

Abstract
The short arms of the five acrocentric human chromosomes harbor sequences that direct the assembly and function of the nucleolus, one of the key functional domains of the nucleus, yet they are absent from the current human genome assembly. Here we describe the genomic architecture of these human nucleolar organizers. Sequences distal and proximal to ribosomal gene arrays are conserved among the acrocentric chromosomes, suggesting they are sites of frequent recombination. Although previously believed to be heterochromatic, characterization of these two flanking regions reveals that they share a complex genomic architecture similar to other euchromatic regions of the genome, but they have distinct genomic characteristics. Proximal sequences are almost entirely segmentally duplicated, similar to the regions bordering centromeres. In contrast, the distal sequence is predominantly unique to the acrocentric short arms and is dominated by a very large inverted repeat. We show that the distal element is localized to the periphery of the nucleolus, where it appears to anchor the ribosomal gene repeats. This, combined with its complex chromatin structure and transcriptional activity, suggests that this region is involved in nucleolar organization. Our results provide a platform for investigating the role of NORs in nucleolar formation and function, and open the door for determining the role of these regions in the well-known empirical association of nucleoli with pathology.
A detailed description of how the genome is organized within the nuclei of human cells to facilitate proper cellular functions is one of the major unsolved problems in biology. While genome-wide technologies are beginning to have an impact (Lieberman-Aiden et al. 2009; The ENCODE Project Consortium 2012), the picture is incomplete since critical regions of the human genome remain unidentified. Prominent among these missing regions is the chromosomal context around which nucleoli form, termed the nucleolar organizer region (NOR) (McClintock 1934). The nucleolus is the largest functional domain within the nucleus and is the site of ribosome biogenesis. It has a distinct structure and houses ribosomal RNA gene (rDNA) transcription, preribosomal RNA (pre-rRNA) processing, and preribosome assembly (Olson 2011). The rDNA repeats encode the major rRNA species and are organized into large head-to-tail tandem arrays located at the NORs (McStay and Grummt 2008). Extensive binding by the nucleolar DNA binding protein, UBF (encoded by UBTF), across the rDNA is responsible for their distinctive appearance on metaphase chromosomes, in which they form secondary constrictions (Mais et al. 2005; McStay and Grummt 2008). Despite ribosome biogenesis being central to cellular biology, many aspects of nucleolar formation, organization, and function remain to be elucidated, and the genomic architecture of NORs has not been described for any vertebrate to date.
In humans, the approximately 300 rDNA repeats are distributed among five NORs on the short arms of the acrocentric chromosomes (HSA13-15, HSA21, and HSA22) (Henderson et al. 1972; Schmickel 1973; Stults et al. 2008). In most human cells, a majority of NORs are active and coalesce to form between one and three nucleoli (Savino et al. 2001). While discrete, nucleoli are not encapsulated and instead seem to be self-organizing, highly dynamic structures (Dundr et al. 2002; Andersen et al. 2005; Sirri et al. 2008) that are spatially isolated from the rest of the nucleoplasm by a shell of heterochromatin (Nemeth and Langst 2011). However, the apparent heterochromatic nature of the regions flanking the rDNA has made them a low priority for genomic analysis (International Human Genome Sequencing Consortium 2004), and thus, the five acrocentric chromosome short arms are missing from the current human genome assembly.
The enormous demand for ribosomes by actively growing cells puts nucleoli at the forefront of cellular growth regulation. Links between nucleoli and growth pathologies date back over a century to observations of abnormal nucleoli in tumor cells (Pianese 1896). Recently, molecular studies have begun to clarify this relationship, with evidence supporting roles for tumor suppressor genes and oncogenes in rDNA transcriptional regulation (Budde and Grummt 1999; Hannan et al. 2000; Grummt 2003; Grandori et al. 2005), and a direct role for increased rDNA transcription in the development of malignancy (Bywater et al. 2012). Surprisingly, recent studies have also revealed that the nucleolus plays roles in many other biological processes, ranging from aging and cell cycle progression to X-chromosome inactivation and viral replication (Visintin et al. 1999; Boisvert et al. 2007; Zhang et al. 2007; Ganley et al. 2009). The central role of the nucleolus in growth regulation, coupled with the potential for the regions adjacent to the rDNA to contribute to NOR function, prompted us to investigate the genomic context in which the rDNA resides. Here we present the identification and characterization of >550 kb of sequence flanking both sides of the rDNA array.
Results
Identification of rDNA flanking regions
To obtain the sequences flanking the rDNA arrays, we made use of preexisting sequences adjacent to the rDNA on the proximal (centromeric; 493 bp) (Sakai et al. 1995) and distal (telomeric; 8.3 kb) sides of the rDNA (Gonzalez and Sylvester 1997). Probes derived from these sequences were used to screen single-chromosome cosmid libraries, and several positive clones were sequenced. Searches of GenBank using the resulting sequences, coupled with BAC walking, ultimately resulted in identification of 15 BAC clones from the distal junction (DJ), five of which include some rDNA sequence (Supplemental Fig. 1). Similar searches identified three BAC clones from the proximal junction (PJ), one of which includes rDNA (Supplemental Fig. 2). These cover 379 kb of DJ sequence and 207 kb of PJ sequence flanking the rDNA (Fig. 1A).
Figure 1.

Human rDNA flanking regions. (A) Schematic human acrocentric chromosome showing telomeric (blue) and centromeric (yellow) regions, and the NOR (black line), expanded below into rDNA, PJ (orange), and DJ (green) regions. Not to scale. (B) DJ and PJ localize distally and proximally to rDNA, respectively, on all acrocentric chromosomes. FISH was performed on normal human metaphase spreads with DJ BAC (red) and rDNA (green) probes (left panels), and PJ BAC (green) and rDNA (red) probes (right panel). Chromosomes are DAPI-stained. (C) DNA combing of HeLa cell nucleolar DNA shows DJ (red) is physically linked to 18S rDNA (green). Three representative images are shown below the hybridization scheme.
We sought evidence that these putative junction sequences adjoin the rDNA. Hybridization of DJ BAC clones to metaphase chromosome spreads places these regions distal to the rDNA on the acrocentric chromosome short arms (Fig. 1B). Further, FISH on combed DNA molecules (Bensimon et al. 1994) clearly shows the DJ region is adjacent to the rDNA (Fig. 1C). Finally, a bioinformatic screen confirmed that the DJ adjoins the rDNA (Supplemental Methods).
Similar evidence was more difficult to obtain for the PJ, particularly with hybridization-based approaches. Metaphase FISH shows the PJ to be centromere proximal to the rDNA, as expected, but additional signals are observed distal to the rDNA and on other chromosomes (Fig. 1B; Supplemental Fig. 3), and clear signals were not observed from DNA combing. These difficulties stem from the high level of PJ segmental duplication (see below). Therefore we sought additional sequence-based evidence for the PJ adjoining the rDNA. First, we sequenced PJ-containing cosmids to show that the PJ is linked to at least 16 kb of rDNA (Supplemental Fig. 4). The PJ contig has previously been identified as a chr21 short-arm pericentromeric region (Lyle et al. 2007). To rule out the possibility that the PJ is adjacent to a large piece of segmentally duplicated rDNA, we compared rDNA external transcribed spacer (ETS) and 18S rRNA coding regions adjoining the DJ (a true junction) and the PJ. If the PJ is adjacent to duplicated, nonfunctional rDNA, it should have accumulated mutations, yet we found these PJ rDNA sequences to be 98.8% identical to the DJ rDNA. This is much higher identity than known 18S/ETS segmental duplicates (maximum of 93.2% identity), suggesting that the PJ-linked rDNA is intact. Finally, we implemented a bioinformatic strategy to look for sequence reads that span the PJ/rDNA junction (Supplemental Fig. 5). The positions of the PJ/rDNA junction in the cosmids and BACs are slightly different, and we found both these PJ junction positions with no evidence for additional PJ junctions (Supplemental Methods). These results suggest that the PJ adjoins the rDNA, although the precise junction position was found to vary slightly among the acrocentric chromosomes (Supplemental Fig. 6).
Interchromosomal conservation of rDNA flanking regions
Previous reports suggested that the sequences distal to the rDNA are conserved across all acrocentric chromosomes (Worton et al. 1988; Gonzalez and Sylvester 1997). Our DJ metaphase FISH results are consistent with this, showing hybridization signals for all the acrocentric chromosomes (Fig. 1B). The FISH also suggests that the PJ is conserved across the acrocentric chromosomes (Fig. 1B). To confirm this and to further validate the integrity of the flanking region sequences, we screened genomic DNA from a panel of mouse somatic cell hybrids, each containing a single human acrocentric chromosome for flanking region sequence. PCR at five intervals across the DJ each gave the expected product for all five acrocentric chromosomes (Fig. 2A). We could not screen across the PJ because only one unique region is present (see below). Nevertheless, this region is also shared among all five acrocentric chromosomes (Fig. 2A). Furthermore, screening genomic DNA from lines containing a chr21 translocation originating within the rDNA confirmed the orientation of the DJ and PJ relative to the rDNA (Fig. 2A) and that the unique PJ region is located exclusively on the proximal side. These results suggest that both the DJ and the PJ are conserved across all five acrocentric chromosomes.
Figure 2.
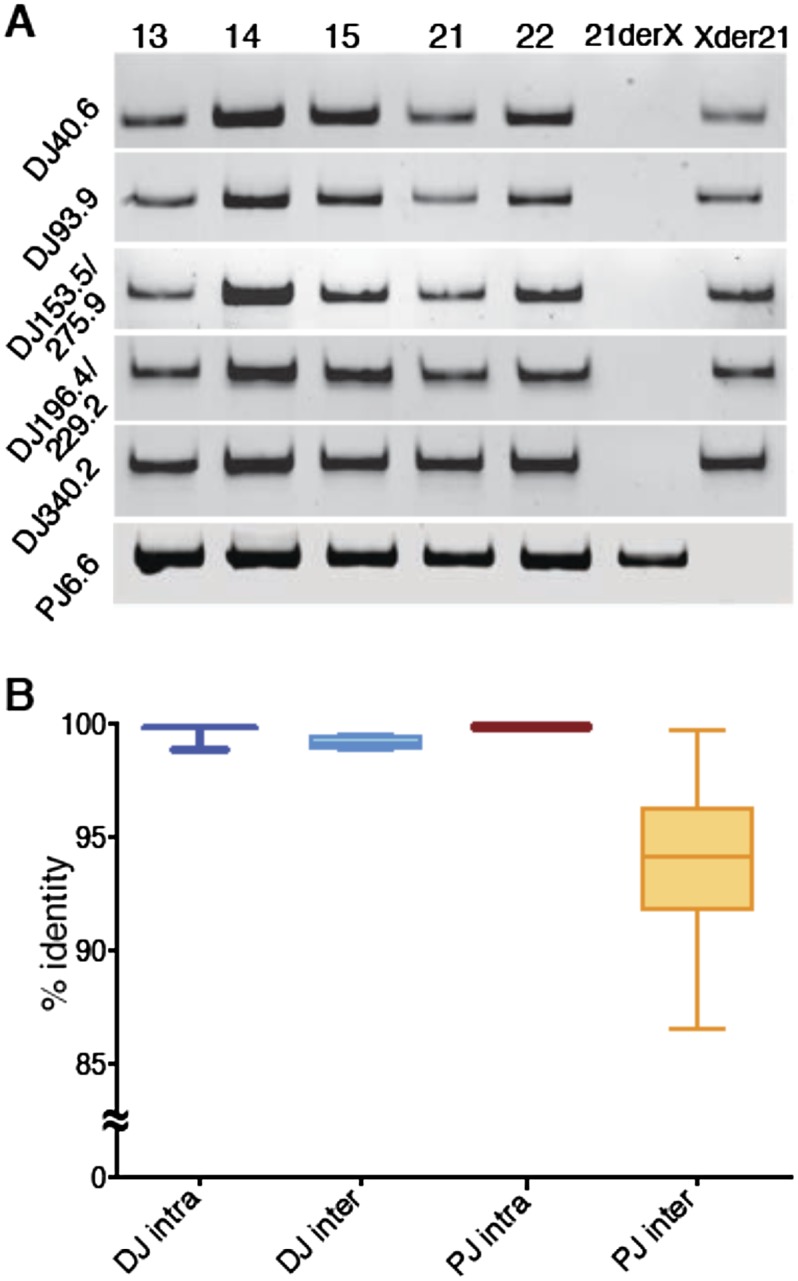
DJ and PJ acrocentric chromosome conservation. (A) PCR performed at increasing distances (left) into the DJ from mouse somatic cell hybrids carrying a single human acrocentric chromosome (indicated above). The right-hand lanes show PCR performed on the reciprocal products (Xder21 and 21derX) of a chr21 translocation that originates in the rDNA, confirming the DJ is located distally to the rDNA. Bottom panel is the same, but uses primers to the single unique PJ region. (B) Average intrachromosomal and interchromosomal DJ and PJ sequence identities from pairwise comparisons of representative BAC and cosmid clones are plotted.
To quantify how similar the DJ and PJ regions are between the different acrocentric chromosomes, we determined BAC and cosmid sequence identities from intra- and interchromosomal pairwise comparisons for PJ and DJ clones (Fig. 2B; Supplemental Data 1). Intrachromosomal sequence identities approach 100% for both DJ and PJ sequences, as expected, with any differences likely resulting from sequence error. DJ interchromosomal identity averages 99.1%, suggesting there is a very active homogenization mechanism that maintains DJ sequence identity between the acrocentric chromosomes. PJ interchromosomal sequence identity is lower, averaging 93.3%, with the variation predominantly arising from interchromosomal polymorphisms in the rDNA junction position and Alu elements (Supplemental Fig. 6). This suggests there is also an active homogenization mechanism that maintains PJ sequences across acrocentric chromosomes (Fig. 2B).
Characterization of DJ and PJ sequences
The DJ and PJ are uncharacterized regions of the human genome, so we next employed a series of bioinformatic approaches to determine whether they harbor genomic features of interest. Consensus DJ and PJ contig sequences (Fig. 3A) were generated from a minimum set of overlapping BACs, using BACs from the same chromosome where possible (Supplemental Fig. 7).
Figure 3.

DJ and PJ sequence characterization. (A) Major genomic features of the PJ (orange) and DJ (green) contigs. BACs used to construct the contigs are shown as black lines, with BAC names and chromosomal origins indicated (chr17 annotation of AC011841 is incorrect). Satellites are shown in blue, the ACRO138 repeats in red, the large DJ inverted repeat as white arrows, and the rDNA array between the PJ and DJ in gray. (B) Segmental duplication analysis. Lines show segmental duplications from PJ (orange) and DJ (green), indicating the location of the duplicate on the human chromosomes (arranged around the flanking regions). Positions of centromeres (yellow) and telomeres (blue) are indicated. Segmental duplicate length is indicated by line color, as defined below.
To determine whether these regions primarily consist of repetitive elements characteristic of constitutive heterochromatin (International Human Genome Sequencing Consortium 2004), we characterized the repeat composition of the DJ and PJ. The transposable repeat element content of both regions is similar to that of the human genome as a whole (Supplemental Fig. 8), except for a lack of known DNA transposons in the DJ. Both regions also contain blocks of satellite repeats, most notably a large (38.6 kb) block of 48-bp satellite repeats at the distal end of the DJ (Fig. 3A). These repeats, initially classed as chr22 pericentromeric repeats (Metzdorf et al. 1988), are now referred to as CER satellites (Jurka et al. 2005). CER blocks are found distal to the rDNA on all acrocentric chromosomes, with additional pericentromeric blocks on chr14 and chr22 (Supplemental Fig. 9). Finally, we looked for novel repeats. Two blocks of a novel 138-bp tandem repeat that we call ACRO138 are present within the DJ (Fig. 3A; Supplemental Fig. 10). Most strikingly, we discovered a large inverted repeat that dominates the DJ (Fig. 3A). Each arm of the inverted repeat is ∼109 kb, and the two arms share an average sequence identity of 80%. Alignment of the two arms reveals that the underlying sequence identity is higher than this but is interrupted by a number of indels (Supplemental Fig. 11), implying either that the inverted repeat is young or that there are mechanisms to maintain sequence identity between the arms.
Although these rDNA flanking regions were thought to be heterochromatic (International Human Genome Sequencing Consortium 2004), we wondered whether they contain any genes. To address this, we designed a gene predication pipeline that integrates ORF prediction, mRNA, EST, and protein data to identify potential gene-coding regions. Eight putative DJ genes and four PJ genes were predicted. These gene models all had support from multiple data sources, but the majority are single exon (Supplemental Fig. 12; Supplemental Tables 1, 2). Experimental validation will be required to determine which of these putative genes are real.
Segmental duplications (duplications of DNA to elsewhere in the genome) are a prominent feature of the human genome and are commonly enriched at centromere boundaries (She et al. 2004; Bailey and Eichler 2006). Given the proximity of the DJ and PJ to centromeres we undertook a segmental duplication analysis. A number of segmental duplications (>1 kb in length and with >85% sequence identity) were found from both regions (Fig. 3B). Interestingly, the DJ and PJ show different segmental duplication patterns. PJ segmental duplicates are more frequent and longer and have greater sequence identity than DJ segmental duplicates (Fig. 3B; Supplemental Tables 3, 4). Furthermore, the majority of PJ segmental duplicates are found in centromeric/pericentromeric regions of the genome as previously observed (Piccini et al. 2001; Lyle et al. 2007), while the majority of DJ segmental duplicates are found in euchromatic/telomeric regions (Fig. 3B). Most strikingly, we found that the level of segmental duplicated DNA is vastly different, with 7.3% of the DJ being segmentally duplicated versus 92.4% for the PJ (Table 1). These results demonstrate that these two rDNA flanking regions have different genomic characteristics in humans: The segmental duplication profile of the PJ resembles pericentromeric regions, while that of the DJ resembles euchromatic regions. The high level of segmental duplication likely explains the problems encountered using hybridization-based approaches with the PJ. Finally, this high level of segmental duplication prevented us from extending the PJ further, as we could not unambiguously assign additional sequences to the PJ region.
Table 1.
Segmental duplication comparison between the DJ and PJ

Perinucleolar positioning of the DJ
To better understand the relationship between these flanking regions and nucleolar architecture, we analyzed DJ and PJ positioning in interphase nuclei of HT1080 cells. 3D immuno-FISH revealed that, despite their close proximity to rDNA, DJ sequences are excluded from the nucleolar interior, instead appearing as discrete foci embedded in the perinucleolar heterochromatin (Fig. 4A). The majority of DJ foci associate with perinucleolar heterochromatin (Supplemental Videos 1, 2). The high level of PJ segmental duplication makes it difficult to determine which FISH signals derive from the PJ versus from unlinked segmentally duplicated regions (Supplemental Fig. 13); therefore, we focused on the DJ for the remainder of this study.
Figure 4.

The DJ forms a perinucleolar anchor for rDNA repeats. (A) 3D immuno-FISH reveals that DJ sequences lie in perinucleolar heterochromatin in HT1080 cells. Nucleoli are visualized with UBF antibodies (red) and DJ with BAC CT476834 (green). The nucleus is DAPI-stained. The extended focus images (left) are stills from Supplemental Videos 1 and 2, while the image on the right shows a single focal plane. (B) Inhibition of rDNA transcription with AMD results in formation of nucleolar CAPs juxtaposed with DJ sequences in perinucleolar heterochromatin. Staining as in A. Two representative cells are shown, one with an enlargement. Cartoon models the transition between active and withdrawn rDNA upon AMD treatment. rDNA (red) retreats from the nucleolus (black) to the DJ (green) that is embedded in perinucleolar heterochromatin (dark blue).
The rDNA is transcribed by RNA polymerase I (pol-I), and inhibition of pol-I transcription by low concentrations of actinomycinD (AMD) induces rapid and remarkable nucleolar reorganization (Hadjiolov 1985). This involves dissociation of nascent rRNA transcripts, resulting in collapse of the rDNA repeats into nucleolar caps that form at the nucleolar periphery and consist of rDNA, pol-I transcription machinery, and processing factors (Supplemental Fig. 14; Prieto and McStay 2007; Sirri et al. 2008). Given the localization of the DJ, we wondered whether it has any relationship to these nucleolar caps. Strikingly, 3D immuno-FISH on AMD-treated cells revealed that nucleolar caps form immediately adjacent to DJ sequences positioned in perinucleolar heterochromatin (Fig. 4B). Although some larger nucleolar caps are bilobed and associated with two DJ signals, it appears that the majority of caps are derived from individual NORs. These results suggest that the DJ anchors the linked rDNA to perinucleolar heterochromatin and that the retreat of the rDNA to the DJ upon AMD treatment provides an explanation for the positioning of nucleolar caps (Fig. 4B).
DJ sequences can target perinucleolar heterochromatin
rDNA arrays regress to perinucleolar heterochromatin in AMD-treated cells, rather than DJ sequences moving toward rDNA foci within the nucleolar interior (Fig. 4). This suggests that elements within the DJ may be responsible for its perinucleolar localization, rather than the DJ being simply linked to the rDNA. To investigate this, we transfected HT1080 cells with a mixture of three BACs that cover the DJ contig. Two stable clones containing large integrated arrays of this BAC mixture were selected for further analysis. FISH with DJ BAC CT476834 and rDNA probes revealed that ectopic DJ BAC arrays had integrated into metacentric (non-NOR bearing) chromosomes (Supplemental Fig. 15A). Quantitative PCR using primer pairs positioned across the DJ contig revealed that the sequence content of the ectopic arrays reflects that of the input BAC mixture (Supplemental Table 5). 3D FISH was performed on cells from these clones to determine whether the ectopic DJ arrays associate with nucleoli. In order to more clearly reveal the boundaries of the nucleolus, cells were treated with AMD (Fig. 5). For both clones, we observe a remarkable degree of association of the ectopic DJ arrays with perinucleolar heterochromatin. Moreover, in the majority of cells, the ectopic DJ array appears to spread through the perinucleolar heterochromatin, covering a significant fraction of the nucleolar surface. Transcription from the small amounts of rDNA derived from BAC AL592188 does not appear to explain this localization, as silver staining (which can detect activity at NORs with comparable rDNA content; A Grob and B McStay, in prep.) shows no activity. Additionally, transcriptionally active ectopic rDNA arrays (neo-NORs) that do not contain any DJ sequences are usually not associated with endogenous nucleoli (A Grob and B McStay, in prep.). Therefore, we conclude that sequences within the DJ contig specify association with perinucleolar heterochromatin even when positioned on metacentric chromosomes. (Supplemental Fig. 15B).
Figure 5.

Ectopic DJ arrays target perinucleolar heterochromatin. Positioning of DJ BAC arrays. 3D FISH was performed on AMD-treated cells from clones 1 and 2 with rDNA (red) and DJ BAC CT476834 (green) probes. The large green hybridization signals identified by arrowheads indicate the ectopic DJ array. Endogenous DJ signals are also visible. Classification of ectopic DJ arrays as nucleolar associated, partially associated, or nonassociated is indicated by white, yellow, and orange arrowheads, respectively, and is quantified below.
Chromatin profiling of the DJ
Our results suggest that the DJ region plays a role in nucleolar organization. Specifically, we hypothesize that DJ sequences provide an anchor point within perinucleolar heterochromatin for the linked rDNA array that is normally present in the nucleolar interior. If so, the DJ may have a chromatin organization that facilitates this role. To profile its chromatin organization, we mapped available histone modification and insulator binding protein CTCF ChIP-seq data sets from the ENCODE Project (Ernst et al. 2011) to the DJ. Discrete patterns of enrichment were observed at specific points across the DJ (Fig. 6A). Integration of these chromatin data sets using ChromHMM (Ernst and Kellis 2012) with multiple cell types revealed a complex chromatin landscape that is largely conserved among cell types (Fig. 6B; Supplemental Fig. 16). Strikingly, chromatin signatures characteristic of promoters are found at regular ∼45-kb intervals across the DJ, interspersed among marks associated with heterochromatin. The periodicity (∼45 kb) of these putative promoters is interesting, as it closely mirrors the size of the rDNA unit. Chromatin marks indicative of promoters (e.g., H3K4me3) coincide with DNase hypersensitive sites and FAIRE signals (Supplemental Fig. 17), and we experimentally confirmed that the H3K4me3 and FAIRE peaks are present in the HT1080 cell line used in our immuno-FISH experiments (Fig. 6C). The open chromatin peaks centered at 138 kb and 290 kb correspond to the ACRO138 repeat blocks identified in the repeat analysis (Fig. 3A). Moreover, chromatin signatures associated with actively transcribed gene bodies (e.g. H3K36me3 and H3K20me1) (Ernst et al. 2011) are observed extending leftward and rightward from the promoters at 187 kb and 238 kb, respectively, (Fig. 6A). CTCF, a multivalent DNA binding protein involved in many cellular processes (Phillips and Corces 2009), has recently been shown to be involved in the transcriptional regulation of ribosomal genes (van de Nobelen et al. 2010) and human nucleolar organization (Hernandez-Hernandez et al. 2012). Multiple CTCF binding peaks were observed across the DJ, coinciding with CTCF consensus sequences (Supplemental Fig. 18). Interestingly, CTCF binding sites are positioned close to the DJ/rDNA boundary and frame the DJ transcription units described above. Together, these results reveal that the DJ has a complex and structured chromatin landscape.
Figure 6.

Chromatin landscape of the DJ. (A) ChIP-seq signals of different chromatin features (right) in H1-hESC cells, normalized to tags per million mapped reads are shown below a schematic of the DJ, including inverted repeats. Asterisks indicate enrichment sites. (Bottom) Control signal is shown in gray. (B) Chromatin states derived from the multivariate HMM analysis for seven different cell types (right). Each colored bar represents a specific chromatin state, as annotated below left. (C) Nucleolar H3K4me3 ChIP-PCR and nucleolar FAIRE-PCR using HT1080 cells validate the presence of H3K4me3 and FAIRE in the DJ. DJ positions of the primers used are shown to the right, and red boxes correspond to peaks of H3K4me3 from A. Genomic DNA (gDNA), input and negative controls (-ve and IgG) are shown.
Transcription profiling of the DJ
The chromatin profiling results suggest that despite being embedded in perinucleolar heterochromatin, DJ sequences are transcriptionally active. To directly investigate transcription in the DJ, we mapped RNA-seq, RNA Pol II, and TAF1 ChIP-seq data (from ENCODE) and mRNA and EST data (from GenBank) onto the DJ (Supplemental Fig. 19). Strong evidence for transcription originating from the majority of putative promoters was obtained, confirming that the DJ is transcriptionally active. The putative promoters at 187 kb and 238 kb in particular are supported by multiple lines of evidence (Fig. 7A), with RNA-seq data indicating that the transcripts from these promoters are spliced, and cDNA clones of these transcripts being present in GenBank (accession nos. AK026938 and BX647680). We used RT-PCR to experimentally confirm the existence of these spliced polyadenylated transcripts, which we term disnor187 and disnor238 (Fig. 7B). In addition, we show using RNA-seq data that they have low to medium expression levels (Supplemental Fig. 20). The largest open reading frames present in disnor187 and disnor238 are 120 and 144 amino acids, respectively. Therefore their size and limited coding capacity suggest that they may function as long noncoding RNAs (lncRNAs). We also confirmed that the ACRO138 repeats within the open chromatin peak at 138 kb are actively transcribed (Supplemental Fig. 21). These three transcripts all lie within the DJ inverted repeat arms and, together with another putative promoter in the ACRO138 repeat block centered at 297 kb, form a symmetrical arrangement of transcriptional units that mirror the inverted repeat structure. Therefore the DJ, rather than being a passive block of heterochromatin, shows a specific pattern of localization, a distinct genomic and chromatin organization, and transcriptional activity.
Figure 7.

DJ transcript profiling. (A) ChIP-seq reveals chromatin features consistent with transcription originating from promoters at 187 kb and 238 kb (boxed) in the DJ. Top four tracks are an enlargement of selected chromatin features from Figure 6A. Bottom two tracks show RNA-seq and cDNA mapping results. Exons are indicated by blocks. These identify spliced transcripts (disnor187 and disnor238) similar to cDNA clones AK026938 and BX647690. (B) RT-PCR using primers to detect disnor187 and disnor238 transcripts in HT1080 cells. Random and oligo(dT)-primed RT-PCR products of the expected sizes for spliced transcripts were produced.
Discussion
NORs were originally defined in 1934 as chromosomal regions that organize formation of the nucleolus (McClintock 1934). Now ∼80 yr later, we can begin to appreciate their genomic architecture. In this study, we have identified >550 kb of sequence from the regions flanking the rDNA array and have performed an in-depth characterization of these regions. We reveal that the sequences flanking the rDNA are conserved across all five acrocentric chromosomes and have a complex sequence feature composition.
Sequences proximal to the rDNA almost entirely consist of segmentally duplicated regions, like those surrounding centromeres (She et al. 2004), and thus are unlikely to contain nucleolus-specific functional elements. The high level of interchromosomal sequence conservation of the PJ, coupled with its high level of segmental duplication, strongly suggests that, far from being a recombinationally inert region of the genome, the PJ experiences frequent and ongoing recombination. This recombination appears to occur predominantly with other peri-/centromeric regions of the genome, implying colocalization of these regions, and may be responsible for Robertsonian translocations that appear to derive from the pericentromeric regions of acrocentric short arms (Therman et al. 1989) and are associated with genetic disorders. Our identification of the sequence of the PJ provides a means to investigate this.
In contrast, the DJ region is replete with unique sequences and displays evidence of functionality. We show that the DJ is localized to perinucleolar heterochromatin, where it appears to anchor the rDNA array to this region, and sequences located within the DJ are likely to be important for this localization. We propose that the DJ acts as a “control panel” for the entire NOR, where it can determine the transcriptional status of the linked rDNA array. In this way, the DJ may be involved in regulating the coalescence of nucleoli around individual NORs. Our model is that active NORs are localized to perinucleolar heterochromatin, where they form nucleoli, while inactive NORs lose this localization and form silent arrays that do not participate in nucleoli.
The sequences we describe begin to close one of the major remaining gaps in the human genome, the short arms of the acrocentric chromosomes, and their identification is an important step toward a complete understanding of nucleolar biology. The level of segmental duplication we observe, particularly of the PJ, suggests that some previously identified nucleolus-associated chromatin domains (NADs) may actually be segmental duplicates (Nemeth et al. 2010; van Koningsbruggen et al. 2010), and our sequences will allow a more refined picture of NADs to be developed. Additionally, the DJ sequence will allow researchers to use hybridization-based approaches to determine whether human nucleoli contain multiple NORs (Supplemental Videos 1, 2), something that has remained difficult to prove. The degree of heterogeneity in nucleolar morphology observed between cancers (Derenzini et al. 2009) suggests that mechanisms other than direct up-regulation of rDNA transcription have a role in the development of malignancy. As acrocentric short arms underpin both nucleolar form and function, we hypothesize that genetic alterations on these chromosome arms contribute to tumorigenesis and other human diseases. Therefore the sequences that we report here lay the foundations for addressing the roles that genetic and epigenetic changes in the DJ and PJ play in human disease, as well as providing a wealth of new tools for studying nucleolar biology.
Methods
Genomic cosmid and BAC clones
Acrocentric chromosome cosmid libraries LA13 NC01, LA14 NC01, LA15 NC01, LL21 NC02, and LL22 NC03 were obtained from the UK HGMP resource center. To obtain cosmids spanning the DJ, libraries were screened with a 638-bp PCR product generated using DJUf/DJUr primers (Worton et al. 1988). To obtain cosmids spanning the PJ, libraries were screened with a 220-bp PCR product generated using PJf/PJr primers (Sakai et al. 1995). Clone names for DJ and PJ cosmids identified and used in this study are shown in Supplemental Figure 4. We identified BAC clones in GenBank representing the DJ and PJ regions flanking the rDNA using BLAST. Cosmids LA14 138F10 and N 29M24 were used as the initial query sequences to search for BACs representing the DJ and PJ, respectively (Supplemental Methods). BAC clones were obtained from BACPAC Resources.
DNA sequencing and assembly
DNA sequencing of cosmid clones was performed through a combination of standard Sanger and next-generation sequencing (NGS). The sequence of the insert in cosmid LA14 138F10 and most of the insert in N 29M24 were determined by Sanger sequencing. The inserts of the remaining cosmids were end sequenced using Sanger sequencing and then subjected to NGS. Indexed libraries were prepared from individual cosmids using a Nextera DNA sample prep kit and Nextera barcodes (Epicentre NGS). NGS was performed on an Illumina Genome Analyzer IIx, using 54-bp singleton processing (Ambry Genetics). Sequences of cosmids were assembled using ABySS v1.2.7 (for parameters, see Supplemental Methods) (Simpson et al. 2009). Velvet v1.101 (Zerbino and Birney 2008) was used to refine the ABySS assemblies.
Cell lines
HeLa cells and HT1080 human fibrosarcoma cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS). RPE-1 cells were maintained in DMEM:F12 medium supplemented with 10% FBS, 2 mM L-glutamine, and 0.348% sodium bicarbonate. Mouse A9 cells containing individual human acrocentric chromosomes were previously described (Sullivan et al. 2001), and those containing X/21 reciprocal translocation products (GM09142 and GM10063) were obtained from Coriell Cell Repositories. To generate cells that contain ectopic DJ arrays, HT1080 cells were cotransfected using a standard calcium phosphate protocol with BACs AL592188, CT476834, and AC011841 together with a blasticidin selection marker in a 200:1 w/w ratio. Stable transfectants were maintained as above but supplemented with 5 μg/mL blasticidin.
FISH and 3D immuno-FISH and RNA FISH
Probes for FISH experiments were labeled using spectrum red or green dUTP (Abbott Molecular). For chromosome mapping experiments, slides of human normal male metaphase chromosome spreads (Applied Genetics) were denatured in 70% formamide/2× SSC for 5 min at 73°C. Slides were then dehydrated through a 70%–100% ethanol series, washed, and air dried. Denatured probe (50 ng/slide) combined with human COT-1 DNA (10 μg/slide) in 20 μL/slide Hybrisol VII (Qbiogene) was then added to the slides and allowed to hybridize for 24–48 h at 37°C in a humidified chamber. For CER satellite detection, hybridizations were performed with a 5′ FITC-labeled oligo and herring sperm DNA. Post-hybridization washes were 0.4× SSC/0.3% NP-40 for 2 min at 74°C followed by 2× SSC/0.1% NP-40 at ambient temperature for 1 min. Slides were air dried and mounted in Vectashield, including DAPI (Vector Laboratories). For 3D immuno-FISH experiments, cells were fixed, denatured, probed, and antibody stained as described previously (Mais et al. 2005; Prieto and McStay 2007). Z-stacks of fluorescent images were captured using a Photometric Coolsnap HQ camera and Volocity 5 imaging software (Improvision) with a 63 Plan Apochromat Zeiss objective mounted on a Zeiss Axioplan2 imaging microscope. In some cases, extended focus projections of deconvolved Z-stacks (iterative restoration) are presented; in other cases, individual focal planes are shown. Movies (Supplemental Videos 1, 2) were prepared from 3D images constructed from deconvolved Z-stacks using Volocity 6 (Improvision). 3D images were rotated to create a series of bookmarks. The movies are an animation of the transitions between these bookmarks.
Nucleolar DNA combing
Nucleoli, prepared from HeLa cells as previously described (Andersen et al. 2002), were resuspended at a concentration of 1 × 106 to 2 × 106 cell equivalents/mL in TE (10 mM Tris at pH 8.0, 1 mM EDTA). Resuspended nucleoli were mixed with an equal volume of 1% low melting point agarose in TE at 50°C. The mixture was pipetted into a plug mould (BioRad, 100 μL/slot). Embedded nucleoli were deproteinized, and encapsulated high-molecular-weight nucleolar DNA was combed onto silanized coverslips as previously described (Caburet et al. 2005) using a Molecular Combing apparatus supplied by Genomic Vision Paris. Coverslips were then hybridized with biotin-labeled DJ cosmid LA14 138F10 and a digoxygenin-labeled 5.8-kb EcoRI restriction fragment that contains 5′ ETS and 18S rRNA from human rDNA. Hybridization and detection were performed as described previously (Caburet et al. 2005).
Bioinformatic analyses
The bioinformatics pipeline that was used to identify potential junction regions from whole-genome sequence data is described in Supplemental Methods, as is the method for determining intra-/interchromosomal sequence identity between DJ and PJ clones.
Construction and analysis of DJ and PJ contigs
To construct the DJ, four BACs were merged (CT476837, CT476834, CU633906, and AC011841). The overlapping regions between BACs CT476837 and CT476834 and BACs CT476834 and CU633906 are 100% identical, but the identity decreases to ∼98% between CU633906 and AC011841 (Supplemental Fig. 7A). To form the PJ, two BACs (CR392039 and CR381535) with an identical overlapping region were merged to obtain a single contig (Supplemental Fig. 7B). The PJ is identical to that previously published (Lyle et al. 2007) and is deposited in GenBank under accession no. NT113958. The repeat content analysis method and the gene identification pipeline that utilizes gene prediction, mRNA, EST, and protein sequence data to identify potential DJ and PJ genes are both presented in Supplemental Methods. Segmental duplicates in the DJ and PJ contigs were detected using a modified BLAST-based detection scheme called the “whole genome assembly comparison” (Bailey et al. 2001). The human genome assembly (hg19) was broken into 400-kb pieces. Repeats in this fragmented human genome and in the DJ/PJ contigs were masked using RepeatMasker (Smit et al. 2010). DJ and PJ contigs were then matched to the fragmented masked genome using BLAST, with a cutoff of ≥85% identity >1 kb. Next, the repeats were reinserted into these matched sequences, and global alignments were created. All steps were performed using a series of Perl scripts (J. Bailey, University of Massachusetts Medical School). Low identity ends of the sequences were identified from the alignments and trimmed. Where the ends of two human genome fragments match a single region or where a fragment is interrupted by repeats, these fragments were merged together. The merged sequences were then aligned again to recalculate the identity.
ENCODE data
ChIP-seq data for 10 chromatin marks (CTCF, H3K4me1, H3K4me2, H3K4me3, H3K36me3, H3K9me3, H3K27me3, H3K9ac, H3K27ac, and H4K20me1) and input were obtained for seven different cell types (GM12878, H1-hESC, HMEC, HSMM, K562, NHEK, and NHLF) from ENCODE Broad Histone (Ernst et al. 2011). DNase-seq and FAIRE-seq data were obtained from ENCODE UNC/Duke (Song et al. 2011). PolyA tailed RNA-seq data were obtained for 11 different cell types (GM12878, H1-hESC, HCT-116, HeLa-S3, HepG2, HSMM, HUVEC, K562, MCF-7, NHEK, NHLF) from ENCODE Caltech RNA-seq. GenBank mRNAs and ESTs data were downloaded from the UCSC Genome Browser (Fujita et al. 2011) on January 1, 2012. These data were mapped to the human genome to which the DJ sequences had been added (Supplemental Methods).
Nucleolar ChIP and FAIRE
ChIP was performed on nucleolar chromatin isolated from HT1080 cells with H4K4me3 antibodies (Millipore, catalog no. 04-745) as described previously (Mais et al. 2005). We adapted a FAIRE protocol (Giresi et al. 2007) for cross-linked nucleolar chromatin. One hundred microliters of HT1080 nucleolar chromatin was extracted using an equal volume of phenol/chloroform. DNA was recovered from the aqueous phase by ethanol precipitation and resuspended in 100 μL of TE buffer. PCR was performed using 2 μL of recovered DNA. DNA recovered from input nucleolar chromatin served as a control for ChIP and FAIRE experiments.
Transcriptome profiling
Paired-end RNA-seq data from the 11 cell types was mapped to the human genome with DJ sequences added using TopHat (v1.2.0) (Trapnell et al. 2009) with mostly default parameters (-r 50 -a 8). We then merged the output alignments from all replicates using SAMtools (Li et al. 2009). The result is used as the input for Cufflinks (v1.3.0), with default parameters, to assemble the transcriptome of the custom genome. Finally we merged all 11 assembled transcriptomes, corresponding to the 11 cell types, using Cuffmerge (Trapnell et al. 2010) to obtain the final transcriptome. We also used this final transcriptome to estimate DJ transcript abundance. We used BLAT (Kent 2002) to map the mRNA and EST data to the DJ using the parameters “-fine -q=rna -minIdentity=95 -maxIntron=70000”, and “-minIdentity=97 -maxIntron=70000”, respectively.
Data access
The DJ contig nucleotide sequence and feature list are available in Supplemental Data 2 and 3. Assembled DJ and PJ cosmid sequences are available from GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession numbers KC876024–KC876030. Raw NGS data have been submitted to the Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/Traces/sra/) under accession number SRP024282.
Acknowledgments
B.M. acknowledges Science Foundation Ireland (PI grant 07/IN.1/B924) for funding work in his laboratory. A.R.D.G. acknowledges the Auckland Medical Research Foundation, the Marsden Fund, the Royal Society of NZ, and the Bio-Protection Research Centre for funding. C.S. thanks Science Foundation Ireland (07/SK/M1211b) for funding. T.T.N. received a PhD fellowship from the Irish Research Council for Science, Engineering and Technology. We also thank Samantha Raggett, Jane Wright, and Mark Anderson for help in library screening and DNA sequencing during the early stages of this work, and Kevin Sullivan and Carol Duffy for comments on the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.157941.113.
Freely available online through the Genome Research Open Access option.
References
- Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI 2002. Directed proteomic analysis of the human nucleolus. Curr Biol 12: 1–11 [DOI] [PubMed] [Google Scholar]
- Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE, Lamond AI, Mann M 2005. Nucleolar proteome dynamics. Nature 433: 77–83 [DOI] [PubMed] [Google Scholar]
- Bailey JA, Eichler EE 2006. Primate segmental duplications: Crucibles of evolution, diversity and disease. Nat Rev Genet 7: 552–564 [DOI] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE 2001. Segmental duplications: Organization and impact within the current human genome project assembly. Genome Res 11: 1005–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Simon A, Chiffaudel A, Croquette V, Heslot F, Bensimon D 1994. Alignment and sensitive detection of DNA by a moving interface. Science 265: 2096–2098 [DOI] [PubMed] [Google Scholar]
- Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI 2007. The multifunctional nucleolus. Nat Rev Mol Cell Biol 8: 574–585 [DOI] [PubMed] [Google Scholar]
- Budde A, Grummt I 1999. p53 represses ribosomal gene transcription. Oncogene 18: 1119–1124 [DOI] [PubMed] [Google Scholar]
- Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, et al. 2012. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 22: 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caburet S, Conti C, Schurra C, Lebofsky R, Edelstein SJ, Bensimon A 2005. Human ribosomal RNA gene arrays display a broad range of palindromic structures. Genome Res 15: 1079–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derenzini M, Montanaro L, Trere D 2009. What the nucleolus says to a tumour pathologist. Histopathology 54: 753–762 [DOI] [PubMed] [Google Scholar]
- Dundr M, Hoffmann-Rohrer U, Hu Q, Grummt I, Rothblum LI, Phair RD, Misteli T 2002. A kinetic framework for a mammalian RNA polymerase in vivo. Science 298: 1623–1626 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M 2012. ChromHMM: Automating chromatin-state discovery and characterization. Nat Methods 9: 215–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, et al. 2011. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res 39: D876–D882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganley AR, Ide S, Saka K, Kobayashi T 2009. The effect of replication initiation on gene amplification in the rDNA and its relationship to aging. Mol Cell 35: 683–693 [DOI] [PubMed] [Google Scholar]
- Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD 2007. FAIRE (formaldehyde-assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res 17: 877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez IL, Sylvester JE 1997. Beyond ribosomal DNA: On towards the telomere. Chromosoma 105: 431–437 [DOI] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ 2005. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol 7: 311–318 [DOI] [PubMed] [Google Scholar]
- Grummt I 2003. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev 17: 1691–1702 [DOI] [PubMed] [Google Scholar]
- Hadjiolov AA. 1985. The nucleolus and ribosome biogenesis. Springer-Verlag, New York. [Google Scholar]
- Hannan KM, Hannan RD, Smith SD, Jefferson LS, Lun M, Rothblum LI 2000. Rb and p130 regulate RNA polymerase I transcription: Rb disrupts the interaction between UBF and SL-1. Oncogene 19: 4988–4999 [DOI] [PubMed] [Google Scholar]
- Henderson AS, Warburton D, Atwood KC 1972. Location of ribosomal DNA in the human chromosome complement. Proc Natl Acad Sci 69: 3394–3398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Hernandez A, Soto-Reyes E, Ortiz R, Arriaga-Canon C, Echeverria-Martinez OM, Vazquez-Nin GH, Recillas-Targa F 2012. Changes of the nucleolus architecture in absence of the nuclear factor CTCF. Cytogenet Genome Res 136: 89–96 [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945 [DOI] [PubMed] [Google Scholar]
- Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467 [DOI] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT: The BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyle R, Prandini P, Osoegawa K, ten Hallers B, Humphray S, Zhu B, Eyras E, Castelo R, Bird CP, Gagos S, et al. 2007. Islands of euchromatin-like sequence and expressed polymorphic sequences within the short arm of human chromosome 21. Genome Res 17: 1690–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mais C, Wright JE, Prieto JL, Raggett SL, McStay B 2005. UBF-binding site arrays form pseudo-NORs and sequester the RNA polymerase I transcription machinery. Genes Dev 19: 50–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B 1934. The relationship of a particular chromosomal element to the development of the nucleoli in Zea Mays. Zeit Zellforsch Mik Anat 21: 294–328 [Google Scholar]
- McStay B, Grummt I 2008. The epigenetics of rRNA genes: From molecular to chromosome biology. Annu Rev Cell Dev Biol 24: 131–157 [DOI] [PubMed] [Google Scholar]
- Metzdorf R, Gottert E, Blin N 1988. A novel centromeric repetitive DNA from human chromosome 22. Chromosoma 97: 154–158 [DOI] [PubMed] [Google Scholar]
- Nemeth A, Langst G 2011. Genome organization in and around the nucleolus. Trends Genet 27: 149–156 [DOI] [PubMed] [Google Scholar]
- Nemeth A, Conesa A, Santoyo-Lopez J, Medina I, Montaner D, Peterfia B, Solovei I, Cremer T, Dopazo J, Langst G 2010. Initial genomics of the human nucleolus. PLoS Genet 6: e1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MOJE. 2011. The nucleolus. Springer, Berlin. [Google Scholar]
- Phillips JE, Corces VG 2009. CTCF: Master weaver of the genome. Cell 137: 1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pianese G 1896. Beitrag zur histologie und aetiologie der carcinoma. Histologische und experimentelle untersuchungen. Beitr Pathol Anat Allgem Pathol 142: 1–193 [Google Scholar]
- Piccini I, Ballarati L, Bassi C, Rocchi M, Marozzi A, Ginelli E, Meneveri R 2001. The structure of duplications on human acrocentric chromosome short arms derived by the analysis of 15p. Hum Genet 108: 467–477 [DOI] [PubMed] [Google Scholar]
- Prieto JL, McStay B 2007. Recruitment of factors linking transcription and processing of pre-rRNA to NOR chromatin is UBF-dependent and occurs independent of transcription in human cells. Genes Dev 21: 2041–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Ohta T, Minoshima S, Kudoh J, Wang Y, de Jong PJ, Shimizu N 1995. Human ribosomal RNA gene cluster: Identification of the proximal end containing a novel tandem repeat sequence. Genomics 26: 521–526 [DOI] [PubMed] [Google Scholar]
- Savino TM, Gebrane-Younes J, De Mey J, Sibarita JB, Hernandez-Verdun D 2001. Nucleolar assembly of the rRNA processing machinery in living cells. J Cell Biol 153: 1097–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmickel RD 1973. Quantitation of human ribosomal DNA: Hybridization of human DNA with ribosomal RNA for quantitation and fractionation. Pediatr Res 7: 5–12 [DOI] [PubMed] [Google Scholar]
- She X, Horvath JE, Jiang Z, Liu G, Furey TS, Christ L, Clark R, Graves T, Gulden CL, Alkan C, et al. 2004. The structure and evolution of centromeric transition regions within the human genome. Nature 430: 857–864 [DOI] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I 2009. ABySS: A parallel assembler for short read sequence data. Genome Res 19: 1117–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirri V, Urcuqui-Inchima S, Roussel P, Hernandez-Verdun D 2008. Nucleolus: The fascinating nuclear body. Histochem Cell Biol 129: 13–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P 2010. RepeatMasker.http://www.repeatmasker.org
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, et al. 2011. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res 21: 1757–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Pierce HH, Pierce AJ 2008. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res 18: 13–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan GJ, Bridger JM, Cuthbert AP, Newbold RF, Bickmore WA, McStay B 2001. Human acrocentric chromosomes with transcriptionally silent nucleolar organizer regions associate with nucleoli. EMBO J 20: 2867–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therman E, Susman B, Denniston C 1989. The nonrandom participation of human acrocentric chromosomes in Robertsonian translocations. Ann Hum Genet 53: 49–65 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL 2009. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Nobelen S, Rosa-Garrido M, Leers J, Heath H, Soochit W, Joosen L, Jonkers I, Demmers J, van der Reijden M, Torrano V, et al. 2010. CTCF regulates the local epigenetic state of ribosomal DNA repeats. Epigenetics Chromatin 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Koningsbruggen S, Gierlinski M, Schofield P, Martin D, Barton GJ, Ariyurek Y, den Dunnen JT, Lamond AI 2010. High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol Biol Cell 21: 3735–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Hwang ES, Amon A 1999. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature 398: 818–823 [DOI] [PubMed] [Google Scholar]
- Worton RG, Sutherland J, Sylvester JE, Willard HF, Bodrug S, Dube I, Duff C, Kean V, Ray PN, Schmickel RD 1988. Human ribosomal RNA genes: Orientation of the tandem array and conservation of the 5′ end. Science 239: 64–68 [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LF, Huynh KD, Lee JT 2007. Perinucleolar targeting of the inactive X during S phase: Evidence for a role in the maintenance of silencing. Cell 129: 693–706 [DOI] [PubMed] [Google Scholar]