

Abstract
Understanding patterns of spontaneous mutations is of fundamental interest in studies of human genome evolution and genetic disease. Here, we used extremely rare variants in humans to model the molecular spectrum of single-nucleotide mutations. Compared to common variants in humans and human–chimpanzee fixed differences (substitutions), rare variants, on average, arose more recently in the human lineage and are less affected by the potentially confounding effects of natural selection, population demographic history, and biased gene conversion. We analyzed variants obtained from a population-based sequencing study of 202 genes in >14,000 individuals. We observed considerable variability in the per-gene mutation rate, which was correlated with local GC content, but not recombination rate. Using >20,000 variants with a derived allele frequency ≤10−4, we examined the effect of local GC content and recombination rate on individual variant subtypes and performed comparisons with common variants and substitutions. The influence of local GC content on rare variants differed from that on common variants or substitutions, and the differences varied by variant subtype. Furthermore, recombination rate and recombination hotspots have little effect on rare variants of any subtype, yet both have a relatively strong impact on multiple variant subtypes in common variants and substitutions. This observation is consistent with the effect of biased gene conversion or selection-dependent processes. Our results highlight the distinct biases inherent in the initial mutation patterns and subsequent evolutionary processes that affect segregating variants.
Mutation is one of the most fundamental processes in biology. It is the ultimate source of genetic variation and one of the driving forces of evolution. Mutation also plays a significant role in the etiology of human diseases. There is considerable interest in understanding the underlying pattern and molecular spectrum of spontaneous mutations. Historically, two approaches were applied to estimate the single-nucleotide mutation rate in humans. The first analyzes divergent sites between humans and another species, typically chimpanzee. According to Kimura's neutral theory, the majority of substitutions are neutral and therefore the extent of between-species divergence can be used to estimate the neutral mutation rate (Kimura 1983). Many groups have applied this approach to estimate the spontaneous mutation rate in humans (Drake et al. 1998; Nachman and Crowell 2000; Kumar and Subramanian 2002; Silva and Kondrashov 2002). However, several forces, including natural selection, biased gene conversion (BGC), and demographic history, can alter fixation probabilities and reshape the spectrum and genomic distribution of between-species substitution patterns. A second, more direct approach, pioneered by Haldane (1935), uses incidence rates of dominant disorders in humans to estimate the mutation rate (Sommer 1995; Sommer and Ketterling 1996; Kondrashov 2003; Lynch 2010). This approach, however, is limited by the fact that only a small subset of new mutations manifest as disease variants (Nachman 2004).
The mutation rates from these studies represent a genome-wide average. However, there is extensive variability among different genes or genomic regions in both between-species divergence and within-species diversity (Wolfe et al. 1989; Nachman and Crowell 2000; Sachidanandam et al. 2001; Smith and Lercher 2002; Kondrashov 2003; Hodgkinson et al. 2009). This suggests that spontaneous mutation rates are not constant throughout the genome, although the reasons behind this variability are unclear.
Local nucleotide composition is a frequently studied feature that could contribute to mutation rate variability. One study showed that AT > GC (an A base replaced with a G or a T base replaced with a C) common variants segregate at a higher frequency in regions with higher GC content (Webster et al. 2003), and others similarly reported increased fixation bias toward GC base pairs in GC-rich regions (Lercher and Hurst 2002a; Lercher et al. 2002). However, analyses of GC content and variant patterns often reported contradicting findings. For example, while some studies showed that GC content is positively correlated with both divergence rates between humans and chimpanzee (Smith et al. 2002; Webster et al. 2003; Arndt and Hwa 2005; Duret and Arndt 2008) and within-human nucleotide diversity (Sachidanandam et al. 2001; Hellmann et al. 2005), another study found a negative correlation (Cai et al. 2009). Furthermore, while some studies reported increasing GC > AT substitution rates with increasing GC content (Smith et al. 2002; Webster et al. 2003), others showed a decrease (Arndt and Hwa 2004; Duret and Arndt 2008). These inconsistencies could be partly explained by differences in the allele frequency, and therefore the evolutionary time scale of the variants analyzed in different studies. Consequently the observed patterns could be the result of confounding factors, such as selection and demography, instead of alterations in the actual mutation rate.
Recombination is known to influence patterns of common variation and substitution rates. Correlations between recombination rate and nucleotide diversity or between species substitution rates have been observed in humans (Nachman et al. 1998; Nachman 2001; Lercher and Hurst 2002b; Hellmann et al. 2003, 2005; Spencer et al. 2006; Duret and Arndt 2008; Cai et al. 2009; Lohmueller et al. 2011), Drosophila (Begun and Aquadro 1992; Begun et al. 2007; Kulathinal et al. 2008), and several plant species (Dvorak et al. 1998; Kraft et al. 1998; Stephan and Langley 1998; Tenaillon et al. 2004). Three major theories exist to explain these observations. First, recombination may be directly mutagenic, leading to increased mutation rates in regions of high recombination and thus higher diversity (Lercher and Hurst 2002b; Hellmann et al. 2003, 2008). Second, while background selection and selective sweeps reduce haplotype diversity, recombination generates new haplotypes by shuffling variants onto different backgrounds, thereby maintaining diversity in regions of high recombination rates (Kaplan et al. 1989; Charlesworth et al. 1993, 1995; Hudson and Kaplan 1995; Nachman 2001). A third explanation is BGC, a recombination-associated process that preferentially repairs AT/GC mismatches produced during recombination to GC bases, leading to preferential fixation of GC alleles (for review, see Duret and Galtier 2009). Over time, the observed effect of BGC can mimic that of natural selection, leading to an excess of “weak” (W) A/T bases converted to “strong” (S) G/C bases as if the latter were under positive selection (Berglund et al. 2009; Galtier et al. 2009; Necsulea et al. 2011). The reports hypothesizing a mutagenic effect of recombination relied on common variants and substitutions (Lercher and Hurst 2002b; Hellmann et al. 2003, 2008). Several lines of evidence argue against the mutagenic recombination theory and instead suggest that a selection-dependent mechanism or BGC can explain the observed correlation between diversity and recombination rate (Duret and Arndt 2008; Berglund et al. 2009; Galtier et al. 2009; Lohmueller et al. 2011).
Previous studies using common variants within humans and substitutions between humans and chimpanzees are effectively dealing with mutations accumulated over many generations. Their patterns, therefore, reflect the cumulative influence of many processes, including natural selection, population demographic history, and BGC. A major challenge in the field is to elucidate the extent to which these forces alter the distribution of variants over time and to distinguish their relative contributions. To minimize the effects of selection, many studies restrict their analysis to noncoding regions of the genome. However, widespread signatures of recent positive selection, even within supposedly neutral regions (Williamson et al. 2007), suggest that noncoding regions may also be influenced by selection.
Rare variants represent a newly available and expanding resource that can overcome some of these limitations. Rare variants are relatively young, predominantly because they are the result of recent mutation events. Therefore, rare variants are typically less affected by population demographic history or natural selection (Messer 2009). Furthermore, as BGC acts only on variants after they have arisen in the population (Duret and Galtier 2009), it does not influence innate mutation rates. Rare variants, therefore, are an appropriate resource for studying the spectrum and genomic distribution of mutations while minimizing the potentially confounding influences. In addition, while family-based whole-genome sequencing has begun to identify de novo mutations that provide more direct measures of mutation rates (The 1000 Genomes Project Consortium 2010; Conrad et al. 2011; Campbell et al. 2012; Kong et al. 2012), the identified mutations sparsely cover the genome. For example, if whole-genome sequencing of each parent-offspring trio yields ∼40 de novo mutations (Conrad et al. 2011), 500 such trios would need to be sequenced to accumulate roughly 20,000 mutations. These mutations, however, would occur once per 150 kb on average, and the data would lack the spatial resolution necessary to detect the effect of local genomic context on a finer scale.
We studied a set of rare variants discovered via targeted resequencing of 202 genes in >14,000 unrelated individuals. We analyzed the per-gene mutation rate as well as the probability of each site to contain a variant of a specific subtype relative to local GC content, recombination rate, and recombination hotspots. In order to compare mutation rate inferences based on rare variants with those obtained by within- and between-species data, we compared rare variant patterns to common variant data from The 1000 Genomes Project Consortium and substitution sites between humans and chimpanzee. These three variant classes cover different evolutionary time scales, and the differences between them allow us to examine the distinct influence of genomic context on the initial mutation process, the subsequent rise of some mutations to become common variants, and eventual fixation.
Results
Variant counts and densities among rare variants, common variants, and substitutions
We obtained rare variants from a previously described sequencing study targeting the exons and flanking intronic regions of 202 genes in >14,000 individuals to a median depth of 27× (Nelson et al. 2012). The genes are drug targets relevant in 12 complex diseases; and the subjects were recruited for genetic association studies of these diseases (Nelson et al. 2012). Several complementary methods were used to assess the quality of rare variants in these data. Among singleton variants, the false positive and negative rates were estimated to be 2.0% and 2.7%, respectively, with lower error rates estimated for more common variants (Nelson et al. 2012). For this study we focused on the 195 autosomal genes, with ∼700 kb targeted regions in ∼2000 targeted exons, which contained a total of 20,053 rare variants with a derived allele frequency (DAF) ≤10−4 in the European subset (N = 12,515). Each variant was categorized into one of seven possible variant subtypes based on the ancestral and derived allele states: AT > GC, GC > AT, CpG GC > AT, AT > CG, GC > TA, AT > TA, and GC > CG (Table 1). The notation of AT > GC indicates a site where the ancestral base A has a G as the derived allele, or ancestral base T has a derived allele C.
Table 1.
Variant counts and conditional variant proportions across variant subtype for rare variants, common variants, and substitutions
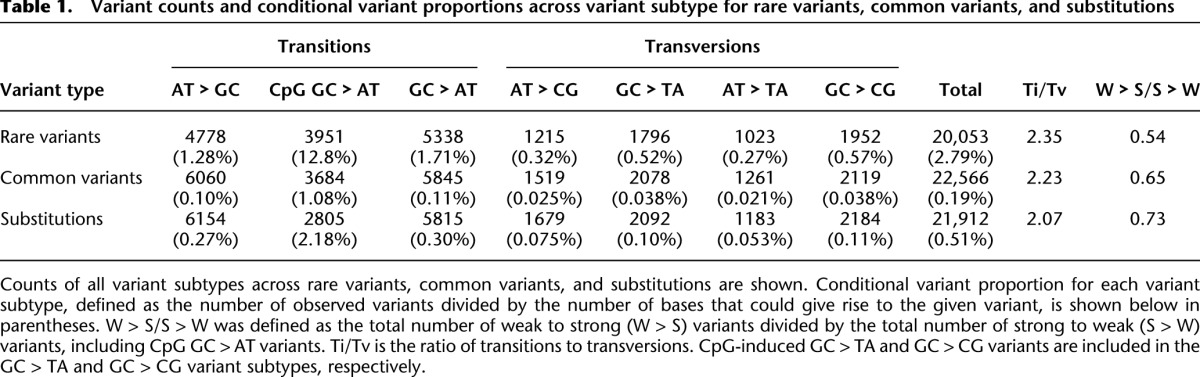
We summarized variant counts by subtype (Table 1). Nearly 13% of CpG sites have a rare GC > AT variant, compared with only 1.71% of non-CpG GC bases, consistent with the known hypermutability of CpG dinucleotides (Nachman and Crowell 2000; Kondrashov 2003; Hwang and Green 2004). Among rare variants, there were nearly twice as many S > W variants (those converting a G/C base pair into an A/T base pair) as the opposite W > S variants (Table 1). This mutational AT bias is consistent with previous observations (Lynch 2010), and can be mainly explained by the relatively high frequency of GC > AT variants at CpG dinucleotides (Table 1).
For comparison, we also analyzed common variants and substitutions. We randomly sampled intergenic regions from the human genome to obtain common variants and substitutions for analysis while matching the genomic context of the rare variant data set (see Methods). Sampling intergenic regions allowed us to minimize effects of selection. To achieve comparable statistical power, we sampled a similar number of common variants and substitutions as the rare variants. In all, we obtained 22,566 variants from the European subset of The 1000 Genomes Project Consortium with a DAF > 5% and 21,912 human-lineage-specific divergent sites between humans and chimpanzee (Table 1).
The relative proportion of variant subtypes differed among the three variant classes. Figure 1 shows the total variant proportion, defined as the number of variants of a given subtype over the total number of variants in that variant class. The relative proportion of AT > GC variants increased progressively from rare variants to substitutions, while CpG GC > AT transitions correspondingly decreased (Fig. 1). Other variant subtypes showed little change across the three variant classes. These observed proportions, however, are influenced by the different sets of sites analyzed for rare variants and for common variants/substitutions, and cannot be directly interpreted as the relative mutation rates across subtypes. For example, mutation rates at CpG sites are affected by methylation status, yet sites near promoters tend to be hypomethylated compared with intergenic regions (Molaro et al. 2011).
Figure 1.

Comparison of total variant proportions of the seven variant subtypes across the three variant classes. The total variant proportion is shown for each of the seven variant subtypes, defined as the number of variants of a given subtype over the total number of variants in that variant class. The three variant classes were rare variants, common variants, and substitutions.
The conditional variant proportion, defined as the number of a given variant subtype divided by the total number of bases that could produce the given subtype, was higher in all rare variant subtypes compared with common variants and substitutions (Table 1). The higher “absolute” conditional variant proportion in rare variants is expected, as the rare variants were discovered in >12,000 individuals. Importantly, the “relative” magnitudes across rare variant subtypes are expected to more closely reflect the relative spontaneous mutation rate than common variants or substitutions. The results for rare variants in Table 1, therefore, provide more accurate estimates of the relative mutation rates among different mutation subtypes.
The per-gene mutation rate was influenced by GC content but not recombination rate
We analyzed the per-gene mutation rate for 193 genes (out of the 195 autosomal genes), calculated previously by Nelson et al. (2012), using the method described by Coventry et al. (2010) and Wakeley and Takahashi (2003). There were considerable fluctuations in the mutation rate across genes (Fig. 2A). To assess the impact of genomic context on this variability, we calculated average GC content and recombination rate within the transcribed region of each gene (Fig. 2B and C, respectively). There was a weak but significant positive correlation between mutation rate and GC content (Pearson's r = 0.22, P = 0.0031) (Fig. 2D, dashed line). Recombination rate, however, was not significantly correlated with mutation rate (Pearson's r = 0.039, P = 0.60) (Fig. 2E, dashed line). To ensure that outliers did not drive these results, we excluded genes that fell outside of two standard deviations from the mean GC content or mutation rate (N = 8) and the recombination rate (N = 10). There was a slight increase in the correlation with GC content and little in the correlation with recombination rate (dotted line in Fig. 2D and 2E, respectively). As previously reported (Kong et al. 2002), GC content and recombination rate themselves are positively correlated (Pearson's r = 0.18, P = 0.017). Multiple linear regression including both GC content and recombination rate as covariates did not change the results from either regression alone, and recombination rate was still not significantly correlated with mutation rate (GC content P-value = 0.002, recombination rate P-value = 0.66).
Figure 2.

Variability of mutation rates across 193 genes and relationships with genomic context. (A) Per-gene mutations rates (×10−7 per base pair per generation) for 193 genes, estimated previously by coalescent modeling (Nelson et al. 2012), are shown ordered from lowest to highest. The black line indicates the average of 193 genes (1.02 × 10−8 per base pair per generation). (B) Per-gene average GC content ordered as in A. (C) Per-gene average recombination rate (log10 cM/Mb) ordered as in A. (D) Relationship between GC content and mutation rate. The dashed line represents the linear regression fitting. After removing outliers (gray filled points), the regression was recalculated (dotted line). (E) Relationship between recombination rate (log10 cM/Mb) and mutation rate. The dashed line represents the linear regression fitting. Outliers were removed (gray filled points) and the regression was recalculated (dotted lines).
Using logistic regression to analyze per-site variant patterns
The per-gene mutation rates analyzed above were calculated using all variant subtypes in aggregate; however, previous studies suggest that GC content and recombination rate may have different effects on specific variant subtypes (Lercher and Hurst 2002a; Arndt et al. 2005; Duret and Arndt 2008; Berglund et al. 2009). Estimating subtype-specific mutation rates on a per-gene or per-exon basis lacks a sufficient number of sites, especially for subtypes with relatively few observed variants (such as transversions). Therefore, we combined the ∼700 K targeted sites over all 195 genes, using a per-site logistic regression strategy to examine the effect of local GC content and recombination rate on the probability of observing a variant of a given subtype (see Methods).
The dependent variable of the logistic regression was obtained by scoring each site as either variant or invariant. If the site was scored as variant, it was further categorized into one of seven variant subtypes based on the ancestral and derived alleles. The log odds of a site being variant was regressed on GC content and recombination rate, calculated in 1-kb windows surrounding each individual site.
GC content affected rare variants differently from common variants and substitutions
Overall, the probability of observing any rare variant was positively influenced by GC content (β = 0.68, P-value < 10−16). However, individual subtypes showed mostly negative or relatively small positive effects of GC content (Fig. 3). The observation that individual subtypes could show opposite regression results to all variants combined may seem counterintuitive, but is an example of Simpson's Paradox, where trends observed in subsets of the data can be the opposite of what is observed in the entire data set (Agresti 2002). CpG-induced GC > AT variants, one of the major variant subtypes, tended to lie in GC-rich regions (50%–65% GC content), whereas AT > GC transitions tended to occur in GC-poor regions (30%–45% GC content) (Supplemental Fig. 1). The unbalanced distribution of GC content across variant subtypes, combined with the much higher mutation rate at CpG dinucleotides, drove the observed positive slope for all variants combined (Supplemental Fig. 1). When all CpG sites (variant or invariant) were removed and the regression run, the relationship between total rare variants and GC content became negative and was no longer significant (β = −0.17, P-value = 0.028). A similar finding was noted previously for substitution data (Duret and Arndt 2008). These results highlight the importance of studying variant subtypes, as analysis of all variants in aggregate could miss the underlying pattern of individual subtypes.
Figure 3.

Regression results for GC content across variant subtypes for rare variants, common variants, and substitutions. The relationship between local GC content and the observed conditional variant proportion for seven variant subtypes: (A) AT > GC, (B) AT > CG, (C) AT > TA, (D) CpG GC > AT, (E) GC > AT, (F) GC > TA, and (G) GC > CG. Filled points show the conditional variant proportions in each GC content bin, scaled by the intercept of the logistic regression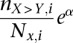 , where
, where 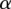 is the intercept calculated in the regression,
is the intercept calculated in the regression, 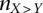 is the count of the given
is the count of the given 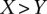 variant subtype, and
variant subtype, and 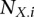 is the number of
is the number of 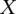 ancestral invariant sites that could produce the given subtype in the
ancestral invariant sites that could produce the given subtype in the 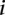 th GC content bin. Symbol size represents the proportion of the given variant subtype falling into a given GC-content bin. The solid lines show the fitted logistic regression curve, where
th GC content bin. Symbol size represents the proportion of the given variant subtype falling into a given GC-content bin. The solid lines show the fitted logistic regression curve, where 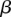 is the slope fitted in the logistic regression and
is the slope fitted in the logistic regression and 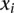 is the GC content in the
is the GC content in the 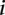 th bin. The gray dashed line represents the baseline of no effect,
th bin. The gray dashed line represents the baseline of no effect, 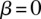 . Legends in each subplot show the regression slope calculated for each variant class and its significance. (***) P-value < 0.0001, (**) P-value < 0.001, (*) P-value < 0.01.
. Legends in each subplot show the regression slope calculated for each variant class and its significance. (***) P-value < 0.0001, (**) P-value < 0.001, (*) P-value < 0.01.
Comparison of rare variants and common variants or substitutions revealed subtype-specific differences among the three variant classes (Fig. 3). For AT > GC and AT > CG rare variants, there was a relatively strong negative relationship between variant proportions and GC content (Fig. 3A,B). These same trends, however, were not observed in AT > GC and AT > CG common variants or substitutions, for which the trends were weaker, and sometimes positive (Fig. 3A,B). In contrast, for GC > AT and GC > TA variants, there were relatively strong negative effects on common variants and substitutions, yet the effects on rare variants were smaller or absent (Fig. 3E,F). Together, these results show that GC-rich regions tend to have fewer W > S rare variants and fewer S > W common variants or substitutions than GC-poor regions. There was a strong negative effect on CpG GC > AT, consistent across rare variants, common variants, and substitutions (Fig. 3D). We also observed consistent negative effects on AT > TA (Fig. 3C) and GC > CG variants (Fig. 3G) across variant classes.
Recombination affects patterns of common variants and substitutions, but not rare variants
The influence of recombination rate on total rare variants (β = 0.15, P-value = 3.58 × 10−4) and individual variant subtypes was relatively small (Fig. 4). In comparison, the effect was much stronger on total common variants (β = 0.95, P-value < 10−16) and total substitutions (β = 0.34, P-value < 10−16), as well as on all variant subtypes (Fig. 4). There was a strong positive effect on W > S common variants and substitutions (Fig. 4A,B), consistent with the expected impact of BGC on variant patterns in the human genome. For the other common variant subtypes (Fig. 4C–G), the effect was positive but weaker than W > S variants. In contrast, the effect on substitutions was negative (Fig. 4C–F) or slightly positive (Fig. 4G). While the positive trends seen in W > S common variant subtypes could be explained solely by BGC, the positive effects in other subtypes suggest that either selective sweep or background selection could also be acting on these variants. Importantly, the lack of effect on rare variants suggests that mutation rates are not altered by recombination rate.
Figure 4.

Regression results for recombination rate across variant subtype for rare variants, common variants, and substitutions. The relationship between local recombination rate (log10 cM/Mb) and the observed conditional variant proportion for seven variant subtypes: (A) AT > GC, (B) AT > CG, (C) AT > TA, (D) CpG GC > AT, (E) GC > AT, (F) GC > TA, and (G) GC > CG (plotted as in Fig. 3). Filled points show the conditional variant proportions, scaled by the intercept of the logistic regression. Symbol size represents the proportion of the given variant subtype falling into a given recombination rate bin. The solid lines show the fitted logistic regression curve, where 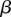 is the slope fitted in the logistic regression and
is the slope fitted in the logistic regression and 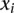 is the recombination rate in the
is the recombination rate in the 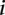 th bin. The gray dashed line represents the baseline of no effect,
th bin. The gray dashed line represents the baseline of no effect, 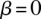 .
.
Since the deCODE recombination map published in 2002 has limited resolution, with 1257 meioses (Kong et al. 2002), we also adopted the higher-resolution deCODE map published in 2010, with 15,257 meioses and a higher marker density (Kong et al. 2010), and reanalyzed the effect of recombination rates on variant subtypes (Supplemental Table 1). The results are largely consistent with the results using the 2002 rates. For rare variants, no subtype was strongly affected by the 2010 recombination rates, just like the results with the 2002 rates. For substitutions, five of the seven subtypes had the same effect direction in the two versions of recombination rate used. For common variants, the regression coefficients were positive for all subtypes, which is what we observed with the 2002 rates. Importantly, the AT > GC and AT > CG subtypes showed the strongest effects, consistent with the influence of BGC.
Recombination hotspots influence common variants, but have little effect on rare variants or substitutions
Previous studies suggested that the distance to a recombination hotspot accounts for most of the observed correlation between nucleotide diversity and recombination rate (Spencer 2006; Spencer et al. 2006). To examine the effect of recombination hotspots, we calculated for each site its absolute distance to the nearest recombination hotspot (DTH) as reported in the population-based estimates from the HapMap Project (McVean et al. 2004; Myers et al. 2005). Median per-site DTH was consistent across all variant classes (median and standard deviation of log-transformed absolute DTH for rare variants: 4.43 ± 0.65, common variants: 4.32 ± 0.60, and substitutions: 4.32 ± 0.60). The regression results using DTH, shown in Figure 5, were largely consistent with those for recombination rate (Fig. 4). We observed relatively weak relationships between DTH and rare variants for total (β = −0.042, P-value = 1.61 × 10−4) and all variant subtypes (Fig. 5). The strongest of these, GC > AT rare variants, had a negative relationship with DTH, but it was weaker than the relationship observed in common variants (Fig. 5E). DTH had a negative effect on total common variants (β = −0.15, P-value < 10−16) and for each of the seven variant subtypes (Fig. 5). For substitutions, however, the negative effects were either weaker than those seen for common variants (Fig. 5A,D,G) or positive (Fig. 5B,C,E,F).
Figure 5.

Regression results for DTH across variant subtypes for rare variants, common variants, and substitutions. The relationship between DTH (log10 bp) and the seven variant subtypes: (A) AT > GC, (B) AT > CG, (C) AT > TA, (D) CpG GC > AT, (E) GC > AT, (F) GC > TA, and (G) GC > CG (plotted as in Fig. 3). Filled points show the conditional variant proportions, scaled by the intercept of the logistic regression. Symbol size represents the proportion of the given variant subtype falling into a given DTH bin. The solid lines are the fitted logistic regression curve, where 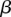 is the slope fitted in the logistic regression and
is the slope fitted in the logistic regression and 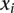 is the DTH in the
is the DTH in the 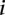 th bin. The gray dashed line represents the baseline of no effect.
th bin. The gray dashed line represents the baseline of no effect.
An alternative approach to assess the effect of recombination hotspots is to compare the variants inside recombination hotspots with those outside. Of the 20,053 rare variants, 1636 are inside hotspots, and the remaining 18,417 are outside. The conditional variant proportion is similar between the inside and outside groups, both for all rare variants combined and for individual subtypes (Supplemental Table 2), directly suggesting that recombination hotspots are not inherently mutagenic. Correspondingly, regression analyses of rare variants using inside versus outside of a recombination hotspot as the independent variable showed very weak effects for all subtypes (Supplemental Table 3). For common variants, AT > GC and AT > CG variants had two of the strongest positive regression results among the common variant subtypes (Supplemental Table 3). This pattern, in addition to the results from the recombination rates, is consistent with the effect of BGC. For substitutions, the strongest effect is in the AT > GC subtype, with a positive regression result, similar to the common variants.
Validation and robustness of regression results
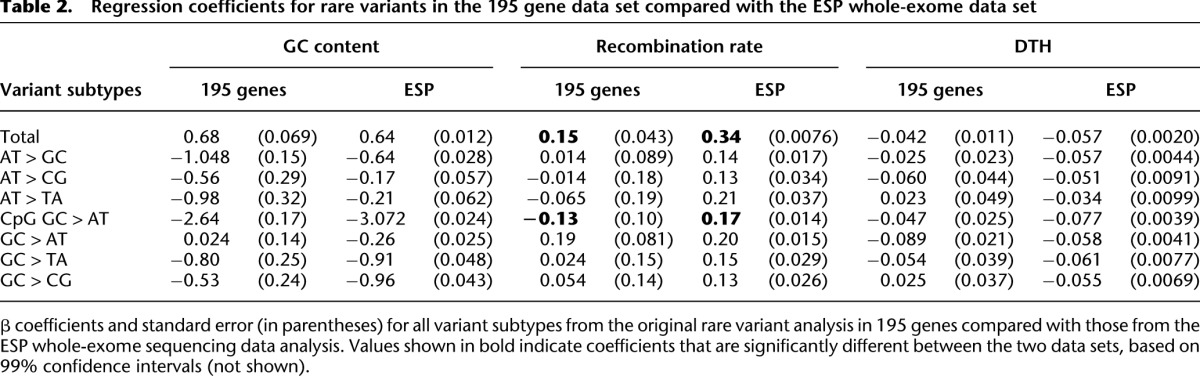
To test how well our results for 195 genes compare with data sets with more complete exome coverage, we analyzed the variants in the European subset (N = 3510) of the Exome Sequencing Project (ESP), recently described by Tennessen et al. (2012). The majority of the regression results from the 195 genes agreed with the results from the whole-exome data (based on 99% confidence intervals) (Table 2). In addition, we used several strategies to test the robustness of our regression results. Since coding variants are more likely to be under selection, we separately analyzed the rare variants in coding exons (N = 8738) and those in flanking intronic regions (N = 4642), and saw little difference in regression results (Supplemental Tables 4, 5). We also calculated GC content in several window sizes and saw little difference in the results (Supplemental Fig. 2). Since logistic regression is based on a specific probability model, we used permutations to confirm that the model-based P-values for rare variants are accurate (Supplemental Table 6), and used subsampling (Supplemental Fig. 3) and bootstrapping (Supplemental Fig. 4) techniques to verify that the magnitude of the rare variant regression results were unbiased. We also jointly tested the effect of genomic context using multivariate regression models and compared the results with those of univariate models for rare variants, common variants, and substitutions (Supplemental Tables 7–9). Additionally, we showed that sequencing coverage has little effect on the regression results for rare variants (Supplemental Table 10). Finally, to assess the impact of potential errors in the ancestral to derived allele orientation, we compared the ancestral sequence definition we adopted, based on a four-species sequence alignment, with the naive method of using the chimpanzee reference allele as the ancestral allele (Supplemental Methods). Permutation analyses showed that even errors on the order of 3%–5%, such as those that would be imposed using this naive method, had little effect on the results (Supplemental Fig. 5). Detailed information regarding these analyses is included in the Supplemental Results and Methods.
Table 2.
Regression coefficients for rare variants in the 195 gene data set compared with the ESP whole-exome data set
Discussion
In this study, we used rare variants as a model to examine mutation patterns of different variant subtypes in the human genome. We also used common variants and human–chimpanzee substitutions to analyze the ongoing biases toward fixation, involving natural selection and neutral evolutionary processes. Our results suggest that both mutation rates and fixation biases are affected by local GC content. However, fixation processes, and not mutation per se, are affected by the recombination rate.
Using rare variants to analyze spontaneous mutation rates was previously suggested in anticipation of the emergence of rare variant data from the next-generation sequencing studies (Messer 2009). Rare variants arose more recently in the population. For example, the variants we analyzed, with DAF ≤ 10−4, arose an average of ∼10 generations in the past, assuming a current population size of 50,000 individuals and a population growth rate of 0.001 (Slatkin 2000). In populations undergoing recent expansion (Coventry et al. 2010) such low-frequency variants will be even younger. As a result, rare variant patterns are primarily governed by mutation itself. Unless the force of selection is strong, natural selection, population demographic history, and BGC will not alter the observed patterns of rare variants.
We considered analyzing synonymous and nonsynonymous variants separately to further minimize the effects of natural selection. However, our logistic regression approach works on individual variant and invariant sites. It is difficult to analyze synonymous and nonsynonymous variants separately because each ancestral allele could mutate to three other nucleotides, and one needs to enumerate the potential synonymous and nonsynonymous variants that could occur at each site. As an alternative, we separately analyzed coding and noncoding rare variants and did not find any significant difference between these two functional classes, consistent with theoretical analysis showing that the effect of selection is attenuated among rare variants (Messer 2009).
The average per-gene mutation rate, based on exome sequence data from 193 genes, was 1.02 × 10−8 per base pair per generation (Nelson et al. 2012), which is similar to recent estimates from family-based sequencing studies (The 1000 Genomes Project Consortium 2010; Campbell et al. 2012; Kong et al. 2012).
Previous studies examining the effect of genomic context on mutation rate relied on local context measures computed in fixed-length genomic windows. This window-based approach is difficult to implement in exome sequencing data, because such data cover short intervals with variable length, representing targeted exons, separated by large gaps, representing introns. This leads to problems in defining window width and estimating average parameter values. In our study, most target regions are small (85% < 500 bp), and calculating rates for low frequency events, such as transversions, in these windows would be highly inaccurate. We therefore adopted a logistic regression approach, using data for individual base positions and aggregating data across sites. This approach has several advantages. It eliminates the need to account for gaps in coverage from intronic and intergenic regions, and provides sufficient numbers of sites to study the effect of genomic context on individual variant subtypes.
Multiple results suggest that recombination rate has a relatively small effect on mutation patterns, but a significant impact on common variants in the population. First, we did not observe a correlation between per-gene mutation rate and recombination rate. Second, the effect of recombination rate on rare variant subtypes was small, especially when compared with the effect on common variants and substitutions. AT > GC and AT > CG common variants and substitutions were both strongly affected by recombination rate, consistent with the role of BGC altering patterns of standing variation in the human genome. BGC has no effect on mutation rates, but over time, is expected to lead to a fixation bias toward GC bases at AT/GC polymorphic sites (Duret and Galtier 2009). A recent study reported a strong bias of W > S substitutions in human accelerated regions and this bias increased with increasing male recombination rate (Berglund et al. 2009). Furthermore, BGC can drive deleterious GC alleles to fixation (Galtier et al. 2009) and lead to the apparent increase in substitution rate with increasing recombination rate (Meunier and Duret 2004; Duret and Arndt 2008; Berglund et al. 2009; Galtier et al. 2009). These conclusions based on local recombination rates were supported by the analysis of recombination hotspots. While our results cannot completely rule out a mutagenic effect due to recombination, they suggest that if such an effect does exist, it is relatively small in comparison to the influence of BGC.
Background selection and selective sweeps can also drive positive correlations between diversity and recombination rate (Smith and Haigh 1974; Kaplan et al. 1989; Charlesworth et al. 1993; Hudson and Kaplan 1995; Cai et al. 2009; Lohmueller et al. 2011). These selection-dependent mechanisms are unlikely to affect rare variants because they are too young in the population. In addition to the impact of recombination rate on AT > GC and AT > CG common variants and substitutions, we also saw relatively strong effects on other variant subtypes. Therefore, we cannot rule out the effect of these other recombination-associated processes.
GC content varies throughout the genome, with long stretches of DNA exhibiting relatively stable GC content, known as isochores (Eyre-Walker and Hurst 2001). Previous studies propose that mutation bias or fixation bias drives the apparent regional variation in GC content and maintenance of isochores (Smith et al. 2002; Webster et al. 2003; Duret and Arndt 2008). Our results are consistent with this hypothesis, suggesting that GC-rich regions of the genome may maintain base composition by simultaneously decreasing GC-enriching, W > S, mutations and reducing the fixation of GC-depleting, S > W, common variants.
Understanding the relationship between local genomic context and mutation processes has several practical implications. More precise estimates of de novo mutation rates can improve genotype calling from short sequencing reads by providing better prior distributions for mutation spectrum. Moreover, our results can help to identify potentially functional de novo mutations by highlighting new variants that are unlikely to arise spontaneously.
Our study, however, has several limitations. We are not able to identify all potential mutations, as some will not be viable in humans. However, truly dominant lethal mutations are extremely rare and other approaches, including direct discovery of de novo variants via trio sequencing, will have similar limitations. Additionally, while rare variants are very young on the evolutionary time scale, they could still be influenced by the same confounding factors that affect common variants and substitutions, albeit to a lesser degree. At present, however, rare variants, especially the extremely rare variants we study here, represent one of the most powerful data sets currently available for high-sensitivity analysis of the rate and molecular spectrum of new mutations. Finally, our data set involves only 195 genes and could generate a biased representation of the genome. Indeed, these genes appear to be under stronger purifying selection than other genes (Nelson et al. 2012). Despite this caveat, we observed strong concordance between the results from the 195 genes and those from an exome-wide data set, indicating that any selection acting on these genes does not influence the relationship with genomic context and that our results are representative of the exome.
In conclusion, our data set of >20,000 rare variants (DAF < 10−4) represent a valuable resource for studying patterns of single-nucleotide mutation in humans. It allows us to take a new step toward differentiating the initial mutation processes from the subsequent forces that act more gradually, affecting fixation processes of segregating variants. Our results reveal a substantial difference in the relative abundance and conditional proportion of variant subtypes between rare variants, common variants, and substitutions. GC content has a strong impact on all variant classes, although the effect is different both among variant classes and among different subtypes. Recombination rate, on the other hand, has relatively little effect on rare variants, but a much stronger effect on AT > GC and AT > CG common variants and substitutions, consistent with BGC acting on existing variants. Future research, aided by deep sequencing data over more genomic targets in larger population samples, will be needed to acquire more precise estimates of such fundamental parameters. Eventually, these studies will help unravel the relative contribution of diverse evolutionary forces acting over different time scales. Such an understanding will also provide the knowledge necessary to study the allelic spectrum of inherited and somatic diseases, as well as the dynamics of human genome variation as it evolves under a variety of environmental and demographic conditions.
Methods
Ethics statement
All study participants in the component studies provided written informed consent for the use of their DNA in genetic studies. A careful review was conducted to verify that the consents were consistent with the activities of this study. In instances where the appropriateness of the informed consent for the current study was not clear, further Institutional Review Board approval was sought and obtained.
Data source and processing
Rare variants
We utilized single-nucleotide variants previously described in Nelson et al. (2012), which can be accessed at http://www.ncbi.nlm.nih.gov/projects/SNP/snp_viewBatch.cgi?sbid=1056695. The variants were discovered from a targeted resequencing study of the exons of 202 potential drug target genes (including 50 bp flanking each exon). For this study, we analyzed 195 autosomal genes, and focused on variants identified in individuals of European descent (N = 12,515). We defined rare variants as those with a DAF ≤ 10−4. We oriented all variants along the human ancestral sequence, as defined by members of The 1000 Genomes Project Consortium (http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase1/analysis_results/supporting/ancestral_alignments/; date accessed: January 3, 2012). The ancestral allele definition relied on the four-way sequence alignment among human, chimpanzee, orangutan, and macaque genomes, and it estimated the ancestral state using a probabilistic phylogenetic model. We analyzed the impact of potential errors in ancestral allele definition on our results and found that even error rates on the order of 3%–5%, such as when naively using the chimpanzee reference allele as the ancestral allele, did not change our results (Supplemental Methods and Results). To minimize the potential confounding effects due to coverage, and to enrich for high-quality variants, we selected variant and invariant sites with ≥10× coverage using per-site coverage data from a random sample of 500 individuals reported by Nelson et al. (2012).
We subdivided variants into seven distinct subtypes based on the ancestral and derived alleles: AT > GC, GC > AT (non-CpG), CpG GC > AT, AT > CG, GC > TA, AT > TA, and GC > CG. GC > AT transitions that occurred at an ancestral CpG site (CpG GC > AT) were analyzed separately from other GC > AT variants because hypermethylation of the cytosine base at CpG dinucleotides leads to spontaneous deamination, resulting in C > T and G > A transitions that occur with substantially higher rates than other subtypes (Nachman and Crowell 2000; Kondrashov 2003; Hwang and Green 2004). In addition, previous studies found that substitution rates at CpG dinucleotides are more strongly negatively correlated with GC content and recombination rate compared with non-CpG-induced GC > AT transitions, suggesting that different molecular mechanisms may be involved (Arndt et al. 2005; Duret and Arndt 2008).
GC > TA and GC > CG variants at CpG sites, which make up the eighth and ninth variant subtypes, were analyzed separately from non-CpG-induced GC > TA and GC > CG variants. They were modeled in the multinomial logistic regressions with CpG as the ancestral base (see below). As there are relatively few observed variants of these two subtypes (∼200 each in our data set), it is difficult to accurately analyze mutation patterns and we did not report these results. These variants, however, are included in the GC > TA and GC > CG variant subtypes presented in Table 1, and included when analyzing all variant subtypes combined.
Per-gene mutation rates and genomic context
We analyzed mutation rates calculated for 193 of the 195 autosomal genes (two genes were excluded due to low numbers of variants), as described previously (Nelson et al. 2012). For each of the 193 genes, we calculated the average GC content and sex-averaged pedigree-based recombination rates (Kong et al. 2002) within the transcribed region of each gene based on definitions in RefGene. Linear regression was performed in R (R Development Core Team 2008).
Sampling of intergenic regions to obtain common variants and substitutions
To sample common variants and substitutions from random genomic intervals with the least selective pressure, we first defined intergenic regions by masking all genic regions ±1 kb of the transcription start and end site of any gene based on RefGene in hg18. We then removed all regions that were not uniquely aligned in the four-way alignments between human, chimpanzee, orangutan, and macaque (ftp://ftp.ensembl.org/pub/release-54/emf/ensembl-compara/epo_4_catarrhini/; date accessed: December 7, 2011). To match the distribution of genomic features with the rare variant data as closely as possible, we sampled 32,279 autosomal regions from all possible regions according to their genomic parameters. Specifically, we matched the size distribution as well as the joint distribution of GC content and recombination rate (Kong et al. 2002) of the selected regions to those of the target regions in the exome sequencing of the 202 genes. We used these regions to sample common variants. Because there were substantially more substitutions in these regions than common variants, we randomly subsampled 12,034 of the 32,279 regions to obtain substitutions. The median and standard deviation of GC content across the assayed regions was 0.49 ± 0.12, 0.47 ± 0.11, and 0.47 ± 0.12 for rare variants, common variants, and substitutions, respectively. The median and standard deviation for recombination rate (log-transformed, in units of cM/Mb) was 0.29 ± 0.17 for rare variants, common variants, and substitutions.
Common variant data
Single-nucleotide variants from the interim phase 1 haplotype data from The 1000 Genomes Project Consortium were used to assemble a data set of common variants. The frequency file for the European subset (N = 381) of the data was downloaded from http://www.sph.umich.edu/csg/abecasis/MACH/download/1000G-PhaseI-Interim.html; date accessed: December 20, 2011. All variants within the selected regions, as described above, were oriented ancestral to derived. Successfully oriented variants with a DAF > 0.05 were categorized into the seven variant subtypes and they form the common variant data set.
Substitution data
Substitutions between human and chimpanzee were obtained using the four-way alignments between human, chimpanzee, orangutan, and rhesus macaque (ftp://ftp.ensembl.org/pub/release-54/emf/ensembl-compara/epo_4_catarrhini/; date accessed: December 7, 2011). To identify substitutions, only regions where there was a unique human, chimp, and orangutan alignment were used. Single-base human–chimpanzee differences were sampled from the 12,034 intergenic regions as described above. All sites were oriented along the ancestral lineage and categorized into the seven variant subtypes. Variant sites where the human lineage base represents the ancestral allele were excluded.
ESP rare variants
Variants from the NHLBI Exome Sequencing Project (ESP) from 5400 individuals were downloaded from the Exome Variant Server (Exome Variant Server, NHLBI ESP, Seattle, WA, URL: http://evs.gs.washington.edu/EVS/; date accessed: December 2, 2011]). We also utilized sequence coverage data downloaded from the Exome Variant Server (date accessed: December 2, 2011 and December 5, 2011) to select sites with ≥10× coverage. Subsequent analysis focused on singleton variants (DAF = 1.4 × 10−4) identified in Europeans (N = 3510). Variants were oriented along the ancestral allele, as before.
Logistic regression analysis
We used a logistic regression framework to model the effect of GC content, recombination rate, and distance to recombination hotspot on the occurrence of rare variants, common variants, and substitutions. We defined GC content at a given site as the percentage of GC bases in a 1-kb window surrounding the site (500 bp upstream, 500 bp downstream) based on the human genome reference sequence (hg18). We calculated the average recombination rate in a 1-kb window surrounding each site using both the 2002 deCODE sex-averaged recombination rates (Kong et al. 2002) and the 2010 sex-averaged recombination maps (Kong et al. 2010). The absolute distance to the center of the nearest recombination hotspot was calculated for each site using recombination hotspot coordinates from Phase II of the HapMap Project (McVean et al. 2004; Myers et al. 2005). These same definitions of recombination hotspots were used to define sites that fell inside and outside of recombination hotspots. We excluded sites if they were within repeats as defined by RepeatMasker. Recombination rates and distances to hotspots were log-transformed to more closely resemble a normal distribution.
To examine the impact on total mutation (all subtypes combined), we regressed the logit of the probability of a site containing a rare variant of any subtype against GC content, recombination rate, or DTH using separate logistic regression models for each genomic context variable. Each logistic regression has the form
where 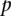 is the probability that the site contains a rare variant and
is the probability that the site contains a rare variant and 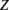 is either GC content, recombination rate, or DTH at that site. We assessed the significance of the regression using a Wald test on the
is either GC content, recombination rate, or DTH at that site. We assessed the significance of the regression using a Wald test on the 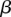 parameter. We fit similar regression models for common variants and substitutions.
parameter. We fit similar regression models for common variants and substitutions.
Next, to analyze the effect of genomic context on specific variant subtypes, we employed a multinomial logistic regression model that jointly analyzes the probability of all possible variant subtypes for a given ancestral state. Additional details are in the Supplemental Methods. Logistic and multinomial regression was performed in R (R Development Core Team 2008), using the mlogit package for multinomial regression.
Acknowledgments
We thank our colleagues who contributed to the generation of the sequencing data and initial analysis (Nelson et al. 2012). We especially thank Jennifer Aponte, Dana Fraser, Keith Nangle, and Andrew Slater for coordinating the preparation and management of the data. V.M.S. was supported by an NIH Genome Sciences Training Grant (HG000040). Funding for D.W. and J.N. was provided by an award from the Searle Scholars Program to J.N. S.Z and M.Z were supported by R01G005855. J.Z.L. was supported by an IMHRO—Johnson & Johnson Rising Star Translational Research Award. We also thank two anonymous reviewers for their insightful comments.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.154971.113.
References
- The 1000 Genomes Project Consortium 2010. A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agresti A. 2002. Categorical data analysis. Wiley-Interscience, New York. [Google Scholar]
- Arndt PF, Hwa T 2004. Regional and time-resolved mutation patterns of the human genome. Bioinformatics 20: 1482–1485 [DOI] [PubMed] [Google Scholar]
- Arndt PF, Hwa T 2005. Identification and measurement of neighbor-dependent nucleotide substitution processes. Bioinformatics 21: 2322–2328 [DOI] [PubMed] [Google Scholar]
- Arndt PF, Hwa T, Petrov DA 2005. Substantial regional variation in substitution rates in the human genome: Importance of GC content, gene density, and telomere-specific effects. J Mol Evol 60: 748–763 [DOI] [PubMed] [Google Scholar]
- Begun DJ, Aquadro CF 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519–520 [DOI] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh YP, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al. 2007. Population genomics: Whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol 5: e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund J, Pollard KS, Webster MT 2009. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol 7: e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai JJ, Macpherson JM, Sella G, Petrov DA 2009. Pervasive hitchhiking at coding and regulatory sites in humans. PLoS Genet 5: e1000336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han L, Vives L, O'Roak BJ, Sudmant PH, Shendure J, et al. 2012. Estimating the human mutation rate using autozygosity in a founder population. Nat Genet 44: 1277–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134: 1289–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B, Morgan MT 1995. The pattern of neutral molecular variation under the background selection model. Genetics 141: 1619–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, Depristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet 43: 712–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coventry A, Bull-Otterson LM, Liu X, Clark AG, Maxwell TJ, Crosby J, Hixson JE, Rea TJ, Muzny DM, Lewis LR, et al. 2010. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nat Commun 1: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF 1998. Rates of spontaneous mutation. Genetics 148: 1667–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Arndt PF 2008. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet 4: e1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet 10: 285–311 [DOI] [PubMed] [Google Scholar]
- Dvorak J, Luo MC, Yang ZL 1998. Restriction fragment length polymorphism and divergence in the genomic regions of high and low recombination in self-fertilizing and cross-fertilizing aegilops species. Genetics 148: 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre-Walker A, Hurst LD 2001. The evolution of isochores. Nat Rev Genet 2: 549–555 [DOI] [PubMed] [Google Scholar]
- Galtier N, Duret L, Glemin S, Ranwez V 2009. GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet 25: 1–5 [DOI] [PubMed] [Google Scholar]
- Haldane JB 1935. The rate of spontaneous mutation of a human gene. J Genet 83: 235–244 [DOI] [PubMed] [Google Scholar]
- Hellmann I, Ebersberger I, Ptak SE, Paabo S, Przeworski M 2003. A neutral explanation for the correlation of diversity with recombination rates in humans. Am J Hum Genet 72: 1527–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann I, Prufer K, Ji H, Zody MC, Paabo S, Ptak SE 2005. Why do human diversity levels vary at a megabase scale? Genome Res 15: 1222–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann I, Mang Y, Gu Z, Li P, de la Vega FM, Clark AG, Nielsen R 2008. Population genetic analysis of shotgun assemblies of genomic sequences from multiple individuals. Genome Res 18: 1020–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson A, Ladoukakis E, Eyre-Walker A 2009. Cryptic variation in the human mutation rate. PLoS Biol 7: e1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL 1995. Deleterious background selection with recombination. Genetics 141: 1605–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang DG, Green P 2004. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci 101: 13994–14001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NL, Hudson RR, Langley CH 1989. The “hitchhiking effect” revisited. Genetics 123: 887–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. 1983. The neutral theory of molecular evolution. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Kondrashov AS 2003. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat 21: 12–27 [DOI] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, et al. 2002. A high-resolution recombination map of the human genome. Nat Genet 31: 241–247 [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Gylfason A, Kristinsson KT, Gudjonsson SA, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467: 1099–1103 [DOI] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Wong WS, et al. 2012. Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft T, Sall T, Magnusson-Rading I, Nilsson NO, Hallden C 1998. Positive correlation between recombination rates and levels of genetic variation in natural populations of sea beet (Beta vulgaris subsp. maritima). Genetics 150: 1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathinal RJ, Bennett SM, Fitzpatrick CL, Noor MA 2008. Fine-scale mapping of recombination rate in Drosophila refines its correlation to diversity and divergence. Proc Natl Acad Sci 105: 10051–10056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Subramanian S 2002. Mutation rates in mammalian genomes. Proc Natl Acad Sci 99: 803–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD 2002a. Can mutation or fixation biases explain the allele frequency distribution of human single nucleotide polymorphisms (SNPs)? Gene 300: 53–58 [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Hurst LD 2002b. Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet 18: 337–340 [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Smith NG, Eyre-Walker A, Hurst LD 2002. The evolution of isochores: Evidence from SNP frequency distributions. Genetics 162: 1805–1810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, Albrechtsen A, Li Y, Kim SY, Korneliussen T, Vinckenbosch N, Tian G, Huerta-Sanchez E, Feder AF, Grarup N, et al. 2011. Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome. PLoS Genet 7: e1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M 2010. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107: 961–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304: 581–584 [DOI] [PubMed] [Google Scholar]
- Messer PW 2009. Measuring the rates of spontaneous mutation from deep and large-scale polymorphism data. Genetics 182: 1219–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Duret L 2004. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol 21: 984–990 [DOI] [PubMed] [Google Scholar]
- Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD 2011. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146: 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324 [DOI] [PubMed] [Google Scholar]
- Nachman MW 2001. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet 17: 481–485 [DOI] [PubMed] [Google Scholar]
- Nachman MW 2004. Haldane and the first estimates of the human mutation rate. J Genet 83: 231–233 [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL 2000. Estimate of the mutation rate per nucleotide in humans. Genetics 156: 297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW, Bauer VL, Crowell SL, Aquadro CF 1998. DNA variability and recombination rates at X-linked loci in humans. Genetics 150: 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Necsulea A, Popa A, Cooper DN, Stenson PD, Mouchiroud D, Gautier C, Duret L 2011. Meiotic recombination favors the spreading of deleterious mutations in human populations. Hum Mutat 32: 198–206 [DOI] [PubMed] [Google Scholar]
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, et al. 2012. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337: 100–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. 2008. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Sachidanandam R, Weissman D, Schmidt SC, Kakol JM, Stein LD, Marth G, Sherry S, Mullikin JC, Mortimore BJ, Willey DL, et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928–933 [DOI] [PubMed] [Google Scholar]
- Silva JC, Kondrashov AS 2002. Patterns in spontaneous mutation revealed by human-baboon sequence comparison. Trends Genet 18: 544–547 [DOI] [PubMed] [Google Scholar]
- Slatkin M 2000. Allele age and a test for selection on rare alleles. Philos Trans R Soc Lond B Biol Sci 355: 1663–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Haigh J 1974. The hitch-hiking effect of a favourable gene. Genet Res 23: 23–35 [PubMed] [Google Scholar]
- Smith NG, Lercher MJ 2002. Regional similarities in polymorphism in the human genome extend over many megabases. Trends Genet 18: 281–283 [DOI] [PubMed] [Google Scholar]
- Smith NG, Webster MT, Ellegren H 2002. Deterministic mutation rate variation in the human genome. Genome Res 12: 1350–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer SS 1995. Recent human germ-line mutation: Inferences from patients with hemophilia B. Trends Genet 11: 141–147 [DOI] [PubMed] [Google Scholar]
- Sommer SS, Ketterling RP 1996. The factor IX gene as a model for analysis of human germline mutations: An update. Hum Mol Genet 5: 1505–1514 [DOI] [PubMed] [Google Scholar]
- Spencer CC 2006. Human polymorphism around recombination hotspots. Biochem Soc Trans 34: 535–536 [DOI] [PubMed] [Google Scholar]
- Spencer CC, Deloukas P, Hunt S, Mullikin J, Myers S, Silverman B, Donnelly P, Bentley D, McVean G 2006. The influence of recombination on human genetic diversity. PLoS Genet 2: e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan W, Langley CH 1998. DNA polymorphism in lycopersicon and crossing-over per physical length. Genetics 150: 1585–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon MI, U'Ren J, Tenaillon O, Gaut BS 2004. Selection versus demography: A multilocus investigation of the domestication process in maize. Mol Biol Evol 21: 1214–1225 [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J, Takahashi T 2003. Gene genealogies when the sample size exceeds the effective size of the population. Mol Biol Evol 20: 208–213 [DOI] [PubMed] [Google Scholar]
- Webster MT, Smith NG, Ellegren H 2003. Compositional evolution of noncoding DNA in the human and chimpanzee genomes. Mol Biol Evol 20: 278–286 [DOI] [PubMed] [Google Scholar]
- Williamson SH, Hubisz MJ, Clark AG, Payseur BA, Bustamante CD, Nielsen R 2007. Localizing recent adaptive evolution in the human genome. PLoS Genet 3: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Sharp PM, Li WH 1989. Mutation rates differ among regions of the mammalian genome. Nature 337: 283–285 [DOI] [PubMed] [Google Scholar]