

Abstract
Ischemic cardiac injury is the leading cause of heart failure and mortality in the US, and a major expense to health care systems. Once the heart is injured, a highly dynamic and coordinated immune response is initiated, which is dependent on both resident and recruited leukocytes. The goal of the inflammatory response is to remove ischemic and necrotic material, and to promote infarct healing. If this system is perturbed, the myocardium heals poorly, leading to significant left ventricular dysfunction. Understanding how inflammatory cells coordinate and interact with each other is required prior to designing therapeutic interventions that target pathological processes at play, and leave untouched those processes that are protective. This review will discuss the intercellular cross talk between cells of the innate immune system following myocardial ischemic injury and how that response is coordinated over time.
Keywords: Myocardial infarction, innate immunity, monocyte, T cell, neutrophil
Introduction
Myocardial ischemic injury is the leading cause of heart failure and a major burden on health care systems worldwide [1]. In the immediate post-infarct period, there are significant biochemical and architectural changes within the myocardium, which leave the damaged segment vulnerable to hemodynamic strain that can lead to adverse cardiac remodeling [2]. Understanding this early period is critical to minimize such pathological changes. A growing body of literature suggests that cells of the innate immune system play a key role in regulating the initial inflammatory response and the subsequent wound healing response. Neutrophils, monocytes, macrophages, mast cells and dendritic cells, and subclasses of T cells are all innate immune cells that have been demonstrated to play a role following cardiac tissue injury. Ischemic injury occurs when there is luminal occlusion of a coronary artery and decreased delivery of oxygen and nutrients to a segment of the myocardium. There is a relatively narrow window of time that myocardial tissue will remain viable and establishing reperfusion within that time frame is critical in order to prevent cardiomyocyte loss. Recent advancements in percutaneous therapies that restore myocardial blood flow have shifted the clinical course from a completed infarct model to one where a majority of patients are subjected to ischemia/reperfusion (I/R) [3]. While reperfusion restores blood flow, it also brings with it I/R injury secondary to the associated intense inflammatory response [4]. Understanding the mechanisms by which innate immune cells are activated following tissue injury, and how these cells coordinate a complex and staged response to cardiac injury is critical if new therapeutic advancements are to be made.
Sterile Inflammation
Inflammation is essential for host defense against invasive pathogens. Following infection, a cascade of signals are released that lead to the recruitment of inflammatory cells, particularly innate immune cells such as neutrophils and macrophages. As the inflammation induced in response to sterile cell death or injury is similar to that observed during microbial infection, host receptors that mediate the immune response to microorganisms may be involved in the activation of sterile inflammation. In the case of infection, the mechanisms by which the inflammatory response is initiated have been well studied. There are several classes of receptors that are important for sensing microorganisms and for the subsequent induction of pro-inflammatory responses. These have been collectively termed pattern recognition receptors (PRRs). These germline-encoded PRRs sense conserved structural moieties that are found in microorganisms and are often called pathogen-associated molecular patterns. More recently it has become clear that these PRRs also recognize molecular patterns of endogenous host material that is released during cellular injury or death, so-called damage associated molecular patterns [5, 6]. This latter observation has provided a potentially important link between tissue injury, activation of pro-inflammatory mediators, and the resulting myocardial response to ischemic injury.
Mast Cells and complement, and then mast cells again
Within the first 15 min following reperfusion, resident cardiac mast cells degranulate and release numerous preformed pro-inflammatory mediates (tumor necrosis factor [TNF, histamine, proteases) which activate a number of targets, including the endothelium, resident monocytes/macrophages and infiltrating neutrophils [7]. Moreover, reperfusion brings with it plasma containing the complement system. It is not entirely clear how complement (in particular the alternative pathway) is triggered, however it is in part regulated by CD59 expression on the myocardium (as reviewed in [8]). The C3a and C5a anaphylaxotoxins are released rapidly during this process, which exacerbate histamine release from mast cells and enhance edema [8]. C5a is a potent neutrophil chemoattractant, inducing firm adhesion of neutrophil to the endothelium through the upregulation of CD11b/CD18 (Mac-1) and subsequent transendothelial migration [9]. Neutrophils exposed to C5a also contribute to oxidative stress via the upregulation of superoxide [10]. This very early response, dominated by resident mast cell activation, complement activation, oxidative stress (from a variety of sources) and proinflammatory cytokine/chemokine production serves to further activate the endothelium and induce the recruitment and complex interplay between a wide variety of innate immune cells.
Interestingly, mast cell density increases with ischemic injury [11]. Mast cell deficient mice (c-kit KO) have demonstrated important, albeit somewhat complicated results. Early after injury (< 12 hrs), there is more viable myocardium in c-kit KO mice [12]. However, after several weeks, c-kit KO mice have increased left ventricular (LV) diameter and decreased scar thickness, suggesting an important protective role in LV remodeling. An interesting study by Ayach et. al. indicated that loss of c-kit in the bone marrow compartment resulted in impaired angiogenesis and wound healing following myocardial infarction, and this was dependent on natural killer (NK) cells (see below) [13]. However, the direct role of mast cells is difficult to assess, insofar as the c-kit deficient mice have deficiencies in other hematopoietic lineages [14]. The subsequent sections will discuss individual innate immune cells, their direct role in cardiac injury and repair, and how interactions between different components of innate immune system are intertwined.
Neutrophils – the first wave
Neutrophils are the most abundant leukocyte in circulation and are recruited into myocardium within hours after injury [15]. Prior to neutrophil infiltration, the endothelium is activated by a variety of cytokines to induce the upregulation of adhesion molecules and production of chemokines that guide neutrophils to the site of injury, which classically include leukotrine B4, platelet activating factor, Gro-α and C5a [15,16]. Interestingly, one of the key cytokines in this process is interleukin-6 (IL-6), which is initially produced by cardiomyocytes in the border zone, where it upregulates ICAM-1 on cardiomyocytes in an autocrine fashion. In addition both neutrophils and subsequent infiltrating mononuclear cells also produce IL-6 [17,18]. Once activated neutrophils infiltrate the myocardium, they bind to ICAM-1 expressing cardiomyocytes (via CD11b/CD18), which triggers an intense oxidative burst and ultimately neutrophil-mediated cardiac injury [10,16,19]. Neutrophils also possess an arsenal of proteases, collagenases and elastase, which contribute to tissue injury [20]. Depletion of neutrophils after short ischemic episodes appears to reduce infarct size, although as the ischemic time increases, and less viable myocardium is present, the depletion of neutrophils losses its protective effects indicating neutrophil-mediated injury is more important in the setting of I/R injury rather than completed infarction [21,22]. This may be due to the fact that neutrophils are contributing to cardiomyocyte death in I/R injury, whereas cell death from completed infarction is the result of prolonged anoxia and nutrient depravation.
Ischemic injury of the myocardium is a model of sterile inflammation. A recent elegant study by McDonald et. al. used spinning disk confocal intravital microscopy to track neutrophils following hepatic necrosis, which is a similar model of sterile inflammation [23]. Release of ATP from necrotic cells activated the NLrp3 inflammasome to generate the necessary milieu to alter patrolling neutrophils to adhere to healthy endothelium at a site fairly remote to injured tissue. In a multi-step process, a chemokine gradient directed transmigrated neutrophils towards injured areas and then ultimately stimulation through formyl-peptide receptors (formyl peptides are released from necrotic cells), directed neutrophils to the non-perfused necrotic area. Although imaging studies such as these are not yet possible within the beating heart, analogous directional queues are likely present in the myocardium, with a spectrum of viable, injured and necrotic tissue that neutrophils most navigate through. Formyl peptides represent ancient motifs similar to bacterial products, which act as “danger signals” when released, and focus inflammatory responses [24]. Oka et. al. demonstrated that hemodynamic overload resulted in the damage of mitochondria and subsequent release of mitochondrial DNA from the myocardium. Mitochondrial DNA was sensed through an endosomal toll like receptor (TLR9)-dependent mechanism, triggering an intense inflammatory response characterized by significant neutrophil and monocyte infiltration, release of IL-1β and IL-6, and ultimately LV dysfunction and death [25]. Release of mitochondrial components following muscle crush injury serve as potent neutrophil chemoattractants that also activate the systemic immune response, again through formyl peptide receptors and TLR9 [26]. Bacterial DNA is similar to mitochondrial DNA, as the mitochondria is an ancient endosymbiont, which helps explain why mitochondrial DNA is such a potent inflammatory trigger. Thus, one of the key inflammatory triggers in sterile tissue injury is the release of conserved inflammatory motifs.
The dynamic bi-directional relationships between neutrophils and myeloid cells
Myeloid cells (monocytes/macrophages) are interesting in that these cells possess a high degree of complexity in phenotype and function. Traditionally, monocytes were thought to be a homogenous population that patrols the vasculature awaiting a signal before they would enter tissue, differentiate into macrophages and subsequently perform effector functions. However, it is monocytes that are the dominant cell type infiltrating the myocardium in the first week following ischemic injury, and it is only recently that we have begun to appreciate the critical and divergent roles monocytes fulfill during the course of infarct healing. Ly-6cHi monocytes are recruited into the myocardium by the chemokine MCP-1 (macrophage chemotactic protein-1) immediately after injury, producing proinflammatory cytokines, secreting lytic enzymes and digesting ischemic/necrotic tissue [27]. In contrast, Ly-6cLow monocytes are recruited by fractalkine, enter the myocardium in large numbers after day 3, and remain in the myocardium for approximately 1–2 weeks where they promote collagen deposition, angiogenesis and wound healing after a completed infarct. We have utilized a closed chest model of ischemia-reperfusion injury to study the recruitment of Ly-6cHi and Ly-6cLow monocytes (as detailed in Figure 1) and originally described by Nossuli et. al. [28]. The advantages of this technique are that 1) the confounding effects of surgical trauma are separated temporally from the induction of ischemia, and 2) this model is a highly analogous system that simulates the contemporary clinical scenario. Initially there is rapid influx of inflammatory Ly-6cHi monocytes, followed by Ly-6cLow monocytes, and contrary to the completed infarction model, there is rapid resolution of cellular infiltrate after 7 days (Figure 1). Importantly, similar subsets exist in humans, and increased monocyte numbers in the blood at the time myocardial infarction (MI) correlates with poor recovery of LV function over time [29].
Figure 1.
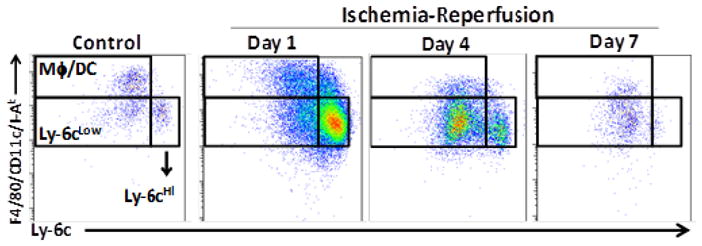
Recruitment dynamics of monocytes following ischemia-reperfusion injury. C57Bl/6 male mice (6–10 weeks) were instrumented via small thoracotomy by placing a suture under the left anterior descending artery. The suture ends were exteriorized through the chest wall and the skin was closed. Animals recovered (7 days) and were then subject to 90 min ischemia using this closed-chest model of ischemia-reperfusion injury [66]. After 1 (D1) and 6 (D6) days, mice were sacrificed, infarcted tissue was minced and digested (collagenase I and VI, hyaluronidase, DNase) for 60 min at 37°C and filtered to produce a single cell suspension that was subject to flow cytometric analysis 1. Cells were initially gated on CD45+ cells (not shown). Monocytes were identified as CD45+CD11b+LIN−(LIN=CD90,B220,NK1.1,CD92, CD49b,Ly-6G)−,(CD11c,I-Ab,F4/80)Int and either Ly-6cHi or Ly-6cLow. M/DC were identified as CD45+CD11b+LIN−(CD11c, I-Ab, F4/80)Hi Ly-6cLow. Neutrophils were CD45+CD11b+LIN+ (CD11c,I-Ab, F4/80)LowLy-6cInt
Nahrendorf et. al. have demonstrated that there is sequential recruitment of Ly-6cHi followed by Ly-6cLow monocytes after infarction, suggesting these cells are recruited separately from the circulation, where they are present in equal numbers [27]. The spleen appears to be a major reservoir of monocytes following ischemic injury and monocytes from the spleen compromise ~50% of the total pool in the myocardium after injury [30]. Interestingly, there is rapid and dynamic recruitment into the myocardium, which is dependent on IL-1β-stimulated extramedullary hematopoeisis of monocytes from myeloid dendritic cell precursors in the spleen [31]. Within the myocardium, there are rapid turnover kinetics (half life ~20 hrs), with very little cell exit and likely substantial local cell death that regulates overall monocytes numbers [31].
Currently, it is not clear what the exact relationship is between Ly-6cHi and Ly-6cLow monocytes entering the myocardium. For example, it is unclear whether there is sequential recruitment or differentiation of Ly-6cHi → Ly-6cLow monocytes, or whether there is an element of proliferation within the myocardium. This is particularly relevant since Ly-6cLow monocytes may arise independently in the bone marrow through the use of the transcription factor Nur77 [32]. Ly-6cHi monocytes are thought to be the immature form, and in addition to the spleen, there are large stores within the bone marrow that are rapidly released with injury or infection [33]. Ly-6cHi monocytes differentiate into Ly-6cLow monocytes in blood after depletion, and through adoptive transfer experiments, in muscle after ischemic tissue injury [34, 35].
The interplay between monocytes and neutrophils in sterile inflammation is highly interdependent and the mechanisms are only now coming to light. In the lung it has been shown that after ischemic injury, there is very rapid recruitment of relatively few Ly-6cHi monocytes that preceded neutrophil infiltration [36]. During I/R, neutrophils cross the endothelial boarder and form dynamic short-lived focal clusters with recruited monocytes. Interestingly, in the absence of monocytes, neutrophils were able to firmly adhere to the endothelium, but could not transmigrate into the interstitium, and in fact obstructed flow, indicating that monocytes may be the source of the chemoattractant and further underscoring the key relationship between monocytes and neutrophils during the early immune response to I/R injury (< 2hrs). This important observation also alters our understanding of the temporal separation of these cellular subsets and identifies a key mechanism by which large numbers of neutrophils are recruited. Understanding these processes can help design therapeutics that avoid inappropriate targeting. For example, depletion of early Ly-6cHi monocyte influx could lead to massive neutrophil accumulation in the vasculature [36]. The relationship between Ly-6cHi monocytes and neutrophils is also bi-directional, in that neutrophils also promote Ly-6cHi monocyte recruitment through a variety of mechanisms, including activation of the local endothelial cells, release of granule components that directly induce the recruitment of monocytes (proteoglycans, cathepsin G), release of neutrophil proteases that digest monocyte chemokines (which can serve to attenuate or enhance chemokine activity) or in the correct context, the direct release of chemokines themselves (as reviewed in [37]).
Traditionally, macrophages are thought to arise from recruitment and rapid differentiation of monocytes in resident tissues. Each tissue has its own resident macrophages that perform specialized function, such as osteoclasts in the bone and kupffer cells in the liver. Several subsets based on function have been described, including M1 (classically activated) macrophages, which mediate defense against pathogens and are activated during inflammatory responses. Alternatively activated macrophages (M2) possess anti-inflammatory functions and aid in tissue regeneration. Regulatory macrophages are thought to secrete IL-10 and promote resolution of inflammation. Macrophages possess many context-dependent functions and have tremendous plasticity. As a result, these different subsets can be thought of as a continuum. Moreover, it may be that macrophages, depending on the stimuli and context, may not arise from monocytes. Recently it has been shown that macrophages can in fact proliferate and greatly increase in number without recruitment, although it is not clear if this can occur in the myocardium [38].
Monocyte/macrophage-mediated phagocytosis of neutrophils is critical in order to limit neutrophil-mediated injury and to begin the healing/resolution phase of the immune response. After the initial phase of neutrophil infiltration, Ly-6cHi monocytes are the dominant cell type after the first day, and the initial “cooperative” interaction between Ly-6cHi monocytes and neutrophils reverses course (Figure 2). Apoptotic neutrophils attract monocytes directly [37]. The process of phagocytosing neutrophils causes monocytes to acquire what has been termed a “suppressor” phenotype, with increased secretion of anti-inflammatory cytokines such as IL-10, nitric oxide, TGF-β AND PGE2, and decreased production of the pro-inflammatory cytokines IL-1β, TNF-α and IL-12 [39,40]. After ischemic injury, depletion of monocytes allows for neutrophil persistence within the infarcted tissue, implicating a key role of monocytes in this process [27].
Figure 2.

Intercellular coordination of the innate immune response to acute cardiac injury. Shortly after I/R injury (A), resident cardiac mast cells rapidly degranulate and release pro-inflammatory cytokines and molecules that activate the endothelium, increase vascular permeability and activate resident monocytes and macrophages. There is an initial recruitment of Ly-6cHi monocytes, which potentiates the massive recruitment of neutrophils. After transendothelial migration, neutrophils follow a multiple chemotactic cues (ATP, chemokines and stimulation through formyl peptide receptors), to reach the site of injury. During this process, neutrophils secrete numerous degradative enzymes that allow them to ultimately reach cardiomyocytes. Neutrophils bind cardiomyocytes through an IL-6 dependent fashion, which triggers an intense oxidative burst, and subsequent cardiomyocyte injury death. As the immune response evolves over time (B), there is a large influx of Ly-6cHi monocytes produced in the spleen that occur in an AngII dependent fashion. Recruitment of Ly-6cHi monocytes is also promoted by multiple chemokines (including MCP-1) and apoptotic neutrophils. Ly-6cHi monocytes phagocytose apoptotic neutrophils and secrete anti-inflammatory cytokines such as IL-10, TGF-β and NO, which limit the inflammatory response. In addition, decreased monocyte/macrophage production of IL-23, which blunts IL-17a production from γδ T cells and other regulator T cells, decreases cardiomyocyte death and neutrophil generation in the bone marrow.
The interplay between monocytes and neutrophils is complex and involves communication between multiple cells types. Several important insights into the pathophysiology stem for observations made under homeostatic conditions. In order to maintain a homeostatic balance of neutrophil numbers at baseline, phagocytosis by resident monocytes and macrophages is required [41]. After neutrophils enter tissues, neutrophil apoptosis ensues (pre-programmed or triggered) and is regulated by a variety of factors, including reactive oxygen species, TNF, Bcl-2 and FasL [42–44]. By phagocytosing neutrophils, monocyte/macrophage production of the pro-inflammatory cytokine IL-23 is blunted, which leads to decreased IL-17a production by a broad group of regulatory and invariant T cells [41]. IL-17a itself drives neutrophil production in the bone marrow though induction of granulocyte/monocyte - colony stimulating factor, demonstrating that neutrophil numbers are regulated by a complex intercellular interaction between monocytes/macrophages and T cells (Figure 2B) [45,46].
Reduced monocyte infiltration correlates with improved LV function after infarct when MCP-1 or CCR2 is targeted, or through the use of angiotensin converting enzyme (ACE) inhibitors or angiotensin II receptor blockers (ARBs), which also reduce monocyte infiltration [30,47,48]. However, when completely depleting monocytes directly, either Ly-6cHi or Ly6cLow (or both), there is increased neutrophil persistence within the infarct, disorganized healing, poor scar formation and ultimately LV rupture [27,49]. While clear roles for monocyte/macrophage depletion have been documented, depletion of dendritic cells specifically, and assessment of their role is more challenging. Depletion of CD11c expressing dendritic cells has been reported to worsen infarct healing, with increased Ly-6cHi monocytes within the myocardium [50]. However, it is very challenging to specifically deplete a single myeloid lineage, and depletion of CD11c expressing cells may also deplete Ly-6cLow monocytes and macrophages, both of which express CD11c at low levels (and data not shown). Moreover, excessive monocyte recruitment also is pathologic, resulting in significant inflammation, fibrosis and LV dysfunction after ischemic injury [33]. Together, these data suggest that while excessive infiltration leads to poor infarct healing, a basal level of monocytes is required to perform necessary effector functions [27,49]. More importantly, it underscores that we still have an incomplete picture about the relationship between monocyte subsets, between monocytes and other cell types, as well as the mechanisms that underlie the ability of monocytes to so drastically modulate infarct healing.
Lymphocytes – the forgotten cells in ischemic injury
Lymphocytes are a diverse group that compromise T cells, B cells and NK cells, and have a wide variety of roles in both innate and adaptive immune responses. However, relatively little attention has been paid to lymphocytes in the setting of ischemic injury, in part because numerically, they are in the minority of influxing cell types. However – regulatory cells often have potent effects despite their relative scarcity.
For example, it is becoming apparent that γδ T cells (which express the invariant γδ T cell receptor [TCR], rather than the typical αβ TCR) are important immunoregulatory cells in infection and autoimmunity through IL-17a and IL-23 dependent mechanisms [51, 52]. IL-17a is involved in the homeostatic regulation of neutrophil numbers (see above), however it also upregulated following ischemic injury in the myocardium [41, 53]. γδ T cells are the major source of IL-17a after injury. Blockade of IL-17a improves LV function, decreases neutrophil recruitment and cardiomyocyte apoptosis [53]. Interestingly, IL-17a possesses multiple targets, and enhances neutrophil-mediated adhesion to endothelium, enhances cardiomyocyte apoptosis both directly, and indirectly through the increased neutrophil infiltration. Defining the role of IL-17a in the myocardium has revealed a previously unknown link between innate T cells, neutrophils, cardiomyocytes and endothelial cells, and highlights the complex interplay between many cells types (Figure 2B).
In addition to γδ T cells, more classical regulatory T cells subsets also play key roles following ischemic injury. CD4 KO mice, MHC-II KO mice and mice with single a TCR, which are therefore functionally deficient unless stimulated with one specific antigen, all have exacerbated monocyte influx and impaired wound healing after ischemic injury, suggesting that CD4 T cells are protective and that an auto-antigen may be presented to CD4 T cells by MHC-II expressing cells (macrophages/dendritic cells), which together form an immunosuppressive axis in the myocardium [54]. Interestingly, it may be that regulatory (CD4+CD25+) T cells, which also promote myocardial recovery via an IL-10 dependent pathway, are the key CD4+ T cell subset involved this process [55,56]. One study demonstrated that RAG KO mice (T cell and B cell deficient) have decreased infarct sizes after ischemic injury and transfer of CD4+ T cells into the RAG KO mice worsened infarct healing to WT levels suggesting that in the absence of other T cells and B cells, total CD4 T cells may be playing a pathogenic role [57]. However, this study did not separate CD4+ T cell into the regulatory subset. In addition, RAG KO mice have decreased circulating antibody (IgG), which is known to act as an immunosuppressant (as reviewed in [58]). CD8 T cells also play a role following ischemic injury. CD8+ T cells express the angiotensin type 2 (AT2R), which is known to be anti- inflammatory [59, 60]. Angiotensin II (AngII) is part of the renin-angiotensin system, long known to be a critical pathologic pathway in heart disease [61, 62]. Classically, AngII is thought to primarily induce cardiomyocyte hypertrophy and increased vascular tone by signaling through AT1R, a process worsened by blockade of AT2R (as reviewed in [63]). After I/R injury CD8+AT2R+ T cells, in an AngII-dependent fashion, inhibit inflammation and decrease infarct size through the production of IL-10 and subsequent dampening of the immune response [59].
While T cells subsets seem play a regulatory role by dampening inflammation, NK cells have a more direct protective role following ischemic injury. NK cells infiltrate the myocardium after ischemia. Adoptively transferring IL-2 activated NK cells into recipients prevents LV dysfunction by enhancing angiogenesis through an apparent contact-dependent interaction with endothelial cells, and also by preventing fibrosis following ischemia [64].
Future directions
Ischemic injury induces a rapid immune response triggered by resident innate immune cells within the myocardium. As summarized in Figure 2, activation of the innate immune system is a highly coordinated and dynamic process, with complex interactions between monocytes, neutrophils and invariant T cells that 1) amplifies the initial inflammatory response; 2) transitions the inflammatory response to a wound healing response; 3) restores homeostasis. The recent studies highlighted in this review reveal the interplay between innate immune cells during the course of infarct healing, however many key questions remain unanswered. Future studies will be focused on identifying the key components that monocytes/macrophages and T cells subsets produce that alter infarct healing (such as IL-17a). Moreover, recent advances in gated imaging are beginning to allow detailed live cell tracking in the myocardium [65], a technique that has so elegantly illuminated neutrophil and monocytes dynamics in other organ systems. To improve myocardial recovery after ischemic injury, we will need to thoroughly dissect the bidirectional signals at play between innate immune cells if we are to develop effective strategies that mitigate cardiomyocyte injury, but at the same time leave untouched the reparative response.
Table 1.
Immune cells in myocardial injury and repair
| Cell | Cell surface molecules | Mechanism of attraction | Role in Inflammation | Role in Repair | Refs |
|---|---|---|---|---|---|
| Mast Cell | C-kit+ | Unknown | Histamine and Proinflammatory cytokine release. Neutrophil recruitment. | Controversial | [7,8] |
| Neutrophil | CD11b+,Ly-6g+ | LTB4, C5a, Gro-a, ATP, mitochondrial DNA | Oxidative burst. Secretion of proteolytic enzymes. Cardiomyocyte death. | None | [10,16,19,23,25] |
| Ly-6cHi monocyte | CD115+,F4/80+, Ly-6cHi | MCP-1 | Facilitates early neutrophil recruitment Digests ischemic and necrotic material Proinflammatory cytokine production | Promotes infiltration of Ly- 6cLow monocytes Phagocytosis of neutrophils thought to terminate inflammation | [27,36,39,40] |
| Ly-6cLow monocyte | CD115+,F4/80+, Ly-6cLow | Fractalkine | None | Secrete pro-angiogenenic and pro-fibrotic cytokines. Fibroblast activation. | [27] |
| Macrophages | F4/80+, CD11b+ | Possibly differentiate from recruited monocytes vs. local proliferation | Secrete proinflammatory cytokines. | Context dependent secretion of pro-angiogenic, pro- fibrotic and anti- inflammatory cytokines. Neutrophil phagocytosis terminates immune responses. | [37] [39,40] |
| γδT cells | CD3+ | Unknown | IL-17a mediated neutrophil activation, cardiomyocyte death. | None | [51–53] |
| Regulatory T cells | CD3+,CD4+, CD25+, FoxP3+ | Unknown | Possibly increase neutrophil recruitment | Blunt excessive monocyte influx. Promote resolution of inflammation via IL-10. | [54–56] |
| CD8 T cells | CD3+, CD8+, AT2R+ | Unknown | None | Promote resolution of inflammation via IL-10 via an AngII dependent fashion | [59] |
| NK cells | NK1.1+, CD11b+ | Unknown | None | IL-2 dependent enhancement of angiogenensis | [64] |
Reference List
- 1.Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PW, Woo YJ. Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation. 2011;123:933–944. doi: 10.1161/CIR.0b013e31820a55f5. [DOI] [PubMed] [Google Scholar]
- 2.Gajarsa JJ, Kloner RA. Left ventricular remodeling in the post-infarction heart: a review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail Rev. 2011;16:13–21. doi: 10.1007/s10741-010-9181-7. [DOI] [PubMed] [Google Scholar]
- 3.Roe MT, Chen AY, Cannon CP, Rao S, Rumsfeld J, Magid DJ, Brindis R, Klein LW, Gibler WB, Ohman EM, Peterson ED. Temporal changes in the use of drug-eluting stents for patients with non-ST-Segment-elevation myocardial infarction undergoing percutaneous coronary intervention from 2006 to 2008: results from the can rapid risk stratification of unstable angina patients supress ADverse outcomes with early implementation of the ACC/AHA guidelines (CRUSADE) and acute coronary treatment and intervention outcomes network-get with the guidelines (ACTION-GWTG) registries. Circ Cardiovasc Qual Outcomes. 2009;2:414–420. doi: 10.1161/CIRCOUTCOMES.109.850248. [DOI] [PubMed] [Google Scholar]
- 4.Kaczorowski DJ, Tsung A, Billiar TR. Innate immune mechanisms in ischemia/reperfusion. Front Biosci (Elite Ed) 2009;1:91–98. doi: 10.2741/E10. [DOI] [PubMed] [Google Scholar]
- 5.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13:114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 6.Ionita MG, Arslan F, de Kleijn DP, Pasterkamp G. Endogenous inflammatory molecules engage Toll-like receptors in cardiovascular disease. J Innate Immun. 2010;2:307–315. doi: 10.1159/000314270. [DOI] [PubMed] [Google Scholar]
- 7.Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998;98:699–710. doi: 10.1161/01.cir.98.7.699. [DOI] [PubMed] [Google Scholar]
- 8.Chakraborti T, Mandal A, Mandal M, Das S, Chakraborti S. Complement activation in heart diseases. Role of oxidants. Cell Signal. 2000;12:607–617. doi: 10.1016/s0898-6568(00)00111-x. [DOI] [PubMed] [Google Scholar]
- 9.Foreman KE, Glovsky MM, Warner RL, Horvath SJ, Ward PA. Comparative effect of C3a and C5a on adhesion molecule expression on neutrophils and endothelial cells. Inflammation. 1996;20:1–9. doi: 10.1007/BF01487740. [DOI] [PubMed] [Google Scholar]
- 10.Tyagi S, Klickstein LB, Nicholson-Weller A. C5a-stimulated human neutrophils use a subset of beta2 integrins to support the adhesion-dependent phase of superoxide production. J Leukoc Biol. 2000;68:679–686. [PubMed] [Google Scholar]
- 11.Engels W, Reiters PH, Daemen MJ, Smits JF, van der V. Transmural changes in mast cell density in rat heart after infarct induction in vivo. J Pathol. 1995;177:423–429. doi: 10.1002/path.1711770414. [DOI] [PubMed] [Google Scholar]
- 12.Bhattacharya K, Farwell K, Huang M, Kempuraj D, Donelan J, Papaliodis D, Vasiadi M, Theoharides TC. Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. Int J Immunopathol Pharmacol. 2007;20:69–74. doi: 10.1177/039463200702000108. [DOI] [PubMed] [Google Scholar]
- 13.Ayach BB, Yoshimitsu M, Dawood F, Sun M, Arab S, Chen M, Higuchi K, Siatskas C, Lee P, Lim H, Zhang J, Cukerman E, Stanford WL, Medin JA, Liu PP. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:2304–2309. doi: 10.1073/pnas.0510997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waskow C, Paul S, Haller C, Gassmann M, Rodewald HR. Viable c-Kit(W/W) mutants reveal pivotal role for c-kit in the maintenance of lymphopoiesis. Immunity. 2002;17:277–288. doi: 10.1016/s1074-7613(02)00386-2. [DOI] [PubMed] [Google Scholar]
- 15.Dreyer WJ, Michael LH, Nguyen T, Smith CW, Anderson DC, Entman ML, Rossen RD. Kinetics of C5a release in cardiac lymph of dogs experiencing coronary artery ischemia-reperfusion injury. Circ Res. 1992;71:1518–1524. doi: 10.1161/01.res.71.6.1518. [DOI] [PubMed] [Google Scholar]
- 16.Entman ML, Youker K, Shappell SB, Siegel C, Rothlein R, Dreyer WJ, Schmalstieg FC, Smith CW. Neutrophil adherence to isolated adult canine myocytes. Evidence for a CD18-dependent mechanism. J Clin Invest. 1990;85:1497–1506. doi: 10.1172/JCI114596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gwechenberger M, Mendoza LH, Youker KA, Frangogiannis NG, Smith CW, Michael LH, Entman ML. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546–551. doi: 10.1161/01.cir.99.4.546. [DOI] [PubMed] [Google Scholar]
- 18.Youker K, Smith CW, Anderson DC, Miller D, Michael LH, Rossen RD, Entman ML. Neutrophil adherence to isolated adult cardiac myocytes. Induction by cardiac lymph collected during ischemia and reperfusion. J Clin Invest. 1992;89:602–609. doi: 10.1172/JCI115626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Entman ML, Youker K, Shoji T, Kukielka G, Shappell SB, Taylor AA, Smith CW. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J Clin Invest. 1992;90:1335–1345. doi: 10.1172/JCI115999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawakami R, Saito Y, Kishimoto I, Harada M, Kuwahara K, Takahashi N, Nakagawa Y, Nakanishi M, Tanimoto K, Usami S, Yasuno S, Kinoshita H, Chusho H, Tamura N, Ogawa Y, Nakao K. Overexpression of brain natriuretic peptide facilitates neutrophil infiltration and cardiac matrix metalloproteinase-9 expression after acute myocardial infarction. Circulation. 2004;110:3306–3312. doi: 10.1161/01.CIR.0000147829.78357.C5. [DOI] [PubMed] [Google Scholar]
- 21.Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016–1023. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 22.Jolly SR, Kane WJ, Hook BG, Abrams GD, Kunkel SL, Lucchesi BR. Reduction of myocardial infarct size by neutrophil depletion: effect of duration of occlusion. Am Heart J. 1986;112:682–690. doi: 10.1016/0002-8703(86)90461-8. [DOI] [PubMed] [Google Scholar]
- 23.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 24.Murphy PM, Ozcelik T, Kenney RT, Tiffany HL, McDermott D, Francke U. A structural homologue of the N-formyl peptide receptor. Characterization and chromosome mapping of a peptide chemoattractant receptor family. J Biol Chem. 1992;267:7637–7643. [PubMed] [Google Scholar]
- 25.Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012 doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, Entman ML. A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol. 2000;278:H1049–H1055. doi: 10.1152/ajpheart.2000.278.4.H1049. [DOI] [PubMed] [Google Scholar]
- 29.Tsujioka H, Imanishi T, Ikejima H, Kuroi A, Takarada S, Tanimoto T, Kitabata H, Okochi K, Arita Y, Ishibashi K, Komukai K, Kataiwa H, Nakamura N, Hirata K, Tanaka A, Akasaka T. Impact of heterogeneity of human peripheral blood monocyte subsets on myocardial salvage in patients with primary acute myocardial infarction. J Am Coll Cardiol. 2009;54:130–138. doi: 10.1016/j.jacc.2009.04.021. [DOI] [PubMed] [Google Scholar]
- 30.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–137. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C− monocytes. Nat Immunol. 2011;12:778–785. doi: 10.1038/ni.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, Libby P, Pittet M, Weissleder R, Nahrendorf M. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–1638. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Capoccia BJ, Gregory AD, Link DC. Recruitment of the inflammatory subset of monocytes to sites of ischemia induces angiogenesis in a monocyte chemoattractant protein-1-dependent fashion. J Leukoc Biol. 2008;84:760–768. doi: 10.1189/jlb.1107756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreisel D, Nava RG, Li W, Zinselmeyer BH, Wang B, Lai J, Pless R, Gelman AE, Krupnick AS, Miller MJ. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A. 2010;107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soehnlein O, Lindbom L, Weber C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood. 2009;114:4613–4623. doi: 10.1182/blood-2009-06-221630. [DOI] [PubMed] [Google Scholar]
- 38.Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 41.Stark MA, Huo Y, Burcin TL, Morris MA, Olson TS, Ley K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi SD, Voyich JM, Braughton KR, DeLeo FR. Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J Immunol. 2003;170:3357–3368. doi: 10.4049/jimmunol.170.6.3357. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi SD, Voyich JM, Somerville GA, Braughton KR, Malech HL, Musser JM, DeLeo FR. An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J Leukoc Biol. 2003;73:315–322. doi: 10.1189/jlb.1002481. [DOI] [PubMed] [Google Scholar]
- 44.Jonsson H, Allen P, Peng SL. Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med. 2005;11:666–671. doi: 10.1038/nm1248. [DOI] [PubMed] [Google Scholar]
- 45.Tan W, Huang W, Gu X, Zhong Q, Liu B, Schwarzenberger P. IL-17F/IL-17R interaction stimulates granulopoiesis in mice. Exp Hematol. 2008;36:1417–1427. doi: 10.1016/j.exphem.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z, Bagby G, Nelson S, Kolls JK. Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol. 2000;164:4783–4789. doi: 10.4049/jimmunol.164.9.4783. [DOI] [PubMed] [Google Scholar]
- 47.Leuschner F, Panizzi P, Chico-Calero I, Lee WW, Ueno T, Cortez-Retamozo V, Waterman P, Gorbatov R, Marinelli B, Iwamoto Y, Chudnovskiy A, Figueiredo JL, Sosnovik DE, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M. Angiotensin-converting enzyme inhibition prevents the release of monocytes from their splenic reservoir in mice with myocardial infarction. Circ Res. 2010;107:1364–1373. doi: 10.1161/CIRCRESAHA.110.227454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, bou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 49.van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJ. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170:818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, Sano M, Yoshikawa T, Okada Y, Koyasu S, Ogawa S, Fukuda K. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. 2012;125:1234–1245. doi: 10.1161/CIRCULATIONAHA.111.052126. [DOI] [PubMed] [Google Scholar]
- 51.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity. 2009;31:321–330. doi: 10.1016/j.immuni.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 53.Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, Yuan J, Jevallee H, Wei F, Shi GP, Cheng X. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol. 2012;59:420–429. doi: 10.1016/j.jacc.2011.10.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, Kerkau T, Frantz S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation. 2012;125:1652–1663. doi: 10.1161/CIRCULATIONAHA.111.044164. [DOI] [PubMed] [Google Scholar]
- 55.Matsumoto K, Ogawa M, Suzuki J, Hirata Y, Nagai R, Isobe M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int Heart J. 2011;52:382–387. doi: 10.1536/ihj.52.382. [DOI] [PubMed] [Google Scholar]
- 56.Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol. 2010;176:2177–2187. doi: 10.2353/ajpath.2010.090759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, French BA, Linden J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation. 2006;114:2056–2064. doi: 10.1161/CIRCULATIONAHA.106.649244. [DOI] [PubMed] [Google Scholar]
- 58.Clynes R. Protective mechanisms of IVIG. Curr Opin Immunol. 2007;19:646–651. doi: 10.1016/j.coi.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 59.Curato C, Slavic S, Dong J, Skorska A, tarche-Xifro W, Miteva K, Kaschina E, Thiel A, Imboden H, Wang J, Steckelings U, Steinhoff G, Unger T, Li J. Identification of noncytotoxic and IL-10-producing CD8+AT2R+ T cell population in response to ischemic heart injury. J Immunol. 2010;185:6286–6293. doi: 10.4049/jimmunol.0903681. [DOI] [PubMed] [Google Scholar]
- 60.Kaschina E, Grzesiak A, Li J, Foryst-Ludwig A, Timm M, Rompe F, Sommerfeld M, Kemnitz UR, Curato C, Namsolleck P, Tschope C, Hallberg A, Alterman M, Hucko T, Paetsch I, Dietrich T, Schnackenburg B, Graf K, Dahlof B, Kintscher U, Unger T, Steckelings UM. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation. 2008;118:2523–2532. doi: 10.1161/CIRCULATIONAHA.108.784868. [DOI] [PubMed] [Google Scholar]
- 61.Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667–1675. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- 62.Dickstein K, Kjekshus J. Effects of losartan and captopril on mortality and morbidity in high-risk patients after acute myocardial infarction: the OPTIMAAL randomised trial. Optimal Trial in Myocardial Infarction with Angiotensin II Antagonist Losartan. Lancet. 2002;360:752–760. doi: 10.1016/s0140-6736(02)09895-1. [DOI] [PubMed] [Google Scholar]
- 63.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 64.Bouchentouf M, Forner KA, Cuerquis J, Michaud V, Zheng J, Paradis P, Schiffrin EL, Galipeau J. Induction of cardiac angiogenesis requires killer cell lectin-like receptor 1 and alpha4beta7 integrin expression by NK cells. J Immunol. 2010;185:7014–7025. doi: 10.4049/jimmunol.1001888. [DOI] [PubMed] [Google Scholar]
- 65.Li W, Nava RG, Bribriesco AC, Zinselmeyer BH, Spahn JH, Gelman AE, Krupnick AS, Miller MJ, Kreisel D. Intravital 2-photon imaging of leukocyte trafficking in beating heart. J Clin Invest. 2012;122:2499–2508. doi: 10.1172/JCI62970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, Entman ML. A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol. 2000;278:H1049–H1055. doi: 10.1152/ajpheart.2000.278.4.H1049. [DOI] [PubMed] [Google Scholar]