington, ON, Canada), 16.5% defined FBS (HyClone, Logan, UT, USA), penicillin (100 units/ml) and streptomycin (100 μg/ml). All mice received MSCs from a single donor and passages 3–8 were utilized.
Cardiac performance
To assess cardiac contractile function, M-mode echocardiography was completed in conscious mice at baseline, seven and 28 days post-MI as previously described [42,43].
Catheterization procedures
Chronic catheterization surgeries were performed 28 days post-ligation (12 weeks HFF) as those previously described for hyperinsulinemic-euglycemic (insulin clamps) [18]. Post-surgery, the mice were housed individually for seven days to ensure the mice were within 10% of pre-surgical weight prior to insulin clamps.
Hyperinsulinemic-euglycemic clamps
Our laboratory has shown that insulin resistance is induced in 12 weeks HFF C57BL/6 mice [44,45]. Insulin clamps were performed with procedural considerations previously described [46] at 36 days post-MI (13 weeks HFF). Mice were fasted at 7:00 a.m., five hours prior to initiation of the insulin clamps. One hour prior to the experiment, the externalized mouse catheters were connected to catheter extensions attached to infusion syringes. Just prior to the onset of the insulin clamp, an arterial blood sample was obtained to evaluate levels of arterial glucose, insulin, non-esterified fatty acids (NEFA) and hematocrit. Each experiment consisted of a continuous infusion (4 mU/kg/min) of insulin. Euglycemia (~7.0 mM) was maintained during the insulin clamps. Venous infusion of saline-washed erythrocytes (5 μl/min) during the insulin clamp prevented a decline in hematocrit due to arterial sampling. Insulin clamp duration averaged 122.07 ± 9.48 minutes until a stable glucose infusion rate (GIR) and euglycemia was achieved for at least 30 minutes. Arterial blood was sampled to determine glucose levels (t = 0 minutes) had achieved the steady state. Immediately following the blood sampling, a bolus containing 2-[14C]deoxyglucose (2-[14C]DG; 481 kBq) was administered into the jugular vein to provide an index of tissue-specific glucose uptake. At t = 2, 5, 10, 15, 20 minutes, arterial blood was sampled to determine glucose and 2-[14C]DG. At t = 30 minutes, arterial blood was taken for the measurement of glucose, experimental insulin, 2-[14C]DG and hematocrit. Plasma was stored at −20°C until analysis. Following the insulin clamp, mice were killed via cervical dislocation and tissues were immediately excised for analysis or stored at −80°C. The heart, soleus, gastrocnemius, superficial vastus lateralis and white adipose tissue from the epididymal fat pad were collected.
Plasma analyses
Arterial insulin was assayed via a double antibody method [47]. Plasma NEFAs (NEFA C kit; Wako Chemicals, Richmond, VA, USA) and glucose were determined spectrophotometrically as previously described [18]. Plasma 2-[14C]DG was assessed as previously outlined [45].
Tissue-specific substrate kinetics
Tissue 2-[14C]DG and tissue phosphorylated 2-[14C]DG (2-[14C]DG-P) were determined as previously described [45]. The metabolic index of glucose (Rg) uptake was calculated [48] and expressed as previously described [49].
Mitochondrial respiration and enzymatic measurements
Cardiac peri-infarct fibers were prepared and saponin-permeabilized as described [50]. High-resolution respirometry (Oroboros Instruments, Innsbruck, Austria) was performed in duplicate at 37°C in MiR05 (Final concentration: 0.5 mM EGTA, 3 mM MgCl2·6H2O, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 1 g/L BSA, 60 mM potassium-lactobionate, 110 mM sucrose, pH 7.1, adjusted at 30°C). Substrates (final concentration) included 10 mM glutamate plus 2 mM malate and 5 mM pyruvate, 5 mM ADP and 10 mM succinate. 10 mM cytochrome c was added to ensure the outer mitochondrial membrane was intact after processing. Peri-infarct citrate synthase activity was determined via previous methods [18].
Immunoblotting
Tissues were homogenized in a lysis buffer (final concentration) containing 20 mM NaCl, 20 mM Tris–HCl, 0.1 mM EDTA, 1% Triton X-100, 0.5% (wt./vol.) sodium deoxycholate, and 0.1% β-Mercapto Ethanol (vol./vol.) (pH 7.4) in the presence of a protease inhibitor cocktail (Sigma-Aldrich, Oakville, ON, Canada) and phosphatase inhibitor cocktail (Thermo Fisher Scientific, Mississauga, ON, Canada). Cardiac (20–30 μg), gastrocnemius (40 μg) and white adipose tissue (40 μg) proteins were resolved on NuPAGE 4-12% (vol./vol.) Bis-Tris gels (Life Technologies) and transferred to a polyvinylidene fluoride membrane. Membranes were probed with peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α; Santa Cruz Biotechnology, Santa Cruz, CA, USA), glucose transporter 4 (GLUT4; Abcam, Cambridge, MA, USA), hexokinase II (HKII; Chemicon, Temecula, CA, USA), UCP3 (Abcam), phospho-Akt(Ser473) (p-Akt; Cell Signaling Technology, Whitby, ON, Canada) and Akt (Cell Signaling Technology) and oxidative phosphorylation complexes I-V (OXPHOS CI-CV; Abcam) antibodies. A goat-anti-mouse secondary antibody (Thermo Fischer Scientific) was used for the OXPHOS CI-CV primary antibody cocktail. A goat-anti rabbit secondary antibody (Cell Signaling Technologies) was used for all other primary antibodies. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Abcam) expression was employed as a control for all immunoblots.
Statistical analyses
ANOVA or two-way repeated measures ANOVA were performed to detect statistical differences (p < 0.05) as appropriate followed by Tukey’s post hoc tests. All data are reported as means ± SEM.
Results
Stem cell therapy promotes improvement in cardiac contractile function
Contractile abnormalities were observed using echocardiography (Table 1) in the MI + PBS group seven and 28 days following a MI as indicated by the depression in ejection fraction (Figure 2a) and fractional shortening (Figure 2b). The MSC-treated animals displayed a comparable reduction in ejection fraction as the MI + PBS group at seven days post-MI (Figure 2a). However, the MSC-treated hearts exhibited a greater ejection fraction than the MI + PBS mice at 28 days following cardiac insult (Figure 2a). The MI + MSC mice, similar to the MI + PBS animals, displayed a depression in fractional shortening at seven and 28 days post-MI (Figure 2b).
Table 1.
Cardiovascular parameters in conscious, high-fat fed, C57BL/6 mice
| |
Baseline |
7 days post-MI |
28 days post-MI |
||||||
|---|---|---|---|---|---|---|---|---|---|
| SHAM | MI + PBS | MI + MSC | SHAM | MI + PBS | MI + MSC | SHAM | MI + PBS | MI + MSC | |
|
HR (bpm) |
647 ± 16 |
662 ± 16 |
689 ± 9* |
720 ± 8 |
699 ± 10 |
713 ± 12 |
710 ± 11 |
720 ± 13 |
731 ± 6 |
|
FS (%) |
49.58 ± 0.75 |
48.88 ± 1.02 |
48.74 ± 0.46 |
49.12 ± 0.78 |
29.88 ± 3.16* |
30.90 ± 2.03* |
48.99 ± 0.70 |
27.34 ± 2.84* |
32.90 ± 2.02* |
|
EF (%) |
82.07 ± 0.72 |
81.37 ± 1.01 |
81.23 ± 0.45 |
81.80 ± 0.76 |
56.51 ± 4.88* |
58.48 ± 2.9* |
81.48 ± 0.74 |
52.80 ± 2.93* |
61.10 ± 2.88*† |
|
IVSd (mm) |
0.77 ± 0.02 |
0.81 ± 0.02 |
0.79 ± 0.01 |
0.76 ± 0.02 |
0.73 ± 0.04 |
0.84 ± 0.01*† |
0.79 ± 0.01 |
0.76 ± 0.03 |
0.85 ± 0.01*† |
|
LVIDd (mm) |
3.19 ± 0.07 |
3.21 ± 0.08 |
3.28 ± 0.04 |
3.02 ± 0.04 |
4.31 ± 0.24* |
4.01 ± 0.13* |
3.23 ± 0.08 |
4.32 ± 0.21* |
4.27 ± 0.13* |
|
LVPWd (mm) |
0.72 ± 0.01 |
0.69 ± 0.02 |
0.80 ± 0.02† |
0.74 ± 0.02 |
0.68 ± 0.07 |
0.86 ± 0.03*† |
0.77 ± 0.02 |
0.71 ± 0.04 |
0.91 ± 0.03 |
|
IVSs (mm) |
0.91 ± 0.02 |
0.91 ± 0.02 |
0.90 ± 0.01 |
0.88 ± 0.02 |
0.87 ± 0.05 |
0.92 ± 0.02 |
0.88 ± 0.01 |
0.84 ± 0.02 |
0.96 ± 0.01*† |
|
LVIDs (mm) |
1.61 ± 0.04 |
1.65 ± 0.06 |
1.68 ± 0.03 |
1.54 ± 0.04 |
3.06 ± 0.28* |
2.79 ± 0.16* |
1.65 ± 0.06 |
3.17 ± 0.26* |
2.89 ± 0.17* |
| LVPWs (mm) | 0.97 ± 0.02 | 0.98 ± 0.04 | 1.04 ± 0.01 | 0.88 ± 0.02 | 0.83 ± 0.09 | 1.01 ± 0.02*† | 0.96 ± 0.02 | 0.82 ± 0.06* | 1.11 ± 0.03*† |
Data are mean ± SEM for n = 8-13 mice per group.
*p < 0.05 vs. SHAM at specified time point.
†p < 0.05 vs. MI + PBS at specified time point.
Abbreviations: HR, heart rate; FS, fractional shortening; EF, ejection fraction; IVSd, interventricular septal thickness in diastole; LVIDd, left ventricle (LV) end-diastolic dimension; LVPWd, LV posterior wall thickness in diastole; IVSs, interventricular septal thickness in systole; LVIDs, LV end-systolic dimension; LVPWs, LV posterior wall thickness in systole.
Figure 2.

Cardiac functional and hypertrophic indices. (a) Cardiac ejection fraction (%) prior to, seven and 28 days following a MI. n = 8-13 mice per group. (b) Cardiac fractional shortening (%) prior to, seven and 28 days following a MI. n = 8-13 mice per group. (c) Heart weight 36 days post-MI surgery. n = 11-14 mice per group. (d) Heart weight-to-body weight ratio 36 days following a MI. n = 11-14 mice per group. (e) Time course of body weight from one week prior to chronic left anterior descending coronary artery ligation to five weeks post-ligation. n = 10-12 mice per group. (f) Tibial length 36 days following a MI. n = 11-14 mice per group. (g) Heart weight-to-tibial length 36 days post-MI. n = 11-14 mice per group. Data are mean ± S.E.M. *p < 0.05 vs. SHAM. †p < 0.05 vs. MI + PBS.
Indices of cardiac structural alterations were also assessed. Heart weight (Figure 2c) and heart-to-body weight (Figure 2d) were increased in MI + PBS mice 36 days post-MI. A comparable elevation in these hypertrophy markers was observed in the MSC-treated mice (Figure 2c,d). Of note, measurement of body weight from one week prior to the ligation surgery to five weeks post-MI indicated that the MI + PBS mice were lower as a result of the MI (Figure 2e). In contrast, mice receiving MSC transplantation displayed a comparable body weight to animals of the SHAM group (Figure 2e). Given the changes in body weight, tibial length was used to normalize heart weight and provide another index of pathological cardiac hypertrophy. Tibial length was unchanged between groups (Figure 2f). Both infarcted groups displayed an elevated heart weight-to-tibial length compared to the SHAM group (Figure 2f) indicating a hypertrophic response to the infarct that was not inhibited by the MSC therapy.
Enhanced insulin-stimulated cardiac glucose utilization in MSC-treated mice
Isotopic glucose (2-[14C]DG) was administered during the insulin clamps to assess tissue-specific glucose uptake. A MI did not influence regional insulin-stimulated glucose uptake (Rg) in the heart. The remote left ventricle (MI + PBS LV) and the peri-infarct region (MI + PBS PI) in the MI + PBS animals were comparable to the SHAM group (Figure 3a,b). In contrast, intramyocardial injection of MSCs enhanced cardiac Rg in response to insulin. The remote left ventricle of the MSC-treated mice (MI + MSC LV) displayed greater Rg compared to that of the MI + PBS mice (Figure 3a). Furthermore, the insulin-mediated response of the peri-infarct region of the MI + MSC hearts (MI + MSC PI) was more apparent. The MI + MSC PI region exhibited a higher Rg than the SHAM hearts and the peri-infarct region of the MI + PBS group (Figure 3b).
Figure 3.

Regional insulin-stimulated cardiac glucose uptake. (a) Metabolic index of glucose uptake (Rg) in the remote left ventricle and (b) peri-infarct region of the left ventricle. Cardiac Rg values are relative to brain Rg. n = 8-9 mice per group. (c) Remote left ventricle and (d) peri-infarct peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1α), glucose transporter 4 (GLUT4) and hexokinase II (HKII) as determined by immunoblotting. (e) Left ventricle and (f) peri-infarct phospho-Akt (p-Akt), Akt and p-Akt-to-total Akt ratio (p-Akt/Akt) as determined by immunoblotting. (g) Representative immunoblotting performed to measure PGC-1α, GLUT4, HKII, p-Akt and Akt. Cardiac proteins are normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content and are relative to the SHAM group. n = 6 mice per group. Data are mean ± S.E.M. *p < 0.05 vs. SHAM. †p < 0.05 vs. MI + PBS.
To identify the mechanisms by which MSC administration promoted cardiac glucose uptake key regulators of glucose metabolism were evaluated. The remote left ventricle and peri-infarct PGC-1α, GLUT4 and HKII were comparable between groups (Figure 3c,d). The insulin signalling pathway was also probed by determining p-Akt(Ser473) and total Akt. Again, the regional p-Akt-to-Akt ratio was similar between groups (Figure 3e,f).
MSC transplantation augments mitochondrial physiology
Given both contractile function and substrate utilization are influenced by energy metabolism, polarographic oxygen flux measurements were performed to assess integrative mitochondrial OXPHOS. The MI + PBS peri-infarct fibers exhibited a reduced basal oxygen consumption (V0) supported by glutamate, malate and pyruvate compared to the SHAM group (Figure 4a). Interestingly, MSC-treated hearts displayed an even further depression in basal oxygen flux compared to the MI + PBS and SHAM peri-infarct regions (Figure 4a). ADP-stimulated oxygen consumption through complex I was reduced in MI + PBS and MI + MSC animals (VMAX-CI; Figure 4a). Similarly, ADP-stimulated oxygen flux via convergent electron flux through mitochondrial complexes I + II was reduced in both infarcted groups (VMAX-CI + CII; Figure 4a). Intriguingly, the respiratory control ratio (RCR) was reduced in the MI + PBS hearts compared with the MI + MSC cardiac fibers (Figure 4b). This suggests a decrease in OXPHOS efficiency in the MI + PBS mice. Citrate synthase activity was assessed to identify differences in mitochondrial content. The MI + PBS group exhibited a decline in citrate synthase activity (Figure 4c). The preservation of cardiac citrate synthase activity in the MI + MSC mice prompted the evaluation of mitochondrial proteins in an effort to identify mechanisms contributing to the reduced V0 but greater RCR. Protein expression of mitochondrial OXPHOS complexes showed the MI + MSC mice to have a lower complex I protein in the remote left ventricle region (Figure 4d,f). Similarly, the MI + PBS and MI + MSC peri-infarct region exhibit a reduced complex I protein compared to the SHAM heart (Figure 4e,f). Surprisingly, there was an added effect of the MSC therapy. The MI + MSC peri-infarct region displayed a lower complex II and complex V compared to the SHAM hearts (Figure 4e,f). Another potential contributor to the lower basal oxygen flux but elevated RCR is the peri-infarct UCP3 that was lower in the MI + MSC mice (Figure 4e,f).
Figure 4.

Cardiac mitochondrial function and characteristics. (a) Peri-infarct permeabilized cardiac fiber basal oxygen consumption supported by glutamate, malate and pyruvate (V0), maximal oxygen consumption (ADP-stimulated) supported by glutamate, malate and pyruvate through complex I (VMAX-CI) and maximal convergent oxygen flux supported by glutamate, malate, pyruvate and succinate (VMAX-CI+CII). n = 8-9 mice per group. (b) Respiratory control ratio (RCR; defined as VMAX-CI/V0). n = 8-9 mice per group. (c) Peri-infarct citrate synthase activity (CSA) (mmol/min/mg protein). n = 8-9 mice per group. (d) Remote Left ventricle and (e) peri-infarct mitochondrial oxidative phosphorylation (OXPHOS) complexes I-V (CI-CV) and uncoupling protein 3 (UCP3) as determined by immunoblotting. (f) Representative immunoblotting of the regional protein levels OXPHOS complexes I-V and UCP3. Cardiac protein was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content and expressed relative to the SHAM group. n = 6 mice per group. Data are mean ± S.E.M. *p < 0.05 vs. SHAM. †p < 0.05 vs. MI + PBS.
Reduced fasting plasma glucose and fatty acids following MSC administration
High-fat diets have been reported to promote an increase in tissue UCP3 protein. More specifically, cardiac UCP3 positively correlates with circulating fatty acid levels [37]. As such, we evaluated fasting plasma non-esterified fatty acid (NEFA) concentration. There was a lowering of NEFA levels in the MI + MSC mice (Table 2). In addition, fasting plasma glucose concentration was reduced following MSC therapy (Table 2). Of note, arterial insulin concentrations in the fasted state and during the insulin clamp were both similar between groups (Table 2).
Table 2.
Biometric characteristics of high-fat fed, C57BL/6 mice
| SHAM | MI + PBS | MI + MSC | |
|---|---|---|---|
|
Fasting Plasma Glucose (mM) |
11.37 ± 0.48 |
10.81 ± 0.43 |
9.06 ± 0.32*† |
|
Fasting Plasma NEFA (mM) |
1.04 ± 0.05 |
1.09 ± 0.06 |
0.8 ± 0.06*† |
|
Fasting Plasma Insulin (μU/ml) |
60.08 ± 13.22 |
50.76 ± 16.15 |
42.46 ± 11.13 |
|
Experimental Plasma Insulin (μU/ml) |
128.02 ± 15.77 |
108.17 ± 8.84 |
160.04 ± 24.32 |
|
Muscle (%) |
68.70 ± 1.76 |
77.75 ± 1.96* |
67.88 ± 2.38† |
|
Fat (%) |
29.53 ± 1.80 |
20.74 ± 2.02* |
30.25 ± 2.42† |
| Free Fluid (%) | 1.77 ± 0.11 | 1.51 ± 0.17 | 1.87 ± 0.27 |
Data are mean ± SEM.
n = 11-12 mice per group for plasma glucose, n = 10-12 for plasma NEFA, n = 8-9 for fasting plasma insulin, n = 8 for experimental plasma insulin, n = 9-13 for body composition.
*p < 0.05 vs. SHAM.
†p < 0.05 vs. MI + PBS.
Systemic insulin sensitivity elevated by MSC injection
The lowering of fasting plasma glucose and fatty acids suggested a systemic effect of the MSC administration. To this end, insulin clamps were performed in the conscious, unrestrained mouse to evaluate whole body glucose disposal in response to insulin in vivo. The GIR required to maintain an arterial glucose concentration of 7.0 mM was elevated in MI + MSC mice throughout the duration of the insulin clamp (Figure 5a). The GIR for the MI + PBS mice was similar to the SHAM group (Figure 5a). Arterial glucose was comparable between groups during the insulin clamp (Figure 5b).
Figure 5.
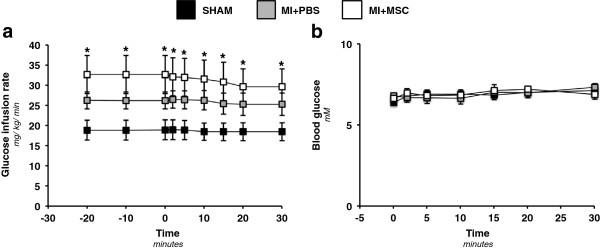
Whole body insulin sensitivity and blood glucose during the hyperinsulinemic-euglycemic (insulin) clamp. (a) Glucose infusion rate (GIR) during the insulin clamp. The GIR is equivalent to the whole body glucose disposal rate in response to venous insulin infusion. The insulin infusion was for approximately 120 minutes prior to 2-[14C]deoxyglucose (2-[14C]DG) administration. The GIR is displayed as a time course commencing twenty minutes prior to administration of 2-[14C]DG (−20 minute time point) to 30 minutes following 2-[14C]DG infusion (30 minute time point). (b) Arterial, blood glucose concentration following 2-[14C]DG for 30 minutes post-administration. Data are mean ± SEM for n = 9-12 mice per group. *p < 0.05 vs. SHAM.
Increased peripheral tissue glucose uptake post-MSC therapy
In agreement with the increased GIR, the MSC administration increased insulin-stimulated Rg in peripheral tissues. The MI + MSC soleus, superficial vastus lateralis, gastrocnemius and white adipose tissue had a greater Rg compared to the same tissues of the SHAM group (Figure 6a-d). The MI + MSC gastrocnemius also exhibited an elevated Rg compared to that of the MI + PBS mice (Figure 6c). The MI + PBS mice displayed a higher Rg in white adipose tissue compared to the SHAM group (Figure 6d). The protein expression of regulators involved in insulin-mediated Rg was evaluated in the gastrocnemius and white adipose tissue. GLUT4 was higher in the MI + MSC gastrocnemius compared to the SHAM group (Figure 6e,f). Also, gastrocnemius p-Akt and the p-Akt-to-Akt ratio were greater in the MI + MSC mice (Figure 6e,f). Both the MI + MSC and MI + PBS groups displayed higher GLUT4 in the white adipose tissue (Figure 6g,h). However, only the MI + MSC exhibited elevated p-Akt relative to the SHAM group in white adipose tissue (Figure 6g,h). Furthermore, the p-Akt-to-Akt ratio in MI + MSC white adipose tissue was greater than that of the SHAM and MI + PBS mice (Figure 6g).
Figure 6.

Insulin-stimulated peripheral tissue glucose utilization. (a) Metabolic index of glucose uptake (Rg) in the soleus. (b) Metabolic index of glucose uptake (Rg) in the superficial vastus lateralis. (c) Metabolic index of glucose uptake (Rg) in the gastrocnemius. (d) Metabolic index of glucose uptake (Rg) in white adipose tissue. Tissue Rg values are relative to brain Rg. n = 9 mice per group. (e) Gastrocnemius glucose transporter 4 (GLUT4), phospho-Akt (p-Akt), Akt and p-Akt-to-total Akt ratio (p-Akt/Akt) as determined by immunoblotting. n = 6-8 mice per group. (f) Representative immunoblotting performed to measure gastrocnemius GLUT4, p-Akt and Akt. (g) White adipose tissue glucose transporter 4 (GLUT4), phospho-Akt (p-Akt), Akt and p-Akt-to-total Akt ratio (p-Akt/Akt) as determined by immunoblotting. n = 6 mice per group. (h) Representative immunoblotting performed to measure white adipose tissue GLUT4, p-Akt and Akt. Protein levels are normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) content and are expressed relative to the SHAM group. Data are mean ± S.E.M. *p < 0.05 vs. SHAM. †p < 0.05 vs. MI + PBS.
Discussion
Numerous studies have reported that cell-based therapeutics can minimize insult to the infarcted heart, in large part, through trophic mechanisms [10,13]. However, as stem cell therapy transfers to clinical trials and ideally to routine use in the clinical setting a clearer understanding as to how the transplanted cells improve outcomes is needed. To gain greater insight into the mechanisms of action, stem cell administration in models of physiological relevance is required. Few studies have included type 2 diabetes and/or its associated metabolic dysregulation on the effect of stem cell transplantation in the infarcted heart. Yan et al. [51] assessed the influence of induced-pluripotent stem (iPS) cell administration in the db/db mouse heart following a MI. Echocardiography revealed that 14 days post-MI left ventricle contractile dysfunction was minimized by iPS cell transplantation [51]. The current study expands on the work of Yan and colleagues [51] by employing a diet-induced insulin resistant C57BL/6 mouse model and assessing the ability of MSC administration to minimize cardiac systolic abnormalities post-infarct. We report the MSC therapy to improve cardiac ejection fraction 28 days post-MI. This indicates that MSC transplantation holds similar potential in restoring left ventricle systolic function in the presence of diet-induced systemic metabolic insults. Furthermore, our experiments investigate beyond cardiac contractile function to evaluate the effect of MSC transplantation on metabolic characteristics that may minimize dysfunction.
The superior contractile function in the MSC-treated mice may be due to the ability of the hearts to more effectively utilize substrates and synthesize ATP. The current study demonstrates intramyocardial administration of MSCs to improve the ability of the heart to utilize glucose in the remote left ventricle and peri-infarct region. This improvement in glucose uptake could assist in energy provision. To provide insight into the means through which the MSC transplantation elevated cardiac glucose uptake molecular regulators of glucose metabolism were evaluated. The current study did not expose alterations in regional cardiac levels of PGC-1α, GLUT4, HKII, p-Akt, Akt and the p-Akt-to-Akt ratio. This suggests key proteins involved in glucose uptake and insulin signalling were not augmented by the MSC treatment in the heart. The absence of changes in these proteins led us to explore mitochondrial function as a possible mechanism by which glucose would be utilized at a higher rate.
Downstream of glucose transport and glycolysis, mitochondria OXPHOS acts as a link between glucose uptake and ATP generation. Early studies evaluating the effect of MSC transplantation show an improvement in the peri-infarct and whole left ventricle phosphocreatine (PCr)-to-ATP ratio [14-16]. The PCr-to-ATP ratio reflects efficiency of myocardial energy provision, and correlates well with left ventricle contractile function [14,52-54]. Furthermore, MSC therapy reduces basal mitochondrial oxygen consumption (V0) and improves cardiac RCR (VMAX-CI/V0) [18]. V0 represents futile respiration that does not contribute to ATP synthesis and a lower basal oxygen flux helps increase RCR; which is indicative of a more efficient OXPHOS. Mitochondrial dysfunction has been theorized to promote insulin resistance [55]. Conversely, improved mitochondrial function may help alleviate impaired glucose uptake and assist in greater ATP synthesis. In the current study, MSC therapy was also able to lower V0 and increase RCR. The ability of MSCs to promote these alterations on mitochondrial function may be due to a direct influence on OXPHOS complexes. Jameel et al. [56] indicate that MSC administration for the infarcted heart reduces gene expression of complex I subunits. We report that in the peri-infarct region, there was a decline in complex I protein levels following a MI. However, MSC transplantation enhanced the reduction in complex I as indicated by the decline induced in the remote left ventricle and peri-infarct region. Complex II and V were also lower in the MSC-treated peri-infarct region. A reduction in OXPHOS complexes I, II and V may explain the diminished V0 as a repressed complex number would subdue electron flux and oxygen consumption. Another potential reason for the lower V0 and elevated RCR is alterations in UCP3. UCP3 has been suggested to function as a potential OXPHOS uncoupler [57]. As such, a reduction in protein may promote a decline in UCP3-mediated uncoupling.
Increases in tissue UCP3 are often more pronounced with high-fat diets and with elevated circulating fatty acids [37,58]. We report that MSC-treated mice exhibited a reduction in fasted plasma NEFAs. This decline would promote a lower tissue expression of UCP3. Further systemic influence of the MSC therapy was observed in fasting plasma glucose. We found plasma glucose levels were diminished in the MSC-treated animals. In agreement, animal studies indicate that MSC therapy is capable of lowering blood glucose levels in both type 1 diabetes [59,60] and type 2 diabetes [61]. A recent pilot clinical study has reported a reduction in blood glucose in individuals with type 2 diabetes following MSC transplantation [62].
The exact means by which MSC exert these effects on metabolism have yet to be elucidated. To date, the majority of studies evaluating stem cell transplantation in diabetes have focused on the ability of the administered cells to regenerate and/or protect the insulin-producing beta cells of the pancreas as a means of providing improved glucose control and reducing diabetic complications [63]. To the authors’ knowledge, only a single study has evaluated the influence of cell-based therapy on peripheral tissue glucose disposal as a mechanism through which the treatment reduces hyperglycemia. Si et al. [61] employed insulin clamps to test whether intravenous MSC administration improves whole-body insulin sensitivity in streptozotocin/high-fat diet-induced diabetes rodents. It was reported that MSC therapy increased GIR but there was not a complete restoration to that of a chow fed animal [61]. We achieved comparable results employing insulin clamps to show the MSC-treated animals display greater systemic insulin sensitivity. Mice receiving MSC displayed an approximately 40% increase in the GIR compared to SHAM mice. Although the MSCs were administered via an intramyocardial injection, previous reports have identified MSC to persist, albeit low in magnitude, in peripheral tissues such as the lungs, spleen and liver [64,65]. As such, a systemic effect was not completely unanticipated. Moreover, the current study provides further support for a system-wide elevation in insulin-mediated glucose uptake by combining the insulin clamp with isotopic techniques to evaluate tissue-specific glucose utilization. In addition to increased cardiac glucose uptake, the soleus, superficial vastus lateralis, gastrocnemius and white adipose tissue exhibited an elevated rate of insulin-stimulated glucose uptake following MSC transplantation. The rise in glucose utilization in a range of insulin-sensitive tissues suggests the stem cell therapy is indeed acting in a systemic fashion rather than solely via tissue-specific mechanisms.
To address the mode by which the MSC treatment promoted glucose uptake in the selected tissues, GLUT4 protein was examined. Both gastrocnemius and white adipose tissue total GLUT4 protein was elevated in the MSC-treated mice. In the current study plasma fasting and experimental insulin concentrations were comparable between groups, however, there was an insulin-dependent influence conferred by the MSC transplantation. The gastrocnemius and white adipose tissue displayed higher p-Akt and an elevated p-Akt-to-Akt ratio. This suggests the cell therapy also augments the PI3K-Akt pathway to enhance glucose uptake.
The comprehensive in vivo approach employed for evaluating insulin sensitivity, substrate uptake and cardiac function following diet-induced insulin resistance and a standardized MI in the conscious mouse provides a greater understanding of the influence of MSC therapy on the diabetic infarcted heart. Despite this, a few readily apparent limitations exist. First, it is not clear if the improved ejection fraction is the result of direct cardio-protection or systemic alterations. The improved insulin sensitivity may give the heart added metabolic flexibility to meet its energetic requirements. Similarly, the reduction in plasma NEFAs may lessen OXPHOS uncoupling for more efficient ATP synthesis. In experimental models, ad libitum feeding of mice a high-fat diet with 55–60% of the caloric content being derived from fatty acids induces cardiovascular irregularities. After 16 weeks of HFF, systolic and diastolic blood pressure is markedly elevated compared to chow fed mice (11% calories by fat) [66]. Park et al. [67] identified C57BL/6 mice to exhibit impaired systolic performance following 20 weeks of HFF as indicated by fractional shortening. The animals evaluated in the current study were HFF for 13 weeks. As such, the therapeutic importance of peripheral glucose uptake and reduced hyperglycemia may increase as the dietary intervention is prolonged.
Another potential limitation of the present study is that the animals were not weight-matched at the time of glucose uptake evaluation. In the infarct-only animals, body weight was significantly lower following the MI event. This was associated with a reduced accumulation of body fat. Many of the metabolic aberrations associated with the HFF model are connected to the induction of obesity (fat deposition) [68]. Thus, the increase in the metabolic index of glucose uptake in the adipose tissue of the MI + PBS group is believed to be the result of the MI insult limiting comparable weight gain in these mice.
Finally, given the metabolic parameters were performed in the peri-infarct region of the heart, differential cell viability may be of modest concern. In the clinical setting, tissue salvage and infarct size reduction is based on the assumption that the core of the infarct is severely ischemic and necrotic [69]. Moreover, this core is surrounded by an area of jeopardized tissue called the peri-infarct region that may be salvaged by reperfusion efforts [69]. In experimental ligation models this is not observed. Rather than a progressive transition from infarct to viable tissue there is an abrupt, but irregular, changeover from ischemic zone to perfused, viable tissue [69,70]. While this is a limitation in matching the clinical setting exactly, it is also an advantage to the studies conducted because it largely minimizes the influence of apoptotic cells in the peri-infarct region following the MI event.
Conclusions
In summary, the novel findings of this study are the MSC-mediated increase in both cardiac glucose uptake and mitochondrial OXPHOS efficiency. These metabolic improvements may assist in greater ATP provision for contractile demands following a MI. Our results also demonstrate MSC transplantation to reduce hyperglycemia, hyperlipidemia and improve peripheral tissue insulin-mediated glucose uptake. These findings differ from the effects of MSC therapy in the absence of type 2 diabetes and/or its metabolic abnormalities. Previous work by the authors evaluating the therapeutic influence of MSC transplantation on cardiac glucose utilization post-MI in chow fed mice did not identify MSC-induced alterations in cardiac glucose metabolism [18]. Furthermore, no apparent whole body effect on insulin sensitivity was mediated by MSC transplantation [18]. By utilizing a combined diet-induced insulin resistant/MI model the current study identifies that not only does MSC transplantation potentially lessen impairment in cardiac contractile performance via augmentation of cardiac-specific metabolism but also holds the ability to reach farther than the heart to dampen systemic dysregulation that may contribute to the maladaptive alterations in cardiac function.
Abbreviations
2-[14C]DG: 2-[14C]deoxyglucose; CI: Mitochondrial electron transport chain complex 1; CII: Mitochondrial electron transport chain complex 2; CIII: Mitochondrial electron transport chain complex 3; CIV: Mitochondrial electron transport chain complex 4; CV: Mitochondrial electron transport chain complex 5; ATP: Synthase; CSA: Citrate synthase activity; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GIR: Glucose infusion rate; HFF: High-fat fed; HKII: Hexokinase 2; LAD: Left anterior descending coronary artery; LV: Remote left ventricle; MI: Myocardial infarction; MSC: Mesenchymal stem cell; OXPHOS: Oxidative phosphorylation; PCr: Phosphocreatine; PGC-1α: Peroxisome proliferator-activated receptor gamma coactivator-1alpha; PI: Peri-infarct region of left ventricle; RCR: Respiratory control ratio; Rg: Metabolic index of glucose uptake; UCP: Uncoupling protein; V0: State 2 respiration supported by complex I substrates malate, glutamate and pyruvate; VMAX-CI: State 3 respiration supported by complex I substrates malate, glutamate and pyruvate; VMAX-CI+CII: State 3 respiration via convergent electron input through complex I and II.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CCH, DHW, JNR and JS did the conception and design of the experiments. CCH, LM, FDJ, DBP and ZW performed the experiments and collected data. CCH and JS analyzed the data. CCH drafted the manuscript. All authors reviewed, edited and approved the final version of this manuscript.
Contributor Information
Curtis C Hughey, Email: cchughey@ucalgary.ca.
Lianli Ma, Email: lianli.ma@Vanderbilt.Edu.
Freyja D James, Email: freyja.james@Vanderbilt.Edu.
Deanna P Bracy, Email: bingle.bracy@Vanderbilt.Edu.
Zhizhang Wang, Email: zhizhang.wang@Vanderbilt.Edu.
David H Wasserman, Email: david.wasserman@vanderbilt.edu.
Jeffrey N Rottman, Email: jeff.rottman@vanderbilt.edu.
Dustin S Hittel, Email: dhittel@gmail.com.
Jane Shearer, Email: jshearer@ucalgary.ca.
Acknowledgements
The authors thank the Vanderbilt University Analytical Resources Core for measuring plasma insulin concentrations. This work was supported by Canadian Institutes of Health Research (CIHR) (C.C. Hughey, J. Shearer), the Killam Trusts (C.C. Hughey), MitoCanada (J. Shearer) and the National Institutes of Health Grant (NIH) DK-054902 (D.H. Wasserman) and DK-59637 (Vanderbilt University Mouse Metabolic Phenotyping Center).
References
- Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79(1):215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- Minicucci MF, Azevedo PS, Polegato BF, Paiva SA, Zornoff LA. Heart failure after myocardial infarction: clinical implications and treatment. Clin Cardiol. 2011;34(7):410–414. doi: 10.1002/clc.20922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henning RJ. Stem cells in cardiac repair. Future Cardiol. 2011;7(1):99–117. doi: 10.2217/fca.10.109. [DOI] [PubMed] [Google Scholar]
- Keats EC, Khan ZA. Vascular stem cells in diabetic complications: evidence for a role in the pathogenesis and the therapeutic promise. Cardiovasc Diabetol. 2012;11:37. doi: 10.1186/1475-2840-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganath SH, Levy O, Inamdar MS, Karp JM. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell. 2012;10(3):244–258. doi: 10.1016/j.stem.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S. Stem cells in the treatment of heart disease. Annu Rev Med. 2010;61:287–300. doi: 10.1146/annurev.med.051508.215152. [DOI] [PubMed] [Google Scholar]
- Penn MS, Ellis S, Gandhi S, Greenbaum A, Hodes Z, Mendelsohn FO, Strasser D, Ting AE, Sherman W. Adventitial delivery of an allogeneic bone marrow-derived adherent stem cell in acute myocardial infarction: phase I clinical study. Circ Res. 2012;110(2):304–311. doi: 10.1161/CIRCRESAHA.111.253427. [DOI] [PubMed] [Google Scholar]
- Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP, Gerstenblith G, DeMaria AN, Denktas AE, Gammon RS. et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. 2009;54(24):2277–2286. doi: 10.1016/j.jacc.2009.06.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Dubois C, Bogaert J, Theunissen K, Deroose C, Desmet W, Kalantzi M, Herbots L, Sinnaeve P, Dens J. et al. Autologous bone marrow-derived stem-cell transfer in patients with ST-segment elevation myocardial infarction: double-blind, randomised controlled trial. Lancet. 2006;367(9505):113–121. doi: 10.1016/S0140-6736(05)67861-0. [DOI] [PubMed] [Google Scholar]
- Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circ Res. 2008;103(11):1204–1219. doi: 10.1161/CIRCRESAHA.108.176826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey TE, Saiget MK, Reinecke H, Murry CE. Systems approaches to preventing transplanted cell death in cardiac repair. J Mol Cell Cardiol. 2008;45(4):567–581. doi: 10.1016/j.yjmcc.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider H, Ashraf M. Strategies to promote donor cell survival: combining preconditioning approach with stem cell transplantation. J Mol Cell Cardiol. 2008;45(4):554–566. doi: 10.1016/j.yjmcc.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollert KC, Drexler H. Cell therapy for the treatment of coronary heart disease: a critical appraisal. Nat Rev Cardiol. 2010;7(4):204–215. doi: 10.1038/nrcardio.2010.1. [DOI] [PubMed] [Google Scholar]
- Feygin J, Mansoor A, Eckman P, Swingen C, Zhang J. Functional and bioenergetic modulations in the infarct border zone following autologous mesenchymal stem cell transplantation. Am J Physiol Heart Circ Physiol. 2007;293(3):H1772–1780. doi: 10.1152/ajpheart.00242.2007. [DOI] [PubMed] [Google Scholar]
- Gnecchi M, He H, Melo LG, Noiseaux N, Morello F, De Boer RA, Zhang L, Pratt RE, Dzau VJ, Ingwall JS. Early beneficial effects of bone marrow-derived mesenchymal stem cells overexpressing Akt on cardiac metabolism after myocardial infarction. Stem Cells. 2009;27(4):971–979. doi: 10.1002/stem.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Hu Q, Wang X, Mansoor A, Lee J, Feygin J, Zhang G, Suntharalingam P, Boozer S, Mhashilkar A. et al. Bioenergetic and functional consequences of bone marrow-derived multipotent progenitor cell transplantation in hearts with postinfarction left ventricular remodeling. Circulation. 2007;115(14):1866–1875. doi: 10.1161/CIRCULATIONAHA.106.659730. [DOI] [PubMed] [Google Scholar]
- Mazo M, Planat-Benard V, Abizanda G, Pelacho B, Leobon B, Gavira JJ, Penuelas I, Cemborain A, Penicaud L, Laharrague P. et al. Transplantation of adipose derived stromal cells is associated with functional improvement in a rat model of chronic myocardial infarction. Eur J Heart Fail. 2008;10(5):454–462. doi: 10.1016/j.ejheart.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Hughey CC, Johnsen VL, Ma L, James FD, Young PP, Wasserman DH, Rottman JN, Hittel DS, Shearer J. Mesenchymal stem cell transplantation for the infarcted heart: a role in minimizing abnormalities in cardiac-specific energy metabolism. Am J Physiol Endocrinol Metab. 2012;302(2):E163–172. doi: 10.1152/ajpendo.00443.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard BV, Rodriguez BL, Bennett PH, Harris MI, Hamman R, Kuller LH, Pearson TA, Wylie-Rosett J. Prevention Conference VI: Diabetes and Cardiovascular disease: Writing Group I: epidemiology. Circulation. 2002;105(18):e132–137. doi: 10.1161/01.CIR.0000013953.41667.09. [DOI] [PubMed] [Google Scholar]
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA: 1979;241(19):2035–2038. doi: 10.1001/jama.1979.03290450033020. [DOI] [PubMed] [Google Scholar]
- Sethi SS, Akl EG, Farkouh ME. Diabetes mellitus and acute coronary syndrome: lessons from randomized clinical trials. Curr Diab Rep. 2012;12(3):294–304. doi: 10.1007/s11892-012-0272-9. [DOI] [PubMed] [Google Scholar]
- Von Bibra H, St John Sutton M. Impact of diabetes on postinfarction heart failure and left ventricular remodeling. Curr Heart Fail Rep. 2011;8(4):242–251. doi: 10.1007/s11897-011-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson E, Sundstrom J, Arnlov J, Zethelius B, Lind L. Insulin resistance and risk of congestive heart failure. JAMA: 2005;294(3):334–341. doi: 10.1001/jama.294.3.334. [DOI] [PubMed] [Google Scholar]
- Lomuscio A, Castagnone M, Vergani D, Verzoni A, Beltrami A, Ravaglia R, Pozzoni L. Clinical correlation between diabetic and non diabetic patients with myocardial infarction. Acta cardiologica. 1991;46(5):543–554. [PubMed] [Google Scholar]
- Glass CE, Singal PK, Singla DK. Stem cells in the diabetic infarcted heart. Heart Fail Rev. 2010;15(6):581–588. doi: 10.1007/s10741-010-9172-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abozguia K, Clarke K, Lee L, Frenneaux M. Modification of myocardial substrate use as a therapy for heart failure. Nat Clin Pract Cardiovasc Med. 2006;3(9):490–498. doi: 10.1038/ncpcardio0583. [DOI] [PubMed] [Google Scholar]
- Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circ Res. 2004;95(2):135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356(11):1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Taha M, Lopaschuk GD. Alterations in energy metabolism in cardiomyopathies. Ann Med. 2007;39(8):594–607. doi: 10.1080/07853890701618305. [DOI] [PubMed] [Google Scholar]
- Van Bilsen M, Van Nieuwenhoven FA, van der Vusse GJ. Metabolic remodelling of the failing heart: beneficial or detrimental? Cardiovasc Res. 2009;81(3):420–428. doi: 10.1093/cvr/cvn282. [DOI] [PubMed] [Google Scholar]
- Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation–a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta. 2011;1813(7):1333–1350. doi: 10.1016/j.bbamcr.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Razeghi P, Young ME, Ying J, Depre C, Uray IP, Kolesar J, Shipley GL, Moravec CS, Davies PJ, Frazier OH. et al. Downregulation of metabolic gene expression in failing human heart before and after mechanical unloading. Cardiology. 2002;97(4):203–209. doi: 10.1159/000063122. [DOI] [PubMed] [Google Scholar]
- Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. Mitochondrial proton and electron leaks. Essays Biochem. 2010;47:53–67. doi: 10.1042/bse0470053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witteles RM, Fowler MB. Insulin-resistant cardiomyopathy clinical evidence, mechanisms, and treatment options. J Am Coll Cardiol. 2008;51(2):93–102. doi: 10.1016/j.jacc.2007.10.021. [DOI] [PubMed] [Google Scholar]
- Wende AR, Symons JD, Abel ED. Mechanisms of lipotoxicity in the cardiovascular system. Curr Hypertens Rep. 2012;14(6):517–531. doi: 10.1007/s11906-012-0307-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes Metab. 2010;36(6 Pt 1):423–435. doi: 10.1016/j.diabet.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet. 2004;364(9447):1786–1788. doi: 10.1016/S0140-6736(04)17402-3. [DOI] [PubMed] [Google Scholar]
- Williams AR, Hare JM. Mesenchymal stem cells: biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res. 2011;109(8):923–940. doi: 10.1161/CIRCRESAHA.111.243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnecchi M, Danieli P, Cervio E. Mesenchymal stem cell therapy for heart disease. Vasc Pharmacol. 2012;57(1):48–55. doi: 10.1016/j.vph.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95(1):9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- Oh JY, Lee RH, Yu JM, Ko JH, Lee HJ, Ko AY, Roddy GW, Prockop DJ. Intravenous mesenchymal stem cells prevented rejection of allogeneic corneal transplants by aborting the early inflammatory response. Mol Ther. 2012;20(11):2143–2152. doi: 10.1038/mt.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottman JN, Ni G, Khoo M, Wang Z, Zhang W, Anderson ME, Madu EC. Temporal changes in ventricular function assessed echocardiographically in conscious and anesthetized mice. J Am Soc Echocardiogr. 2003;16(11):1150–1157. doi: 10.1067/S0894-7317(03)00471-1. [DOI] [PubMed] [Google Scholar]
- Exil VJ, Roberts RL, Sims H, McLaughlin JE, Malkin RA, Gardner CD, Ni G, Rottman JN, Strauss AW. Very-long-chain acyl-coenzyme a dehydrogenase deficiency in mice. Circ Res. 2003;93(5):448–455. doi: 10.1161/01.RES.0000088786.19197.E4. [DOI] [PubMed] [Google Scholar]
- Fueger PT, Lee-Young RS, Shearer J, Bracy DP, Heikkinen S, Laakso M, Rottman JN, Wasserman DH. Phosphorylation barriers to skeletal and cardiac muscle glucose uptakes in high-fat fed mice: studies in mice with a 50% reduction of hexokinase II. Diabetes. 2007;56(10):2476–2484. doi: 10.2337/db07-0532. [DOI] [PubMed] [Google Scholar]
- Shearer J, Fueger PT, Bracy DP, Wasserman DH, Rottman JN. Partial gene deletion of heart-type fatty acid-binding protein limits the severity of dietary-induced insulin resistance. Diabetes. 2005;54(11):3133–3139. doi: 10.2337/diabetes.54.11.3133. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, McGuinness OP, Wasserman DH. Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes. 2006;55(2):390–397. doi: 10.2337/diabetes.55.02.06.db05-0686. [DOI] [PubMed] [Google Scholar]
- Morgan CR, Lazarow A. Immunoassay of pancreatic and plasma insulin following alloxan injection of rats. Diabetes. 1965;14(10):669–671. doi: 10.2337/diab.14.10.669. [DOI] [PubMed] [Google Scholar]
- Shearer J, Fueger PT, Wang Z, Bracy DP, Wasserman DH, Rottman JN. Metabolic implications of reduced heart-type fatty acid binding protein in insulin-resistant cardiac muscle. Biochim Biophys Acta. 2008;1782(10):586–592. doi: 10.1016/j.bbadis.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottman JN, Bracy D, Malabanan C, Yue Z, Clanton J, Wasserman DH. Contrasting effects of exercise and NOS inhibition on tissue-specific fatty acid and glucose uptake in mice. Am J Physiol Endocrinol Metab. 2002;283(1):E116–123. doi: 10.1152/ajpendo.00545.2001. [DOI] [PubMed] [Google Scholar]
- Hughey CC, Hittel DS, Johnsen VL, Shearer J. Respirometric oxidative phosphorylation assessment in saponin-permeabilized cardiac fibers. J Vis Exp: JoVE. 2011;48:1–7. doi: 10.3791/2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan B, Abdelli LS, Singla DK. Transplanted induced pluripotent stem cells improve cardiac function and induce neovascularization in the infarcted hearts of db/db mice. Mol Pharm. 2011;8(5):1602–1610. doi: 10.1021/mp2003576. [DOI] [PubMed] [Google Scholar]
- Hamman BL, Bittl JA, Jacobus WE, Allen PD, Spencer RS, Tian R, Ingwall JS. Inhibition of the creatine kinase reaction decreases the contractile reserve of isolated rat hearts. Am J Physiol. 1995;269(3 Pt 2):H1030–1036. doi: 10.1152/ajpheart.1995.269.3.H1030. [DOI] [PubMed] [Google Scholar]
- Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS. et al. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96(7):2190–2196. doi: 10.1161/01.CIR.96.7.2190. [DOI] [PubMed] [Google Scholar]
- Zhang J, McDonald KM. Bioenergetic consequences of left ventricular remodeling. Circulation. 1995;92(4):1011–1019. doi: 10.1161/01.CIR.92.4.1011. [DOI] [PubMed] [Google Scholar]
- Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nat rev Endocrinol. 2012;8(2):92–103. doi: 10.1038/nrendo.2011.138. [DOI] [PubMed] [Google Scholar]
- Jameel MN, Li Q, Mansoor A, Qiang X, Sarver A, Wang X, Swingen C, Zhang J. Long-term functional improvement and gene expression changes after bone marrow-derived multipotent progenitor cell transplantation in myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;298(5):H1348–1356. doi: 10.1152/ajpheart.01100.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costford SR, Seifert EL, Bezaire V, M FG, Bevilacqua L, Gowing A, Harper ME. The energetic implications of uncoupling protein-3 in skeletal muscle. Appl Physiol Nutr Metab. 2007;32(5):884–894. doi: 10.1139/H07-063. [DOI] [PubMed] [Google Scholar]
- Azzu V, Jastroch M, Divakaruni AS, Brand MD. The regulation and turnover of mitochondrial uncoupling proteins. Biochim Biophys Acta. 2010;1797(6–7):785–791. doi: 10.1016/j.bbabio.2010.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezquer FE, Ezquer ME, Parrau DB, Carpio D, Yanez AJ, Conget PA. Systemic administration of multipotent mesenchymal stromal cells reverts hyperglycemia and prevents nephropathy in type 1 diabetic mice. Biol Blood Marrow Transplant. 2008;14(6):631–640. doi: 10.1016/j.bbmt.2008.01.006. [DOI] [PubMed] [Google Scholar]
- Lee RH, Seo MJ, Reger RL, Spees JL, Pulin AA, Olson SD, Prockop DJ. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci U S A. 2006;103(46):17438–17443. doi: 10.1073/pnas.0608249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Y, Zhao Y, Hao H, Liu J, Guo Y, Mu Y, Shen J, Cheng Y, Fu X, Han W. Infusion of mesenchymal stem cells ameliorates hyperglycemia in type 2 diabetic rats: identification of a novel role in improving insulin sensitivity. Diabetes. 2012;61(6):1616–1625. doi: 10.2337/db11-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Han Z, Zhuo G, Qu X, Li X, Wang X, Shao Y, Yang S, Han ZC. Transplantation of placenta-derived mesenchymal stem cells in type 2 diabetes: a pilot study. Front Med. 2011;5(1):94–100. doi: 10.1007/s11684-011-0116-z. [DOI] [PubMed] [Google Scholar]
- Couri CE, De Oliveira MC, Simoes BP. Risks, benefits, and therapeutic potential of hematopoietic stem cell transplantation for autoimmune diabetes. Current diab Rep. 2012;12(5):604–611. doi: 10.1007/s11892-012-0309-0. [DOI] [PubMed] [Google Scholar]
- Hou D, Youssef EA, Brinton TJ, Zhang P, Rogers P, Price ET, Yeung AC, Johnstone BH, Yock PG, March KL. Radiolabeled cell distribution after intramyocardial, intracoronary, and interstitial retrograde coronary venous delivery: implications for current clinical trials. Circulation. 2005;112(9 Suppl):I150–156. doi: 10.1161/CIRCULATIONAHA.104.526749. [DOI] [PubMed] [Google Scholar]
- Campbell NG, Suzuki K. Cell delivery routes for stem cell therapy to the heart: current and future approaches. J Cardiovasc Transl Res. 2012;5(5):713–726. doi: 10.1007/s12265-012-9378-3. [DOI] [PubMed] [Google Scholar]
- Kobayasi R, Akamine EH, Davel AP, Rodrigues MA, Carvalho CR, Rossoni LV. Oxidative stress and inflammatory mediators contribute to endothelial dysfunction in high-fat diet-induced obesity in mice. J Hypertens. 2010;28(10):2111–2119. doi: 10.1097/HJH.0b013e32833ca68c. [DOI] [PubMed] [Google Scholar]
- Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, Dey A, Rothermel B, Kim YB, Kalinowski A. et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54(12):3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech. 2010;3(9–10):525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearse DJ. Models and problems in the study of myocardial ischemia and tissue protection. Eur Heart J. 1983;4 Suppl C:43–48. doi: 10.1093/eurheartj/4.suppl_c.43. [DOI] [PubMed] [Google Scholar]
- Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol. 2008;103(6):501–513. doi: 10.1007/s00395-008-0743-y. [DOI] [PubMed] [Google Scholar]