

Abstract
Genome-wide association studies (GWAS) are useful for nominating candidate genes, but typically are unable to establish disease causality or differentiate between the effects of variants in linkage disequilibrium (LD). Additionally, some GWAS loci might contain multiple causative variants or genes that contribute to the overall disease susceptibility at a single locus. However, the majority of current GWAS lack the statistical power to test whether multiple causative genes underlie the same locus, prompting us to adopt an alternative approach to testing multiple GWAS genes empirically. We used gene targeting in a disease-susceptible rat model of genetic hypertension to test all six genes at the Agtrap-Plod1 locus (Agtrap, Mthfr, Clcn6, Nppa, Nppb, and Plod1) for blood pressure (BP) and renal phenotypes. This revealed that the majority of genes at this locus (five out of six) can impact hypertension by modifying BP and renal phenotypes. Mutations of Nppa, Plod1, and Mthfr increased disease susceptibility, whereas Agtrap and Clcn6 mutations decreased hypertension risk. Reanalysis of the human AGTRAP-PLOD1 locus also implied that disease-associated haplotype blocks with polygenic effects were not only possible, but rather were highly plausible. Combined, these data demonstrate for the first time that multiple modifiers of hypertension can cosegregate at a single GWAS locus.
Genome-wide association studies (GWAS) are able to nominate loci for complex diseases, but are largely unable to identify the causative variant(s) for multiple reasons (Zuk et al. 2012; Chatterjee et al. 2013). First, GWAS loci typically contain several candidate genes and are unable to distinguish between the effects of SNPs in linkage disequilibrium (LD). Second, the possibility exists that multiple causative SNPs influence the overall phenotypic variance at a single GWAS locus (Yang et al. 2012); however, most GWAS lack the statistical power to test this possibility due to sample size (Zuk et al. 2012). For example, Zuk et al. (2012) estimated that association of a single SNP at a relatively strong GWAS locus requiring 4900 test subjects (50% power and genome-wide significance level of α = 5 × 10−8) would require ∼450,000 individuals to achieve the same power when analyzing two SNPs simultaneously. Other estimates suggest that the full predictive power of many GWAS might not be met until sample sizes increase to >106 individuals (Chatterjee et al. 2013). Of the handful of loci that do show multi-SNP effects (Galarneau et al. 2010; Lango Allen et al. 2010; Ripke et al. 2011; Sanna et al. 2011; Sklar et al. 2011; Yang et al. 2012), all are of readily quantifiable traits with high heritability and low experimental variability (e.g., height), whereas none have been identified in more complex disease phenotypes (e.g., hypertension and renal disease). This raises the question whether genetic interactions within GWAS loci could be far more pervasive, but are simply missed by underpowered GWAS or lost in the noise of more complex disease phenotypes.
Here, we aimed to push past the limitations of current GWAS to identify causative gene(s) and test whether multiple genes within a GWAS locus contribute to the overall disease variance. We phenotyped rats with mutations in the Agtrap-Plod1 locus, which has been associated with blood pressure (BP) or renal disease in 11 human studies (Supplemental Table 1; Kato et al. 2000; Jiang et al. 2004; Zhang et al. 2005; Levy et al. 2009; Newton-Cheh et al. 2009a,b; Chen et al. 2010; Tomaszewski et al. 2010; Johnson et al. 2011; Liu et al. 2011; Fung et al. 2012), yet the genetic mechanism(s) underlying this locus were completely unknown. We used a recent technological advancement—zinc-finger nuclease (ZFN) mutagenesis (Geurts et al. 2009)—to introduce damaged alleles into each of the six Agtrap-Plod1 genes (Agtrap, Mthfr, Clcn6, Nppa, Nppb, and Plod1) in a rodent model of genetic hypertension, the SS (SS/JrHsdMcwi) rat. The uniform genetic background allowed us to assign (+), (−), or (=) phenotypic effects to each of the genes at the Agtrap-Plod1 locus, which is not possible using mouse knockout models with mixed genetic backgrounds that confound inter-strain comparisons due to genetic heterogeneity (Hunter 2012). Thus, by leveraging the genetic homogeneity of our SS strains, we were able to attribute the phenotypic contributions of multiple genes at the Agtrap-Plod1 locus that were previously unattainable in human population studies. We then retrospectively analyzed human data sets (e.g., HapMap, 1000 Genomes, and ENCODE) to demonstrate that interplay between multiple causative genes within the human AGTRAP-PLOD1 locus were not only possible, but rather were highly plausible.
Results
Strain generation and phenotyping strategy
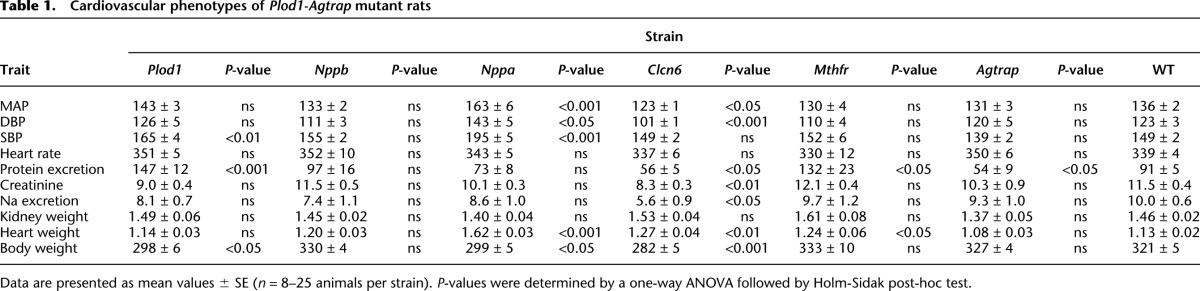
We generated six mutant SS strains (Agtrap, Mthfr, Clcn6, Nppa, Nppb, and Plod1) by ZFN mutagenesis for phenotypic comparison with wild-type (WT) littermates. The target sequences for ZFNs, the sequence of mutant alleles, and the predicted functional consequences are provided in Supplemental Table 2. All ZFN-targeted alleles were predicted to cause deleterious mutations by frameshift and/or by disrupting a functional domain of the protein (Supplemental Table 2). Strains were tested for eight cardiovascular disease (CVD)-associated phenotypes following a 10-d high-salt challenge (see Methods and Table 1). Because BP and renal phenotypes are most commonly reported in human studies (Kato et al. 2000; Jiang et al. 2004; Zhang et al. 2005; Levy et al. 2009; Newton-Cheh et al. 2009a,b; Chen et al. 2010; Tomaszewski et al. 2010; Johnson et al. 2011; Liu et al. 2011; Fung et al. 2012), we focused specifically below on the (+), (−), and (=) effects of mutant alleles on BP and urinary protein excretion (a commonly used index for renal damage).
Table 1.
Cardiovascular phenotypes of Plod1-Agtrap mutant rats
Blood pressure phenotypes
To test BP, 8- to 9-wk-old males from each gene-targeted strain and WT littermates were placed on a 4% NaCl diet for 10 d, and BP was recorded by radiotelemetry. Of note, all rat strains developed hypertension (>140/90 mmHg) in response to a 4% NaCl diet (Fig. 1). Only Plod1, Nppa, and Clcn6 mutations significantly modified BP (i.e., the severity of hypertension) in the SS rat on a 4% NaCl diet compared with WT (Fig. 1A–C). The Nppa mutation increased mean arterial pressure (MAP) (Δ + 27 mmHg, P < 0.001), systolic BP (SBP) (Δ + 46 mmHg, P < 0.001), and diastolic BP (DBP) (Δ + 20 mmHg, P < 0.05) compared with WT. The BP changes coincided with increased heart weight (1.62 ± 0.03 g vs. 1.13 ± 0.03 g, P < 0.001), but did not significantly change renal phenotypes (Table 1). This is consistent with increased MAP and heart weight in Nppa knockout mice (John et al. 1995) and with decreased circulating NPPA levels associated with human hypertension (Marques et al. 2010). Compared with WT, Plod1 mutation significantly increased only SBP (Δ + 17 mmHg, P < 0.01), whereas Clcn6 mutation decreased DBP (Δ − 22 mmHg, P < 0.001) and MAP (Δ − 14 mmHg, P < 0.05) but did not significantly affect SBP (Fig. 1A–C). These data indicate that NPPA, PLOD1, and CLCN6 could have additive or subtractive effects on BP in human, depending on the mode of inheritance and functional consequence of the causal variant(s) (i.e., gain or loss of function).
Figure 1.

Mean arterial pressure (MAP) (A), systolic blood pressure (SBP) (B), and diastolic blood pressure (DBP) (C) measured by radiotelemetry in conscious WT and mutant SS rat strains on 4% NaCl diet (n = 8–25 per strain). Data are presented as mean BP ± SEM. (*) P < 0.05, (**) P < 0.01, and (***) P < 0.001 vs. WT as determined by one-way ANOVA followed by a Holm-Sidak post-hoc test. (D) Rats were placed in metabolic cages overnight to acclimate, followed by a 24-h urine collection. Data are presented as mean protein excretion ± SEM (n = 8–25 rats per strain). (*) P < 0.05 and (***) P < 0.001 vs. WT as determined by one-way ANOVA followed by a Holm-Sidak post-hoc test. (E) BP and renal damage are linked with multiple AGTRAP-PLOD1 locus genes in human association studies and mutant SS rat strains. For human, genes were considered linked to a phenotype if an associated SNP or SNPs in LD (r2 > 0.6) with an associated SNP caused nonsynonymous mutations or were significantly associated with expression of a gene. Blue lines indicate decreased BP and/or proteinuria associated with a gene in the rat. Red lines indicate increased BP and/or proteinuria associated with a gene in the rat.
Renal phenotypes
The AGTRAP-PLOD1 locus has also been linked with renal function (Supplemental Table 1) but the causative gene(s) are unknown, prompting us to determine if renal phenotypes were secondary to hypertension or independent but residing within the same LD block. On a 4% NaCl diet, urinary protein excretion was increased by mutation of Plod1 (147 ± 12 mg/day, P < 0.001) and Mthfr (132 ± 23 mg/day, P < 0.05) compared with WT (91 ± 5 mg/day), whereas mutation of Clcn6 (56 ± 5 mg/day, P < 0.05) and Agtrap (54 ± 9 mg/day, P < 0.05) attenuated proteinuria (Fig. 1D). Compared with WT, the changes in proteinuria in Plod1 and Clcn6 mutant rats are likely secondary to BP (Fig. 1A–C). In contrast, the BP in Mthfr and Agtrap mutant strains did not differ significantly from WT on a 4% NaCl diet (Fig. 1), indicating that the significant changes in proteinuria (relative to WT) were independent of BP in these strains. Of these genes, only Mthfr has been suggested as a mediator of renal damage, largely in response to homocysteine-mediated oxidative stress in the kidney (Yi and Li 2008). Plod1 is a lysyl hydroxylase that mediates collagen cross-linking, and Plod1 knockout mice are prone to aortic aneurism (Takaluoma et al. 2007); however, Plod1 has not yet been associated with renal phenotypes. This is also the first report of a protective effect on proteinuria by Agtrap, a mediator of the renin-angiotensin system (Oppermann et al. 2010).
Functional annotation of the human AGTRAP-PLOD1 locus
Our data show that multiple genes in the Agtrap-Plod1 locus modify BP and renal phenotypes in the SS rat (Fig. 1A–D), prompting us to test whether similar mechanisms could be inherited at the homologous human AGTRAP-PLOD1 locus. First, we determined whether SNPs, genes, and reported phenotypes could be connected by LD in human using data from HapMap (The International HapMap Consortium 2003) and 1000 Genomes (The 1000 Genomes Project Consortium 2012). Genes were considered linked to the reported phenotype if an associated lead SNP or a SNP in LD (r2 > 0.6, HapMap CEU population) caused a nonsynonymous mutation or was associated with a change in gene expression (Supplemental Table 3). When taking into account all 226 SNPs in LD, we found that four out of six genes (MTHFR, CLCN6, NPPA, and NPPB) can be linked to BP or renal phenotypes at the AGTRAP-PLOD1 locus (Fig. 1E). Collectively, this strengthens the possibility that multiple modifiers of BP (NPPA, PLOD1, and CLCN6) and renal phenotypes (MTHFR, PLOD1, CLCN6, and AGTRAP) can cosegregate at the same GWAS locus.
Using ENCODE data (The ENCODE Project Consortium 2011), we tested more specifically whether CVD-associated haplotypes were likely to influence multiple genes at the AGTRAP-PLOD1 locus, rather than the “best-fit” candidate gene that is typically reported. Across nine different haplotype blocks, three (1.3%) SNPs caused nonsynonymous mutations (nSNPs), 13 (5.8%) SNPs were linked to gene expression (eSNP), and 16 (7.1%) SNPs were predicted to interrupt a functional motif (fSNP), whereas 197 SNPs (85.8%) had limited functional evidence or no annotated function (Fig. 2A; Supplemental Table 3). Out of the nine total haplotype blocks, six were linked with differential expression or nSNPs in multiple genes (Fig. 2B; Supplemental Table 3). One BP-associated haplotype block (led by rs5063) contained two nSNPs, two eSNPs, and five fSNPs that are linked to NPPA, NPPB, CLCN6, and MTHFR (Fig. 2C). Another haplotype block contained two lead SNPs (rs1801131 and rs4846049) that were independently associated with BP and renal phenotypes (Fig. 2D). This haplotype block included one nSNP in MTHFR and five fSNPs that were associated with MTHFR and CLCN6 expression (Supplemental Table 3). One fSNP in particular (rs12121543) is predicted to disrupt STAT1 binding in proximity to MTHFR and CLCN6 regulatory regions (Fig. 2D), fitting with previously reported enrichment of STAT1 binding at the AGTRAP-PLOD1 locus (Johnson et al. 2011).
Figure 2.

(A) Proportions of functional consequences of all SNPs in LD (r2 > 0.6) with the SNPs associated with BP and renal phenotypes at the AGTRAP-PLOD1 locus (see Supplemental Table 1). (B) Genes linked with lead SNPs of haplotypes associated with BP and renal phenotypes at the AGTRAP-PLOD1 locus. A lead SNP was considered linked to a gene if the lead SNP or SNPs in LD (r2 > 0.6) with a lead SNP caused nonsynonymous mutations or were significantly associated with expression of a gene. (C) An example of a BP-associated nSNP in NPPA (rs5063) that is in LD (r2 > 0.6) with an nSNP in MTHFR (rs2274976). Both are also in LD with fSNPs correlated with the expression of MTHFR, CLCN6, and NPPB. Note that a total of two nSNPs (boxed), two eSNPs (blue), and five fSNPs (green) are in LD with rs5063. (D) An example of an fSNP (rs12121543) that is in LD with two SNPs (rs1801131 and rs484609) associated with BP and renal phenotypes. An overview of the region between genes MTHFR and CLCN6 shows that rs12121543 falls in a transcriptional active region (vertical black bar overlapping histone modification, DNase hypersensitivity, and ChIP-seq peaks). rs12121543 is predicted to change a conserved nucleotide in a consensus STAT1-binding site and is associated with significant differences in MTHFR and CLCN6 expression compared with the major allele. (A,C,D) nSNP, nonsynonymous SNP; eSNP, expression SNP; and fSNP, functional SNP.
Discussion
The goal of this study was to identify mechanism(s) that underlie changes in BP and proteinuria at a GWAS locus (AGTRAP-PLOD1) with high genetic complexity, as indicated by multiple independent associations with BP and renal phenotypes in the human population (Kato et al. 2000; Jiang et al. 2004; Zhang et al. 2005; Levy et al. 2009; Newton-Cheh et al. 2009a,b; Chen et al. 2010; Tomaszewski et al. 2010; Johnson et al. 2011; Liu et al. 2011; Fung et al. 2012). By combining rat phenotype data (Fig. 1; Table 1) with multiple human data sets (Supplemental Tables 1, 3), we were able to draw previously unforeseen conclusions, foremost that multiple genes at a single GWAS locus can influence clinically relevant hypertension phenotypes. Although this has been implied statistically by GWAS for other traits previously (Yang et al. 2012), it had not been tested experimentally in a physiological setting for hypertension. Specific to this locus, we present the first evidence that Nppa, Clcn6, Mthfr, Plod1, and Agtrap mutations cause divergent CVD phenotypes and have the ability to modify renal phenotypes independently of BP (Fig. 1). Combined, these data suggest that the “best-fit” candidate gene interpretations of GWAS are in some cases only a simplified view of the complex genetic architecture underlying individually associated loci. Moreover, the complex combinations of alleles and haplotypes, due to genetic heterogeneity in human, would have the potential to impact the overall disease susceptibility and association of the AGTRAP-PLOD1 locus with BP and increase the challenge of finding all causative variants.
Challenges to identifying hypertension loci with multiple causative variants
GWAS loci that contain multiple interacting alleles have not yet been reported for human hypertension. This is likely attributed to several confounding factors that limit the sensitivity of GWAS for multi-SNP associations at a single locus. Firstly, current hypertension GWAS are likely too small (i.e., underpowered) to detect interplay between multiple causative genes at the same locus. Power limitations are due in part to a multi-SNP association requiring >10 times the sample size to achieve the same statistical power of a single SNP association (Zuk et al. 2012). This can be further complicated by frequency and effect size of the individual SNPs (e.g., rare SNPs with large effect sizes will have more combined power). Secondly, BP measurements are notoriously variable due to patient stress (e.g., “white coat effect”) and other environmental factors (Mancia et al. 2009), which might artificially inflate or deflate BP readings and obscure true SNP associations. We propose that this “phenotypic noise” likely masks the detection of multi-SNP hypertension GWAS loci, and therefore multiple causative SNPs cannot be ruled out in the absence of systematic experimental testing, as we have demonstrated here.
Gene editing in disease sensitive rat strains—alternate approach to characterizing GWAS loci
We were able to test a “multi-SNP hypothesis” by limiting variability using inbred disease-susceptible ZFN rat strains in a controlled experimental setting. By introducing target gene mutations on an inbred SS background by gene editing with ZFNs, our rat strains are isogenic except for the target-gene mutation. Because these strains are otherwise identical, we are able to compare (+), (−), or (=) effects of target-genes on BP and renal phenotypes, without the potential influence of other genetic heterogeneity. In contrast, existing mouse KO models are on a variety of strain backgrounds and therefore carry substantial heterogeneity in addition to the target gene, which can influence comparison of phenotypes between models (Hunter 2012). Using gene editing on a disease-susceptible rat background also provides multiple endogenous variants that are required for pathogenesis of complex diseases, such as hypertension. In other words, all strains developed salt-sensitive hypertension due to the SS background, but to varying degrees and with differences in end-organ damage depending on the modifying effects of the Agtrap-Plod1 locus (Fig. 1; Table 1).
Having first identified the causative genes underlying a GWAS locus by traditional ZFN-mediated gene editing (Fig. 1), we postulate that gene-editing technology (e.g., ZFN or TALEN) could then be used to ask more specific questions in future studies. Testing-combined haplotype effects could be achieved by editing multiple genes in the same pedigree. This would require sequential gene editing over multiple generations, as it is prohibitive to generate “multi-mutants” by cross-breeding together single mutants in close proximity. For example, recombination between two genes separated by only 150 kb (the approximate size of the Agtrap-Plod1 locus) would require >1000 offspring (assuming 1 Mb = 1 cM in the rat). It is also possible to humanize alleles using gene editing in the rat, which would likely be more representative of the hypomorphic alleles and smaller effect sizes observed in human GWAS. Thus, gene-editing technology could be used to test humanized alleles as single variants or whole haplotype blocks, which would likely be the gold standard to predicting human haplotype function in an experimental model of a complex disease.
Gene editing in the rat could also be used to explore additional factors that are generally treated as covariates in large GWAS, including genetic background, gender, and dietary or environmental stressors. To date, we have demonstrated the ability to edit genes on multiple backgrounds (e.g., FHH, SHR, and SS-BN13 consomic) (http://rgd.mcw.edu/wg/physgenknockouts), enabling the possibility of testing GWAS-nominated haplotypes in experimental models with inherently different disease etiologies (e.g., high renin hypertension in SHR vs. low renin hypertension in SS). Testing other experimental parameters, such as diet (e.g., high fat, high glucose, or caloric restriction), environmental stressors (e.g., sleep deprivation), response to pharmacological inhibitors (e.g., ACE inhibitor or diuretics), and biomarkers (e.g., circulating Nppa and homocysteine) will offer significant insight into the molecular underpinnings of GWAS loci. Finally, gender differences in genetic hypertension are becoming increasingly apparent, but not yet fully understood. Thus, future comparisons between males and females of gene-edited rat strains will also likely offer significant insight into gender discrepancies in hypertension risk that is associated with GWAS loci.
Conclusions
Our data demonstrated for the first time that multiple causative alleles for BP and renal phenotypes can cosegregate at the same hypertension GWAS locus. Of the six Agtrap-Plod1 genes, only three (Agtrap, Nppa, and Mthfr) were previously reported to have BP or renal phenotypes (John et al. 1995; Yi and Li 2008; Wakui et al. 2013), but none had been systematically examined in a uniform genetic background that is susceptible to hypertension. Using a ZFN rat strategy, we found that Nppa, Clcn6, Mthfr, Plod1, and Agtrap mutations caused divergent CVD phenotypes and have the ability to modify renal phenotypes independently of BP (Fig. 1). Additionally, Clcn6 was identified here as a key mediator of BP and proteinuria for the first time. Differentiating this role from MTHFR was not previously possible using only human association data, because both genes share a common promoter that separates them by only 47 bp (Supplemental Fig. 1). Analysis of disease-associated human haplotypes suggested that several human haplotypes likely carry multiple causative SNPs. Combined, these data offer new insight into regulation of BP and renal phenotypes at the Agtrap-Plod1 locus and provide rationale for empirically testing the individual and combined haplotype effects of human SNPs on AGTRAP-PLOD1 gene function and overall association with CVD risk.
Methods
Generation of ZFN-mutated rat strains
Mutant SS rat strains were generated by microinjection of custom CompoZr ZFNs (Sigma) into one-cell SS/JrHsdMcwi (SS) rat embryos and then implanted into pseudopregnant Sprague Dawley females, as described previously (Geurts et al. 2010). Supplemental Table 2 provides the strain designations, target sites, DNA recognition helices of ZFNs, and description of gene mutation. F0 generation animals were screened for ZFN-induced mutations by PCR amplification with the vendor-supplied primers (Supplemental Table 2) and assayed with the SURVEYOR Mutation Detection kit (Transgenomic, Inc.). Mutant alleles were then confirmed by Sanger sequencing. F0 animals were back-crossed to the parental strain, and multiple pairs of N2 offspring were intercrossed to establish a breeding colony, thereby reducing any chance of off-target effects to <1%. Expression of mutant and WT alleles were assessed by RT-PCR using primers provided in Supplemental Figure 2.
Blood pressure phenotyping
All strains were maintained on a 14/10-h light-dark cycle with ad libitum access to normal chow (Teklad Low Salt diet #7034, Harlan Laboratories, Inc.) and water. MAP of 8- to 9-wk-old male rats that had been placed on a 4% NaCl diet for 10 d were recorded by radiotelemetry at 10-sec intervals, every 2 min, and averaged over 4 h. Telemeters (TA11PA-C40, Data Sciences) were subcutaneously implanted with a catheter inserted into the abdominal aorta via the femoral artery.
Renal function and serum biochemistry
Immediately after MAP recordings, rats were placed in metabolic cages and allowed to acclimate for 24 h, followed by a 24-h urine collection. Urine samples were then cleared of insoluble particulate by centrifugation at 3500 rpm and measured for total protein by Bradford assay (Bio-Rad). Sodium and potassium were measured by flame photometry (IL943, Instrumentation Laboratory); creatinine was measured by autoanalyzer (VET ACE Alera, Alfa Wassermann) using the Jaffe method.
Analysis of human SNPs and haplotype blocks
LD (defined here as r2 > 0.6) for human SNPs from the CEU cohort from the HapMap (http://hapmap.ncbi.nlm.nih.gov/; The International HapMap Consortium 2003) and 1000 genome (http://browser.1000genomes.org/index.html; The 1000 Genomes Project Consortium 2012) projects were calculated using SNAP (http://www.broadinstitute.org/mpg/snap/ldsearch.php; Johnson et al. 2008). SNPs associated with gene regulatory mechanisms were curated from the ENCODE study (The ENCODE Project Consortium 2011) using the RegulomeDB database (http://www.regulomedb.org/; Boyle et al. 2012).
Statistical analysis
Statistical analyses were performed using Sigma Plot 11.0 software. Data are presented as mean ± SE. BP and renal phenotypes of mutant strains were compared with WT (control) by one-way ANOVA followed by a Holm-Sidak post-hoc test. Because MAP, SBP, DBP, and proteinuria failed the equal variance test, these data were log-transformed before one-way ANOVA followed by a Holm-Sidak post-hoc test. For all statistical tests, P < 0.05 was considered significant.
Competing interest statement
The Medical College of Wisconsin could one day receive royalties on sales of genetically modified rat strains through a license agreement with Sigma Advanced Genetic Engineering (SAGE).
Acknowledgments
We thank R. Schilling, J. Klotz, M. Grzybowski, S. Kalloway, J. Foeckler, C. Hansen, and M. Casati for excellent technical support. We also acknowledge all members of the PhysGen Knockout team. M.J.F. is supported by an NHLBI training grant (5T32HL007792). A.M.G. is supported by a NIH Director's New Innovator Award (DP2OD008396). This study was supported by NHLBI grant 5RC2HL101681 to H.J.J.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.160283.113.
References
- The 1000 Genomes Project Consortium 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, Karczewski KJ, Park J, Hitz BC, Weng S, et al. 2012. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22: 1790–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Wheeler B, Sampson J, Hartge P, Chanock SJ, Park JH 2013. Projecting the performance of risk prediction based on polygenic analyses of genome-wide association studies. Nat Genet 45: 400–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Chen X, Guo Y, Shi R, Zhang G 2010. Brain natriuretic peptide rs198388 polymorphism and essential hypertension in Hunan Han people. Zhong Nan Da Xue Xue Bao Yi Xue Ban 35: 1207–1213 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2011. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol 9: e1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung MM, Salem RM, Lipkowitz MS, Bhatnagar V, Pandey B, Schork NJ, O'Connor DT 2012. Methylenetetrahydrofolate reductase (MTHFR) polymorphism A1298C (Glu429Ala) predicts decline in renal function over time in the African-American Study of Kidney Disease and Hypertension (AASK) Trial and Veterans Affairs Hypertension Cohort (VAHC). Nephrol Dial Transplant 27: 197–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G 2010. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 42: 1049–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, et al. 2009. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, Menoret S, Jacob HJ, Anegon I, Buelow R 2010. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol Biol 597: 211–225 [DOI] [PubMed] [Google Scholar]
- Hunter KW 2012. Mouse models of cancer: Does the strain matter? Nat Rev Cancer 12: 144–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium 2003. The International HapMap Project. Nature 426: 789–796 [DOI] [PubMed] [Google Scholar]
- Jiang S, Hsu YH, Xu X, Xing H, Chen C, Niu T, Zhang Y, Peng S 2004. The C677T polymorphism of the methylenetetrahydrofolate reductase gene is associated with the level of decrease on diastolic blood pressure in essential hypertension patients treated by angiotensin-converting enzyme inhibitor. Thromb Res 113: 361–369 [DOI] [PubMed] [Google Scholar]
- John SW, Krege JH, Oliver PM, Hagaman JR, Hodgin JB, Pang SC, Flynn TG, Smithies O 1995. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science 267: 679–681 [DOI] [PubMed] [Google Scholar]
- Johnson AD, Handsaker RE, Pulit S, Nizzari MM, O'Donnell CJ, de Bakker PIW 2008. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 24: 2938–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T, Gaunt TR, Newhouse SJ, Padmanabhan S, Tomaszewski M, Kumari M, Morris RW, Tzoulaki I, O'Brien ET, Poulter NR, et al. 2011. Blood pressure loci identified with a gene-centric array. Am J Hum Genet 89: 688–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Sugiyama T, Morita H, Nabika T, Kurihara H, Yamori Y, Yazaki Y 2000. Genetic analysis of the atrial natriuretic peptide gene in essential hypertension. Clin Sci (Lond) 98: 251–258 [PubMed] [Google Scholar]
- Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, Willer CJ, Jackson AU, Vedantam S, Raychaudhuri S, et al. 2010. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467: 832–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, et al. 2009. Genome-wide association study of blood pressure and hypertension. Nat Genet 41: 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li H, Qi Q, Lu L, Gan W, Loos RJ, Lin X 2011. Common variants in or near FGF5, CYP17A1 and MTHFR genes are associated with blood pressure and hypertension in Chinese Hans. J Hypertens 29: 70–75 [DOI] [PubMed] [Google Scholar]
- Mancia G, Bombelli M, Facchetti R, Madotto F, Quarti-Trevano F, Polo Friz H, Grassi G, Sega R 2009. Long-term risk of sustained hypertension in white-coat or masked hypertension. Hypertension 54: 226–232 [DOI] [PubMed] [Google Scholar]
- Marques FZ, Campain AE, Yang YH, Morris BJ 2010. Meta-analysis of genome-wide gene expression differences in onset and maintenance phases of genetic hypertension. Hypertension 56: 319–324 [DOI] [PubMed] [Google Scholar]
- Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, et al. 2009a. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet 41: 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton-Cheh C, Larson MG, Vasan RS, Levy D, Bloch KD, Surti A, Guiducci C, Kathiresan S, Benjamin EJ, Struck J, et al. 2009b. Association of common variants in NPPA and NPPB with circulating natriuretic peptides and blood pressure. Nat Genet 41: 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M, Gess B, Schweda F, Castrop H 2010. Atrap deficiency increases arterial blood pressure and plasma volume. J Am Soc Nephrol 21: 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin DY, Duan J, Ophoff RA, Andreassen OA, et al. 2011. Genome-wide association study identifies five new schizophrenia loci. Nat Genet 43: 969–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna S, Li B, Mulas A, Sidore C, Kang HM, Jackson AU, Piras MG, Usala G, Maninchedda G, Sassu A, et al. 2011. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet 7: e1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar P, Ripke S, Scott LJ, Andreassen OA, Cichon S, Craddock N, Edenberg HJJ, Nurnberger JI, Rietschel M, Blackwood D, et al. 2011. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 43: 977–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaluoma K, Hyry M, Lantto J, Sormunen R, Bank RA, Kivirikko KI, Myllyharju J, Soininen R 2007. Tissue-specific changes in the hydroxylysine content and cross-links of collagens and alterations in fibril morphology in lysyl hydroxylase 1 knock-out mice. J Biol Chem 282: 6588–6596 [DOI] [PubMed] [Google Scholar]
- Tomaszewski M, Debiec R, Braund PS, Nelson CP, Hardwick R, Christofidou P, Denniff M, Codd V, Rafelt S, van der Harst P, et al. 2010. Genetic architecture of ambulatory blood pressure in the general population: Insights from cardiovascular gene-centric array. Hypertension 56: 1069–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakui H, Tamura K, Masuda SI, Tsurumi-Ikeya Y, Fujita M, Maeda A, Ohsawa M, Azushima K, Uneda K, Matsuda M, et al. 2013. Enhanced angiotensin receptor-associated protein in renal tubule suppresses angiotensin-dependent hypertension. Hypertension 61: 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ferreira T, Morris AP, Medland SE, Madden PA, Heath AC, Martin NG, Montgomery GW, Weedon MN, Loos RJ, et al. 2012. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44: 369–375, S361–S363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Li PL 2008. Mechanisms of homocysteine-induced glomerular injury and sclerosis. Am J Nephrol 28: 254–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Mao G, Zhang Y, Tang G, Wen Y, Hong X, Jiang S, Yu Y, Xu X 2005. Association between human atrial natriuretic peptide Val7Met polymorphism and baseline blood pressure, plasma trough irbesartan concentrations, and the antihypertensive efficacy of irbesartan in rural Chinese patients with essential hypertension. Clin Ther 27: 1774–1784 [DOI] [PubMed] [Google Scholar]
- Zuk O, Hechter E, Sunyaev SR, Lander ES 2012. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci 109: 1193–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]