

Abstract
Selective breeding of dogs by humans has generated extraordinary diversity in body size. A number of multibreed analyses have been undertaken to identify the genetic basis of this diversity. We analyzed four loci discovered in a previous genome-wide association study that used 60,968 SNPs to identify size-associated genomic intervals, which were too large to assign causative roles to genes. First, we performed fine-mapping to define critical intervals that included the candidate genes GHR, HMGA2, SMAD2, and STC2, identifying five highly associated markers at the four loci. We hypothesize that three of the variants are likely to be causative. We then genotyped each marker, together with previously reported size-associated variants in the IGF1 and IGF1R genes, on a panel of 500 domestic dogs from 93 breeds, and identified the ancestral allele by genotyping the same markers on 30 wild canids. We observed that the derived alleles at all markers correlated with reduced body size, and smaller dogs are more likely to carry derived alleles at multiple markers. However, breeds are not generally fixed at all markers; multiple combinations of genotypes are found within most breeds. Finally, we show that 46%–52.5% of the variance in body size of dog breeds can be explained by seven markers in proximity to exceptional candidate genes. Among breeds with standard weights <41 kg (90 lb), the genotypes accounted for 64.3% of variance in weight. This work advances our understanding of mammalian growth by describing genetic contributions to canine size determination in non-giant dog breeds.
Domestic dogs exhibit the greatest diversity in body size of any land mammal. Mastiffs can be 50 times heavier than Chihuahuas, and Great Danes five times taller than Pekingese. Dog breeds are all descended from the gray wolf (Wayne 1993; Lindblad-Toh et al. 2005) and are the product of artificial selection that began between 15,000 and 100,000 yr ago (Vilà et al. 1997; Sablin and Khlopachev 2002; Savolainen et al. 2002; Germonpré et al. 2009; Pang et al. 2009; Ovodov et al. 2011). However, the majority of the modern dog breeds were developed within the past 300 yr (American Kennel Club 1998; Parker et al. 2004). More than 400 breeds now exist worldwide, including 175 that are recognized in the United States by the American Kennel Club (AKC; www.akc.org).
Modern domestic dog breeds are codified by standards, which apply persistent selective pressure on fixed phenotypes that are often breed defining, such as coat color, skull shape, leg length, and body size. This pressure reduces phenotypic and genetic heterogeneity within breeds, yet enormous phenotypic diversity exists across breeds (Parker et al. 2004, 2007; vonHoldt et al. 2010). These factors, along with the genetic isolation of breeds, have established domestic dog breeds as an excellent genetic system for the study of complex traits, including skeletal size and shape variation (Chase et al. 2002; Shearin and Ostrander 2010).
Loci determining size have strong signatures of selection (Akey et al. 2010; Boyko et al. 2010; Vaysse et al. 2011). The first association studies of canine body size found an influential locus in spite of sparse marker density (Chase et al. 2002; Jones et al. 2008). Chase et al. (2002) used genotypes at ∼500 microsatellites to analyze the genetic basis for canid morphological variation in Portuguese water dogs, a breed with significant variation in skeletal size (Chase et al. 2002), and identified multiple quantitative trait loci (QTLs) related to canine body size. A locus on canine chromosome 15 (CFA15) was observed to be highly associated with measures of skeletal size. Further investigation by our collaborative group led to the identification of a single haplotype composed of 20 single-nucleotide polymorphisms (SNPs) that was shared among all small breeds (<9 kg [20 lb]), but was nearly absent from giant breeds (>30 kg [66 lb]) (Sutter et al. 2007). The haplotype spans the insulin-like growth factor 1 (IGF1) gene, which is known to regulate skeletal size in both mice and humans (Baker et al. 1993; Woods et al. 1996).
A subsequent study by Jones et al. (2008) extended these findings and pioneered the use of breed-defined phenotypes (“stereotypes”) to identify associated markers, a method which is also used in the present study. Jones et al. (2008) tested the association of genotypes in 2801 dogs representing 147 breeds at 1536 SNPs with several breed stereotypes including weight, limb length, and height. They identified several new body size loci as well as replicating findings from previous studies (Chase et al. 2002, 2005), thus further supporting the use of breed standard measures, rather than individual measurements on each dog, in genetic studies of canine morphology.
Subsequent studies performed by our collaborative group on a much larger data set of 915 dogs from 80 breeds genotyped using 60,968 SNPs (the “CanMap project”) highlighted a number of phenotype-associated loci (Boyko et al. 2010). Among these were loci important in body size, some of which had been previously identified (Chase et al. 2002, 2005; Jones et al. 2008). Associations at four of the size-associated loci were replicated in data released by a subsequent study of 509 dogs from 46 breeds genotyped with 170,000 SNPs (Vaysse et al. 2011). Finally, the CanMap data set was used by Hoopes et al. (2012) to identify a new dog body size locus on CFA3 at the insulin-like growth factor 1 receptor gene (IGF1R).
Here we describe the combinatorial effects of genetic variation at six loci on determining body size in dog breeds. At four autosomal loci previously found to be associated with canine body size (Boyko et al. 2010), the critical intervals resulting from our fine-mapping revealed excellent candidate genes, including growth hormone receptor (GHR), high mobility group AT-hook 2 (HMGA2), stanniocalcin 2 (STC2), and SMAD family member 2 (SMAD2). We genotyped the most highly associated marker(s) at each locus, together with highly associated markers from the IGF1 and IGF1R genes in a large set of dogs representing the entire range of canine body size. The resulting analysis shows that approximately half of the variance of the weights of dog breeds can be explained by polymorphisms at just these six loci.
Results
We fine-mapped four body size QTLs identified in a previous genome-wide association study (GWAS) (Boyko et al. 2010). Initial critical intervals were selected based on association scores in the CanMap study at the following positions in CanFam3.1 coordinates: CFA10 (8,454,499, P = 7.06 × 10−09), CFA4 (39,200,720, P = 9.10 × 10−09 and 67,026,055, P = 2.58 × 10−07), and CFA7 (43,865,905, P = 1.05 × 10−06).
Standard breed weight (SBW) was used as a surrogate for body size, as has been done previously (Boyko et al. 2010). Specifically, when a weight was specified as part of an AKC breed standard, that value was used as the SBW for each dog of the breed in the data set. For breeds with no specified weight, values from other authorities were used (Methods; Supplemental Table 1). Where a range or different weights for male and female were given, an average was used. Since the phenotypic basis of this study is the standard weights of AKC breeds, which are specified and widely referred to in lb units, results are reported in lb as well as kg.
Fine-mapping the size loci
Fine-mapping of the four autosomal loci validated the scan associations and revealed critical intervals that include the excellent candidate genes GHR, HMGA2, STC2, and SMAD2 (see Supplemental Results, Supplemental Figs. 1–3, and Supplemental Tables 2–5 for details on the fine-mapping experiments). The most highly associated variants at each locus were two nonsynonymous SNPs in GHR, one SNP in the 5′ UTR of HMGA2, one SNP 20-kb downstream from STC2, and one deletion 24-kb downstream from SMAD2 (Fig. 1A–D; Table 1). Here we refer to each variant by the name of the proximal gene. The two nonsynonymous SNPs in GHR are termed GHR(1) and GHR(2).
Figure 1.

Fine-mapping of four loci associated with canine body size. (A–D) Regional plots of the four fine-mapped loci: CFA4:67 Mb (A), CFA10:8 Mb (B), CFA4:39 Mb (C), and CFA7:43 Mb (D). Each plot includes the following tracks, from top to bottom: P-values of the genotyped SNPs in the CanMap data set (Boyko et al. 2010) (with coordinates updated to CanFam 3.1 genome assembly); the regions of the genome covered during fine-mapping (green and blue; amplicons for marker discovery and SNP positions for SNPlex, respectively); genes (orange; see Methods for identifiers); and the most highly associated marker(s) identified in each region (red).
Table 1.
Size-associated markers
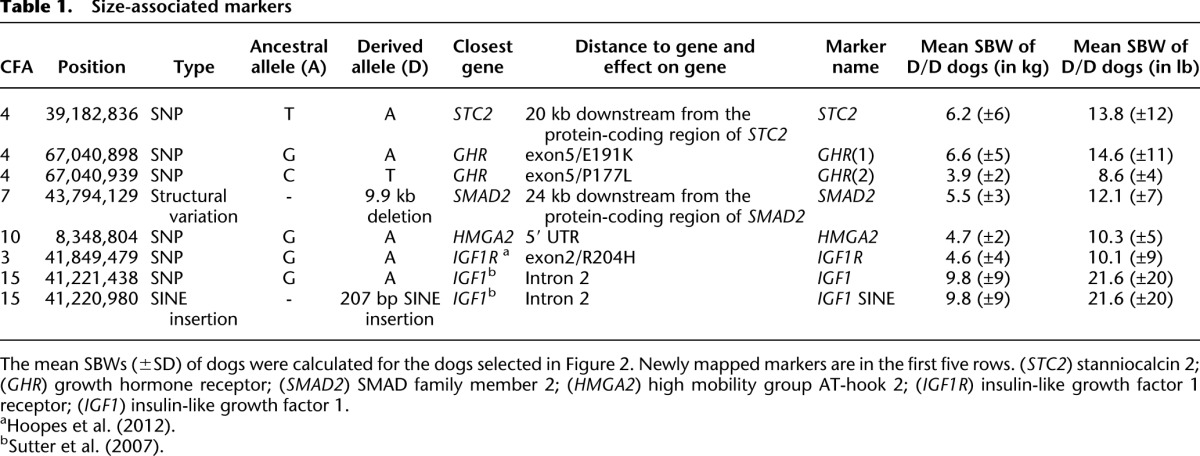
Frequency of derived alleles at size-associated markers in 500 dogs
In order to determine the effective contributions of variants in or around IGF1, IGF1R, GHR, HMGA2, SMAD2, and STC2 on body size, the allele frequencies of tagging markers for each locus were determined from a large, physically diverse set of dogs representing 93 breeds.
We added previously described and highly associated markers at IGF1 (Sutter et al. 2007; Gray et al. 2010) and IGF1R (Hoopes et al. 2012) to the panel of size-associated markers identified by fine-mapping, for a total of seven markers (Table 1). Of note, a SNP (CFA15:41,221,438) and a SINE insertion (CFA15:41,220,980) in intron 2 of IGF1 were genotyped on DNA from 500 dogs and found to be in complete LD, which is consistent with previous reports (Sutter et al. 2007; Gray et al. 2010). Consequently, all future references to the IGF1 variant refer to the SNP, but the conclusions apply to the SINE element as well. The IGF1R SNP marker (CFA3:41,849,479) codes for a missense mutation, as we described previously (Hoopes et al. 2012).
We genotyped DNA from 500 dogs, representing 93 AKC-recognized breeds, at each of the seven markers (genotyping results are in Supplemental Table 6). Breeds span the entire range of canine weights. All dogs are unrelated at the grandparent level, and at least two males and two females were genotyped from each breed.
To determine the ancestral allele for each marker, we genotyped a set of wild canids, including 26 geographically diverse gray wolves, two red wolves, and two coyotes. The genotypes in the red wolves and coyotes were all homozygous, defining the ancestral alleles (Table 1; Supplemental Table 7). In gray wolves, the ancestral alleles greatly predominated (Supplemental Table 7).
The SBWs of dogs with different genotypes were compared (Fig. 2). To ensure that no single breed was overrepresented, we randomly selected only two males and two females from each breed for this analysis.
Figure 2.

Body size is tightly regulated in dogs homozygous for the derived alleles. (A) The standard breed weight (SBW) of each dog (y-axis) is plotted by genotype at each marker (x-axis). The SBWs of dogs homozygous for the derived allele (D/D) at the IGF1 marker are significantly smaller than dogs that are heterozygous (A/D) or homozygous for the ancestral allele (A/A), as determined by Kolmogorov-Smirnov and Mann-Whitney-Wilcoxon tests. (***) P < 0.001. The distribution of SBWs for a given genotype/marker combination is generally less for homozygous D/D dogs than for other genotypes (the median and first and third quartiles are indicated by the boxplots). Statistics for each genotype/marker combination are summarized in B. SBWs of genotype classes are reported as mean ± SD. Two females and two males were randomly selected from each breed for this analysis. The SBWs of all selected dogs are plotted in the leftmost column. Points were randomly scattered on the x-axis within each column to facilitate visualization.
Genotypes at each marker corresponded to differences in size. Reflecting the similarity of size between larger dogs and gray wolves, the ancestral alleles of each variant were always those more commonly found in larger dogs. For each variant, SBWs of dogs homozygous for the derived allele (D/D) were significantly less than SBWs of dogs homozygous for the ancestral allele (A/A). Moreover, SBWs of D/D dogs were also significantly less than the SBWs of heterozygotes (A/D) at four of seven markers (Fig. 2).
When comparing across loci, we observed similar trends. At all loci except IGF1, the mean SBW of the D/D dogs was 4–7 kg (8–15 lb). For most pairs of loci, the SBWs of dogs homozygous for the derived allele at one locus had a distribution similar to the SBWs of dogs homozygous for the derived allele at each of the other loci (boxplots) (Fig. 2A). However, dogs that were homozygous for the derived allele at IGF1 had a greater size range and a higher mean SBW (9.8 kg [21.6 lb]) than D/D dogs at any other locus (Fig. 2B).
The relationship of D/D and A/D dogs was more complicated at HMGA2, IGF1R, and GHR(2), in part because fewer heterozygotes were observed. HMGA2 was the most extreme, with only 16 A/D dogs and 87 D/D dogs (Fig. 2B). This ratio (16:87) was smaller than that observed at any other locus. By comparison, the small number of heterozygotes at IGF1R and GHR(2) was due in part to the low frequencies of the derived alleles (7.5% and 7.3%, respectively), which were found almost exclusively in the smallest breeds. Dogs with the D/D genotype at IGF1R or GHR(2) had a breed mean weight of 4–4.5 kg (9–10 lb), which was consistent with our previously reported findings (Hoopes et al. 2012). The frequency of genotypes did not differ between male and female dogs at any of the loci (no P-value <0.6).
Allelic trends among dogs of similar weights
A step-like pattern was apparent in the allele frequencies found in 5-lb bins (2.3 kg) (Fig. 3). Overall, as body size decreased the derived allele frequency increased, as did the number of markers with derived alleles. Considering each variant separately (Fig. 3, columns), in most cases allele frequencies changed gradually across body sizes, as represented by the gradient from yellow to red. By comparison, the incidence of the HMGA2 derived allele dropped abruptly in dogs with an SBW of 4.5–9.1 kg (10–20 lb).
Figure 3.

Derived allele frequencies increase at multiple loci as body weight decreases. The frequency of the derived allele in 5-lb weight classes is represented on a color scale. The smallest dogs (bottom row) are consistently red at all markers except IGF1R, while the largest dogs rarely carry a derived allele, as observed in weight classes of 90–95 (40.8–43.1 kg), 95–100 (43.1–45.4 kg), and above 105 (47.6 kg). The high frequency of the IGF1 derived allele in the 100–105 class represents the only breed we tested in the class, Rottweilers. Dogs with an SBW above 105 lb are collapsed in a single category due to the lack of genotype variation in the group at these markers. This analysis includes all 500 dogs genotyped.
While most derived alleles are observed in smaller breeds, the IGF1 derived allele is observed surprisingly frequently in several larger breeds. Notably, nine of the 10 Rottweilers (the only breed in the data set between 45.4 and 47.6 kg [100 and 105 lb]) were homozygous D/D at IGF1.
More typically, all dogs ≥40.8 kg (90 lb) were either homozygous for ancestral alleles at all markers or carried derived alleles at only one marker, usually IGF1. Among dogs <11.3 kg (25 lb), 90% carried derived alleles at three or more markers, and 98% carried the derived allele at IGF1.
Combinations of genotypes
Many allelic combinations were observed when all seven markers were considered. To define the combination present in a given dog, we recorded the markers at which the dog carried the derived allele (A/D or D/D). While 128 possible combinations exist, only 39 were observed in this data set (Fig. 4A). Thirteen combinations were common, as defined by their presence in 10 or more dogs. In the most frequent combination, both alleles at every marker were ancestral. This combination was observed most frequently in large breeds (Fig. 4B), but was also noted infrequently in breeds with an SBW as low as 15.9 kg (35 lb) (Supplemental Table 1). Combinations were generally not breed-specific. Of the 31 combinations that occur more than once, only two are limited to a single breed. Rare combinations of alleles were also identified. For instance, eight combinations were found in only one dog each, suggesting that other low-frequency combinations exist in the population at large.
Figure 4.
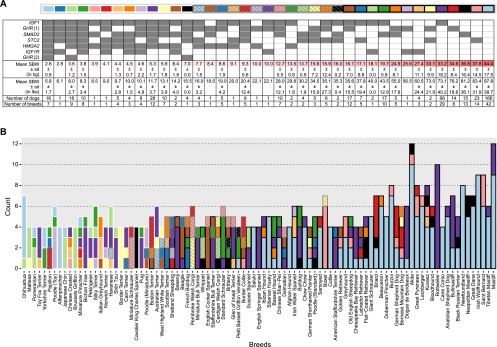
Multiple combinations of genotypes are observed in most breeds. We assessed combinations of genotypes in individual dogs (A). The presence of a derived allele (whether heterozygous or homozygous) is indicated by a filled square. The first column represents the combination with derived genotypes at each marker; the mean weight of dogs with this combination is less than the mean weight of any other combination. The percent standard deviations for a given combination are typically smaller than the percent standard deviations of dogs sharing only a genotype at a single marker (which are reported in Fig. 2). The combinations observed in each breed are uniquely identified by the pairing of fill and outline color in B. Breeds are sorted by SBW. This analysis includes all 500 dogs genotyped.
There is one set of combinations that is unlikely to exist in any dog. Of four possible haplotypes at the two nonsynonymous GHR markers (GC, GT, AC, and AT), only three were observed. The missing haplotype contains the allele associated with large dogs at GHR(1) and the allele associated with tiny dogs at a marker 41 bases away, GHR(2). In essence, we found haplotypes corresponding to “large + not tiny” (GC), “small + not tiny” (AC), and “small + tiny” (AT), but never “large + tiny” (GT). Since the GHR(2) marker T allele occurs at very low frequency and the two markers are in close proximity, we believe the GT haplotype is unlikely to exist in the general population. This suggests that selection of the GHR(2) derived variant occurred among dogs that were already carriers of the GHR(1) derived allele.
In one of the few widely observed combinations, a derived allele was present only at the IGF1 locus. Dogs presenting this combination belonged to two broad categories of breeds. The first group contained breeds with SBWs <31.8 kg (70 lb) and included Basenjis, English Springer Spaniels, and American Staffordshire Terriers (with SBWs of 10.2, 20.4, and 29.0 kg, respectively [22.5, 45, and 64 lb]). The second were breeds with SBWs ≥40.8 kg (90 lb) and included Mastiffs and related breeds such as Tibetan Mastiffs, Bullmastiffs, Dogues de Bordeaux, Rottweilers, and Black Russian Terriers (Fig. 4).
The mean SBW of dogs with a given combination was calculated (Fig. 4). As expected, the combination with the lowest mean SBW had derived alleles at all markers, and the heaviest combination had no derived alleles. In some cases, breeds that vary substantially in size shared the same combination, such as Papillons, Boston Terriers, and Border Collies (2.8, 7.9, and 17.9 kg, respectively [6.1, 17.5, and 39.5 lb]). Nevertheless, the standard deviation of SBWs for dogs sharing a combination was generally lower than that observed for weight groups defined by genotypes at a single marker (Fig. 2), indicating that combinations of genotypes explain body size differences better than any single genotype.
Unifying model
Since derived allele frequencies among the seven profiled markers clearly corresponded to progressive diminution, we sought to quantify how well alleles at these markers accounted for differences in body size. We used breed-averaged allele frequencies to calculate the proportion of phenotypic variance that these seven markers explained in a linear model. In order to determine which components should be present in the model, we first tested the mode of inheritance for each marker. We found that both HMGA2 (P = 0.0094) and GHR(2) (P = 0.0366) have a significant dominance component, consistent with the log of the mean SBW of heterozygotes deviating from the mean of the homozygotes (Supplemental Fig. S4).
The resulting model related the log-transformed SBW to the allele frequencies at each of the seven size-associated markers and the breed-average of the dominance component for HMGA2 and GHR(2). Derived allele frequencies at each marker accounted for 86.0% of SBW variance for the 93 breeds, as measured by the adjusted R-squared (Fig. 5A). The terms corresponding to all markers except GHR(2) were significant by ANOVA (P < 0.05).
Figure 5.

Allele frequencies at size markers explain 86% of size variation before correction for population structure (A) and 52.5% after (B). (A) A linear model was generated to assess the power of breed-averaged allele frequencies to explain variance in standard breed weights (SBWs). SBWs in lb (in parentheses) were transformed by natural log to approximate a normal distribution as was done in previous studies (Boyko et al. 2010). The black line indicates perfect equality of the fitted values with the SBWs. The cluster of breeds with a fitted weight of 90 lb (40.8 kg) reflects the lack of informativeness of these loci for large breeds. Small amounts of scatter (≤0.05) were added to plotted values to reduce overplotting (n = 93). (B) A correction for population structure was performed by regressing the SBW on breed-averaged, genome-wide principal components (PCs). More than half (52.5%) of the variance in the residuals of this regression, the corrected SBWs (cSBWs), was explained by allele frequencies at the seven size markers. Since PCs were calculated from the CanMap data set, cSBWs could only be calculated for the 65 breeds that were present in both our data set and the CanMap data set.
Because dog breeds do not represent a randomly mating population, we investigated the role of population structure in the explanatory power of the allele frequencies. In order to use them to correct for population structure, we calculated breed-averaged principal components (PCs) from genome-wide SNP profiles for each of the 65 breeds that were present in both our data set and the CanMap data set (Boyko et al. 2010). We then compared the SBW variance explained with and without terms representing PCs, using only PCs that are significantly predictive of SBW and the 65 breeds that have PCs. Genotypes alone account for 85.8% of variance; PCs alone, 44.2%; and PCs and genotypes together, 90.0%. These variances are not additive, but rather they indicate the upper limit that each could contribute in our model. Taken together, our uncorrected and population-corrected models show that genotypes at these seven loci account for between 45.8% and 85.8% of SBW variance.
As an alternate approach to accounting for population structure, corrected SBW (cSBW) values were determined by taking the residuals of a linear regression of SBW on PCs. Allele frequencies would then explain 52.5% of the variance in the resulting cSBWs (Fig. 5B). The two numbers, 46% and 52.5%, bracket a conservative estimate of the variance of SBW explained by genotypes at these markers.
The seven markers are less informative in large and giant dog breeds. Allele frequencies accounted for 64.3% of cSBW variance in dogs with SBWs <40.8 kg (90 lb), but only 8.4% of cSBW among dogs with SBW ≥40.8 kg. This is reflected in the cluster of fitted values around 90 lb in Figure 5, A and B. These points represent breeds that were homozygous for the ancestral allele at all markers and reflect the lack of relevance of these markers to differences in size among large and giant breeds.
The genotype–phenotype relationships were subjected to further analysis. We found no significant interactions between markers (Supplemental Results). We also found that the model is unlikely to be overfitting the data, since 42.1% of cSBW variance in a test set could be accounted for using coefficients calculated by a training set. The comparable number in the full 65-breed cSBW data set is 52.5%. The applicability of our findings to individuals was also assessed. Tests with 124 individually weighed dogs showed that 74.4% of the variance of their uncorrected weight could be accounted for using individual allele frequencies with coefficients derived from calculations based on uncorrected SBW, in which 86% of SBW variance was explained with allele frequencies averaged by breeds. The cross-validation and the model's ability to explain size variance in individuals underscore the substantial nature of the effects we describe.
Discussion
We have identified the source of approximately half of size variation in domestic dog breeds by genotyping DNA from 500 dogs at seven markers, five of which we identified by fine-mapping, and two of which we identified previously (Sutter et al. 2007; Hoopes et al. 2012). The dog breeds we analyzed were selected to represent the full range of canine body size, and by analyzing the relationship of the standard breed weight with genotype, the underlying pattern was revealed: For each variant, the derived allele corresponds to reduced body size relative to the ancestral gray wolf, and the presence of derived alleles at multiple variants further reduces body size. In a linear model, allele frequencies account for ∼86% of variance in SBW without correction and, conservatively, 46%–52.5% after correcting for population structure, a degree of explanatory power rarely seen in genetic studies. This strong statistical relationship of genotypes with phenotype is compelling evidence of functional effects by variants in LD with these markers, if not the markers themselves.
Modern dog breeds are defined by rigorous standards, which describe the ideal representatives of the breed. For genetic studies of these strongly selected traits, the fixed phenotype can be used as a proxy for the individual's genetically determined phenotype, as we (Sutter et al. 2007, 2008; Jones et al. 2008; Boyko et al. 2010) and others (Vaysse et al. 2011) have done previously, and as we have done here. By leveraging AKC breed standards and averaged measurements from registered dogs, we can reduce the effect of environment, thus targeting genes underlying strongly selected traits, which often reflect the defining features of a breed. In this study, the approach resulted in the identification of variants under strong selection by breeders that correspond to major differences in overall breed body size. By virtue of our study design, intrabreed body size variation is discounted and genes that contribute exclusively to it will not be identified. Indeed, given that the participants in our study are mostly show animals that compete for breed standard conformation titles, we expect the genetic contribution of intrabreed size variants to be minor compared with those that are the major contributors to interbreed size differences.
The starting point of this study was our earlier multibreed GWAS, which used breed standard weight to identify QTLs associated with dog body size variation (Boyko et al. 2010), a study which found that six size-associated SNP chip markers explained 72% of variance of SBWs without correction for population structure. Our fine-mapping experiments were designed to identify the most highly associated and diagnostic variants. Each variant is potentially causal, with compelling cases for three of the variants: the two protein-altering SNPs in GHR and the SNP in the 5′ UTR of HMGA2. The SMAD2 variant is a large deletion (9.9 kb) that appears to be in complete LD with a neighboring 5.7-kb deletion. Although the deletions are more than 15 kb from the gene, they could potentially affect transcription efficiency, as predicted by the loss of a transcription factor binding site cluster (Supplemental Fig. 3). The STC2 SNP is the least likely to be the causal variant, as it only affects a single base and is 20 kb from the gene. However, it is highly associated and therefore remains an excellent marker. There are no better-associated markers in the exons of the studied genes, yet the possibility remains that there are better-associated markers within the extensive range of regulatory effect. As high-throughput sequencing is applied to more dog genomes, further information about the potentially functional role of regions far from genes will be available.
Several of the genes reported in this study are known to be involved in size regulation in other organisms. IGF1, IGF1R, and GHR participate in the GH/IGF1 pathway, which is required for normal stature in humans. Mutations in the GH/IGF1 pathway genes have been associated with human growth disorders (Walenkamp and Wit 2006; Rosenfeld et al. 2007; David et al. 2011). The interdependence of these three proteins—GHR, IGF1, and IGF1R—is well documented (David et al. 2011), but we see no strong evidence for statistical interactions in the effects of the variants studied here.
GHR is an attractive candidate for canine size regulation because it is implicated in human body size (Amselem et al. 1989; Ayling et al. 1997) and affects IGF1 signal transduction (David et al. 2011). Human studies suggest a mechanism by which the GHR variants identified here could cause reduced body size. The GHR SNPs selected in this study are located in the extracellular domain of the canine GH receptor. In the syntenic human exon, three disease-associated SNPs have been reported <25 amino acids away. These SNPs affect growth hormone binding and are believed to cause a human growth hormone insensitivity disorder termed Laron Syndrome (Wojcik et al. 1998).
HMGA2 has been associated with height determination in multiple human GWAS (Weedon et al. 2007, 2008; Gudbjartsson et al. 2008; Lettre et al. 2008; Sanna et al. 2008; Soranzo et al. 2009; N'Diaye et al. 2011; Carty et al. 2012). HMGA2 is a transcription factor expressed during embryonic and fetal development (Rogalla et al. 1996; Gattas et al. 1999). Hmga2 knockout mice have a pygmy phenotype, characterized by reduced birth weight and growth retardation (Benson and Chada 1994; Zhou et al. 1995).
Neither STC2 nor SMAD2 have been implicated in size determination in humans. However, STC2, a secreted glycoprotein hormone inhibits growth in mice independently of the GH/IGF1 pathway (Gagliardi et al. 2005; Chang et al. 2008). Although no SMAD2-mediated size phenotype has been reported, it is a transcription factor known to transduce signals from members of the transforming growth factor beta (TGF-beta) superfamily (Moustakas and Heldin 2009; Wu and Hill 2009). An appealing possibility is that the deletion identified proximal to SMAD2 is acting in cis to alter this gene's expression in developmental processes such as myogenesis, chondrogenesis, or osteogenesis (Sartori et al. 2009; Song et al. 2009; Chen et al. 2012).
While it is not surprising that genes with a conserved role in mammalian size determination might have variants in both humans and dogs, both the population structure and the study methods complicate comparisons. GWAS in humans have identified 180 loci significantly associated with height (Gudbjartsson et al. 2008; Lettre et al. 2008; Sanna et al. 2008; Weedon et al. 2008; Soranzo et al. 2009; Kim et al. 2010; Lango Allen et al. 2010; N'Diaye et al. 2011; Carty et al. 2012). However, even together, these loci account for only ∼10% of the adult human height variation (Lango Allen et al. 2010), although the heritability of height is ∼80% (Silventoinen 2003; Visscher et al. 2006; Perola et al. 2007). Unlike our approach, which uses SBWs, human studies have focused on individual measurements of subpopulations, enabling partitioning of variance attributable to environment and capturing intra-group variation. However, methodological approaches are unlikely to explain the entire difference in study results (six genes explaining ∼50% of SBW in dogs vs. 180 loci explaining ∼10% of individual height in humans). Dogs are under intense artificial selection and have a much greater range of sizes than humans. The relative subtlety of height regulation in humans may be more typical of species subjected to many thousands of generations of natural selection, such as wolves. We hypothesize that the variants of large effect in dogs that we have found are superimposed on a subtler size-regulation system inherited from wolves.
In addition to explaining ∼65% of variance in dogs <40.8 kg (90 lb), this study defines two substantial types of body size variation that remain to be explained: 35% of body size variation in dogs <40.8 kg, and ∼90% of the body size variation among dogs weighing ≥40.8 kg. Some of the unexplained variation in dogs <40.8 kg is evident in breeds like Shih Tzu and Pugs. Shih Tzu weigh 20% less than Pugs, but most individuals belonging to either breed have identical genotypes at the seven size variants studied here. To investigate size determination on a finer scale, individual dog weights and perhaps measurements will be necessary. Individual weights and measurements may also permit the elucidation of epistatic relationships, which have been observed in other domesticated species (Carlborg et al. 2006).
Although typically found in small dogs, we found the IGF1 derived allele in Rottweilers, consistent with previous reports (Sutter et al. 2007), and in other large mastiff-related breeds (Fig. 4). We offer two explanatory hypotheses. First, it is possible that neither of the two IGF1 variants genotyped in this study (a SNP and a SINE insertion) are causal; rather they tag the ancestral haplotype on which the causal variant first emerged. Thus, some very large breeds could carry the tagging variants and yet lack the causal variant. Alternatively, epistasis of a yet-unidentified locus may reduce the effects of the IGF1 small allele in some large dog breeds.
The genotypes of dogs from breeds with an SBW ≥40.8 kg (90 lb), represented by 18 breeds in our study, allow us to distinguish them from small and medium dogs, but not from each other (Fig. 5A). The genotypes at the seven size markers account for <9% of differences among dogs over 40.8 kg. Clearly, other loci that contribute to large body size in dogs remain to be found, and further analysis of these giant breeds is warranted.
Size determination in large and giant dogs probably shares features of size determination observed in small and medium-sized dogs. Several size-associated intervals on the X chromosome have been identified, but not studied further (Boyko et al. 2010; Vaysse et al. 2011). In a predictive model that considered breeds of all sizes, adding the locus at 104 Mb on the X chromosome to a model with only the IGF1 locus increased the amount of variance explained from 47.6% to 57.8%, without correction for population structure (Boyko et al. 2010). However, our ongoing efforts indicate that fine-mapping the chromosome X loci is extremely challenging, as LD on this chromosome extends over megabases (M Rimbault, unpubl.) and includes dozens of genes.
Size determination could also be more wolf-like in large dogs than in small dogs. Compared with small dogs, the sizes of large dogs overlap more with the sizes of wolves. Wolves vary substantially in size, with the weights of adult male wolves in Yellowstone National Park alone ranging from 38 to 66 kg (85 to 145 lb) (MacNulty et al. 2009). Size determination in wolves may be more similar to height determination in humans than to an artificially selected group like domestic dogs and result from the collective effects of many variants of small effect (Lango Allen et al. 2010).
In this study, we identified markers at loci that define the major size ranges in domestic dogs and show how combinations of alleles produce the extensive range of dog sizes present in modern breeds. It remains to be seen how size is regulated on a finer scale, within breeds, between sexes, and among giant dogs. Some of these studies will require individual measurements and perhaps larger numbers to compensate for noise due to environmental effects. It will also be valuable to extend our existing findings by identifying the functional consequences of size-determining variants. It is our hope that these studies can shed light on growth-related health issues in dogs and humans.
Methods
All coordinates refer to the CanFam3.1 dog genome assembly (Sept. 2011). Unless otherwise noted, analysis was performed using the software program R (R Development Core Team 2012), and figures were generated with R base graphics and the plotting package ggplot2 (Wickham 2009). The genes and identification numbers in Figure 1 are: SEPP1 (NM_001115118), GHR (NM_001003123), NKX2-5 (NM_001010959), STC2 (ENSCAFG00000031727), SMAD2 (ENSCAFG00000017567), MSRB3 (ENSCAFG00000029740), and HMGA2 (BLAT results of KC529658).
Sample collection and DNA extraction
Blood samples were collected from dogs belonging to AKC-registered breeds at AKC-sanctioned dog shows, specialty events, breed clubs, and veterinary clinics. Samples were collected as whole blood into ACD or EDTA anticoagulant tubes after obtaining written consent from dog owners. Genomic DNA was isolated from whole blood using a standard proteinase-K/phenol:chloroform extraction protocol (Maniatis et al. 1982). All procedures were reviewed and approved by the NHGRI Animal Care and Use Committee at the National Institutes of Health.
Phenotype assignment
Standard breed weights were obtained from several publications. If the AKC specified a weight for a breed, it was used (American Kennel Club 1998). If separate values were listed for males and females, those values were averaged. When the AKC did not specify a weight or if only an upper or lower limit was specified, we used data from The Encyclopedia of the Dog (Fogle 1995). If no weight was specified, we utilized data from Atlas of Dog Breeds of the World (Wilcox and Walkowicz 1995). We also considered weights recorded in our NHGRI database of individual dogs. These owner-reported weights were collected at AKC-sanctioned dog shows, breed specialty events, and breed club meetings. If there were more than six adult dog weights listed for a breed in our database, we removed the maximum and minimum weights listed and compared the mean of the remaining weights with the published breed standard weight. If the weights differed by >20%, we used the mean breed weight from our database. A list of the breeds and of the standard breed weight used in this study can be found in Supplemental Table 1. Because the phenotype of interest is size, we treated the three varieties of poodles as separate breeds.
Genotyping of the highly associated markers
The highly size-associated markers (Table 1) were genotyped on an additional set of samples consisting of 500 dogs, termed the validation set, from 93 AKC-recognized breeds representing the full range of canine body size (Supplemental Table 6). Dogs were unrelated to one another at the grandparent level. Forty-one percent of the 500 dogs had also been included in the CanMap data set (Boyko et al. 2010). The validation set was not fully independent from this study's discovery set either: 13 dogs, six small and seven large, were used for both marker discovery and validation experiments. Wild canids, including 26 geographically diverse gray wolves from North America, Europe, and Asia (10 females and 16 males), two coyotes (one female and one male), and two red wolves (two males) were also genotyped (Supplemental Table 7).
Three hundred and eighty-four dogs were genotyped at the following markers using a GoldenGate genotyping assay (Illumina): IGF1, IGF1R, GHR(1), and STC2. GoldenGate genotypes at one position, GHR(1), were all validated by Sanger sequencing with 100% concordance. The remaining dogs and variants were genotyped by PCR and Sanger sequencing (see Supplemental Methods for reaction conditions). The SINE insertion in intron two of IGF1 was genotyped by PCR amplification, and PCR products were analyzed after migration on 1% agarose gels to determine the presence or absence of the insertion. To genotype the 9.9-kb deletion downstream from the SMAD2 gene on CFA7, PCR products from two different primer pairs were analyzed on 1% agarose gels to determine the absence or the presence of the deletion. A list of the primers and PCR conditions are given in Supplemental Table 8.
Model
In all models, we used the natural log of weight in lb to approximate a normal distribution, as was done previously (Boyko et al. 2010). Twenty principal components were calculated on the CanMap data set using SmartPCA from the Eigensoft package (Patterson et al. 2006; Price et al. 2006). We used a pruned data set that excludes individuals with >10% missing genotype data, SNPs in high LD as defined by pairwise genotypic r2 > 0.8 within sliding windows of 50 SNPs, and SNPs that were within 2 Mb of the most strongly size-associated markers at each of the six loci. Outliers of more than six ∑ were excluded, as were breeds with fewer than four dogs remaining after individual outliers were excluded. The breed average of each PC was calculated. Ninety-three breeds were represented in the pool of dogs genotyped at all seven markers; PC values were available for 65 of those breeds (Supplemental Table 9). The PCs predictive of SBW (PC2, PC4, PC6, PC10, PC11, and PC18 at P < 0.05) were used for subsequent corrections for population structure.
Significant dominance components were identified by applying a nested ANOVA to each marker with and without a partial dominance term. PCs that were significant for weight were included in both equations.
For the purposes of testing our model on individual dogs, we used individual measurements from dogs in our own database. For each dog, we calculated the mean and standard deviation of the other dogs in the breed. If a dog's Z-score relative to those numbers exceeded 1.5, the dog was excluded.
Data access
The sequence containing the first exon of canine HMGA2 has been submitted to the National Center for Biotechnology Information (NCBI) GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession number KC529659. The mRNA sequence of HMGA2 has been submitted to NCBI GenBank under accession number KC529658.
Acknowledgments
We thank the American Kennel Club–Canine Health Foundation, the Intramural Program of the National Human Genome Research Institute of the National Institutes of Health (E.A.O., M.R., H.C.B., and J.J.S.), and Cornell University (N.B.S. and J.J.A.) for supporting this work. J.J.S. was funded by an NIGMS PRAT postdoctoral fellowship. Sabbatical support for B.C.H. was provided by a grant from the Research Council of Colgate University. R.K.W. is supported by grants NSF-DEB 1021397 and 0733033, and N.B.S. by NIH grant 5R21HG006051-02. We thank Drs. John Novembre, Heidi Parker, and Jonine Figueroa for their helpful insights and feedback. We thank the NIH Intramural Sequencing Center staff for valuable technical and computational assistance. We thank Dr. Shelley Hoogstraten-Miller and Irene Ginty for assistance in blood draws. Finally, we are grateful to the many dog owners and breeders who generously provided DNA samples for this study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.157339.113.
Freely available online through the Genome Research Open Access option.
References
- Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, Nicholas TJ, Neff MW 2010. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci 107: 1160–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Kennel Club. 1998. The complete dog book. Howell Book House, New York. [Google Scholar]
- Amselem S, Duquesnoy P, Attree O, Novelli G, Bousnina S, Postel-Vinay MC, Goossens M 1989. Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med 321: 989–995 [DOI] [PubMed] [Google Scholar]
- Ayling RM, Ross R, Towner P, Laue Von S, Finidori J, Moutoussamy S, Buchanan CR, Clayton PE, Norman MR 1997. A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nat Genet 16: 13–14 [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A 1993. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 75: 73–82 [PubMed] [Google Scholar]
- Benson KF, Chada K 1994. Mini-mouse: Phenotypic characterization of a transgenic insertional mutant allelic to pygmy. Genet Res 64: 27–33 [DOI] [PubMed] [Google Scholar]
- Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, Zhao K, Brisbin A, Parker HG, vonHoldt BM, et al. 2010. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol 8: e1000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlborg Ö, Jacobsson L, Åhgren P, Siegel P, Andersson L 2006. Epistasis and the release of genetic variation during long-term selection. Nat Genet 38: 418–420 [DOI] [PubMed] [Google Scholar]
- Carty CL, Johnson NA, Hutter CM, Reiner AP, Peters U, Tang H, Kooperberg C 2012. Genome-wide association study of body height in African Americans: The Women's Health Initiative SNP Health Association Resource (SHARe). Hum Mol Genet 21: 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AC-M, Hook J, Lemckert FA, McDonald MM, Nguyen M-AT, Hardeman EC, Little DG, Gunning PW, Reddel RR 2008. The murine stanniocalcin 2 gene is a negative regulator of postnatal growth. Endocrinology 149: 2403–2410 [DOI] [PubMed] [Google Scholar]
- Chase K, Carrier DR, Adler FR, Jarvik T, Ostrander EA, Lorentzen TD, Lark KG 2002. Genetic basis for systems of skeletal quantitative traits: Principal component analysis of the canid skeleton. Proc Natl Acad Sci 99: 9930–9935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase K, Carrier DR, Adler FR, Ostrander EA, Lark KG 2005. Interaction between the X chromosome and an autosome regulates size sexual dimorphism in Portuguese Water Dogs. Genome Res 15: 1820–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Deng C, Li Y-P 2012. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8: 272–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David A, Hwa V, Metherell LA, Netchine I, Camacho-Hübner C, Clark AJL, Rosenfeld RG, Savage MO 2011. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev 32: 472–497 [DOI] [PubMed] [Google Scholar]
- Fogle B. 1995. The encyclopedia of the dog. DK Publishing, Inc., New York. [Google Scholar]
- Gagliardi AD, Kuo EYW, Raulic S, Wagner GF, Dimattia GE 2005. Human stanniocalcin-2 exhibits potent growth-suppressive properties in transgenic mice independently of growth hormone and IGFs. Am J Physiol Endocrinol Metab 288: E92–E105 [DOI] [PubMed] [Google Scholar]
- Gattas GJ, Quade BJ, Nowak RA, Morton CC 1999. HMGIC expression in human adult and fetal tissues and in uterine leiomyomata. Genes Chromosomes Cancer 25: 316–322 [PubMed] [Google Scholar]
- Germonpré M, Sablin MV, Stevens RE, Hedges REM, Hofreiter M, Stiller M, Després VR 2009. Fossil dogs and wolves from Palaeolithic sites in Belgium, the Ukraine and Russia: Osteometry, ancient DNA and stable isotopes. J Archaeol Sci 36: 473–490 [Google Scholar]
- Gray MM, Sutter NB, Ostrander EA, Wayne RK 2010. The IGF1 small dog haplotype is derived from Middle Eastern grey wolves. BMC Biol 8: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Walters GB, Thorleifsson G, Stefansson H, Halldorsson BV, Zusmanovich P, Sulem P, Thorlacius S, Gylfason A, Steinberg S, et al. 2008. Many sequence variants affecting diversity of adult human height. Nat Genet 40: 609–615 [DOI] [PubMed] [Google Scholar]
- Hoopes BC, Rimbault M, Liebers D, Ostrander EA, Sutter NB 2012. The insulin-like growth factor 1 receptor (IGF1R) contributes to reduced size in dogs. Mamm Genome 23: 780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Chase K, Martin A, Davern P, Ostrander EA, Lark KG 2008. Single-nucleotide-polymorphism-based association mapping of dog stereotypes. Genetics 179: 1033–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-J, Lee H-I, Park T, Kim K, Lee J-E, Cho NH, Shin C, Cho YS, Lee J-Y, Han B-G, et al. 2010. Identification of 15 loci influencing height in a Korean population. J Hum Genet 55: 27–31 [DOI] [PubMed] [Google Scholar]
- Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, et al. 2010. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467: 832–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G, Jackson AU, Gieger C, Schumacher FR, Berndt SI, Sanna S, Eyheramendy S, Voight BF, Butler JL, Guiducci C, et al. 2008. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat Genet 40: 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438: 803–819 [DOI] [PubMed] [Google Scholar]
- MacNulty DR, Smith DW, Mech LD, Eberly LE 2009. Body size and predatory performance in wolves: Is bigger better? J Anim Ecol 78: 532–539 [DOI] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J 1982. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- Moustakas A, Heldin C-H 2009. The regulation of TGFβ signal transduction. Development 136: 3699–3714 [DOI] [PubMed] [Google Scholar]
- N'Diaye A, Chen GK, Palmer CD, Ge B, Tayo B, Mathias RA, Ding J, Nalls MA, Adeyemo A, Adoue V, et al. 2011. Identification, replication, and fine-mapping of loci associated with adult height in individuals of African ancestry. PLoS Genet 7: e1002298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovodov ND, Crockford SJ, Kuzmin YV, Higham TFG, Hodgins GWL, van der Plicht J 2011. A 33,000-year-old incipient dog from the Altai Mountains of Siberia: Evidence of the earliest domestication disrupted by the last glacial maximum. PLoS ONE 6: e22821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J-F, Kluetsch C, Zou X-J, Zhang A-B, Luo L-Y, Angleby H, Ardalan A, Ekström C, Sköllermo A, Lundeberg J, et al. 2009. mtDNA data indicate a single origin for dogs south of Yangtze River, less than 16,300 years ago, from numerous wolves. Mol Biol Evol 26: 2849–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker HG, Kim LV, Sutter NB, Carlson S, Lorentzen TD, Malek TB, Johnson GS, DeFrance HB, Ostrander EA, Kruglyak L 2004. Genetic structure of the purebred domestic dog. Science 304: 1160–1164 [DOI] [PubMed] [Google Scholar]
- Parker HG, Kukekova AV, Akey DT, Goldstein O, Kirkness EF, Baysac KC, Mosher DS, Aguirre GD, Acland GM, Ostrander EA 2007. Breed relationships facilitate fine-mapping studies: A 7.8-kb deletion cosegregates with Collie eye anomaly across multiple dog breeds. Genome Res 17: 1562–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D 2006. Population structure and eigenanalysis. PLoS Genet 2: e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perola M, Sammalisto S, Hiekkalinna T, Martin NG, Visscher PM, Montgomery GW, Benyamin B, Harris JR, Boomsma D, Willemsen G, et al. 2007. Combined genome scans for body stature in 6,602 European twins: Evidence for common Caucasian loci. PLoS Genet 3: e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909 [DOI] [PubMed] [Google Scholar]
- R Development Core Team. 2012. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Rogalla P, Drechsler K, Frey G, Hennig Y, Helmke B, Bonk U, Bullerdiek J 1996. HMGI-C expression patterns in human tissues. Implications for the genesis of frequent mesenchymal tumors. Am J Pathol 149: 775–779 [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld RG, Belgorosky A, Camacho-Hubner C, Savage MO, Wit JM, Hwa V 2007. Defects in growth hormone receptor signaling. Trends Endocrinol Metab 18: 134–141 [DOI] [PubMed] [Google Scholar]
- Sablin MV, Khlopachev GA 2002. The earliest ice age dogs: Evidence from Eliseevichi 1. Curr Anthropol 43: 795–799 [Google Scholar]
- Sanna S, Jackson AU, Nagaraja R, Willer CJ, Chen W-M, Bonnycastle LL, Shen H, Timpson N, Lettre G, Usala G, et al. 2008. Common variants in the GDF5-UQCC region are associated with variation in human height. Nat Genet 40: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M 2009. Smad2 and 3 transcription factors control muscle mass in adulthood. AJP: Cell Physiology 296: C1248–C1257 [DOI] [PubMed] [Google Scholar]
- Savolainen P, Zhang Y-P, Luo J, Lundeberg J, Leitner T 2002. Genetic evidence for an East Asian origin of domestic dogs. Science 298: 1610–1613 [DOI] [PubMed] [Google Scholar]
- Shearin AL, Ostrander EA 2010. Canine morphology: Hunting for genes and tracking mutations. PLoS Biol 8: e1000310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silventoinen K 2003. Determinants of variation in adult body height. J Biosoc Sci 35: 263–285 [DOI] [PubMed] [Google Scholar]
- Song B, Estrada KD, Lyons KM 2009. Smad signaling in skeletal development and regeneration. Cytokine Growth Factor Rev 20: 379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soranzo N, Rivadeneira F, Chinappen-Horsley U, Malkina I, Richards JB, Hammond N, Stolk L, Nica A, Inouye M, Hofman A, et al. 2009. Meta-analysis of genome-wide scans for human adult stature identifies novel Loci and associations with measures of skeletal frame size. PLoS Genet 5: e1000445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter NB, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones PG, et al. 2007. A single IGF1 allele is a major determinant of small size in dogs. Science 316: 112–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter NB, Mosher DS, Gray MM, Ostrander EA 2008. Morphometrics within dog breeds are highly reproducible and dispute Rensch's rule. Mamm Genome 19: 713–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaysse A, Ratnakumar A, Derrien T, Axelsson E, Rosengren Pielberg G, Sigurdsson S, Fall T, Seppälä EH, Hansen MST, Lawley CT, et al. 2011. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet 7: e1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilà C, Savolainen P, Maldonado JE, Amorim IR, Rice JE, Honeycutt RL, Crandall KA, Lundeberg J, Wayne RK 1997. Multiple and ancient origins of the domestic dog. Science 276: 1687–1689 [DOI] [PubMed] [Google Scholar]
- Visscher PM, Medland SE, Ferreira MAR, Morley KI, Zhu G, Cornes BK, Montgomery GW, Martin NG 2006. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2: e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vonHoldt BM, Pollinger JP, Lohmueller KE, Han E, Parker HG, Quignon P, Degenhardt JD, Boyko AR, Earl DA, Auton A, et al. 2010. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464: 898–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walenkamp MJE, Wit JM 2006. Genetic disorders in the growth hormone—insulin-like growth factor-I axis. Horm Res 66: 221–230 [DOI] [PubMed] [Google Scholar]
- Wayne RK 1993. Molecular evolution of the dog family. Trends Genet 9: 218–224 [DOI] [PubMed] [Google Scholar]
- Weedon MN, Lettre G, Freathy RM, Lindgren CM, Voight BF, Perry JRB, Elliott KS, Hackett R, Guiducci C, Shields B, et al. 2007. A common variant of HMGA2 is associated with adult and childhood height in the general population. Nat Genet 39: 1245–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon MN, Lango H, Lindgren CM, Wallace C, Evans DM, Mangino M, Freathy RM, Perry JRB, Stevens S, Hall AS, et al. 2008. Genome-wide association analysis identifies 20 loci that influence adult height. Nat Genet 40: 575–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2009. ggplot2: Elegant graphics for data analysis (use R!). Springer, New York. [Google Scholar]
- Wilcox B, Walkowicz C 1995. Atlas of dog breeds of the world. T.F.H. Publications, Neptune City, NJ [Google Scholar]
- Wojcik J, Berg MA, Esposito N, Geffner ME, Sakati N, Reiter EO, Dower S, Francke U, Postel-Vinay MC, Finidori J 1998. Four contiguous amino acid substitutions, identified in patients with Laron syndrome, differently affect the binding affinity and intracellular trafficking of the growth hormone receptor. J Clin Endocrinol Metab 83: 4481–4489 [DOI] [PubMed] [Google Scholar]
- Woods KA, Camacho-Hübner C, Savage MO, Clark AJ 1996. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med 335: 1363–1367 [DOI] [PubMed] [Google Scholar]
- Wu MY, Hill CS 2009. Tgf-β superfamily signaling in embryonic development and homeostasis. Dev Cell 16: 329–343 [DOI] [PubMed] [Google Scholar]
- Zhou X, Benson KF, Ashar HR, Chada K 1995. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C. Nature 376: 771–774 [DOI] [PubMed] [Google Scholar]