

Abstract
Variation in protein output across the genome is controlled at several levels, but the relative contributions of different regulatory mechanisms remain poorly understood. Here, we obtained global measurements of decay and translation rates for mRNAs with alternative 3′ untranslated regions (3′ UTRs) in murine 3T3 cells. Distal tandem isoforms had slightly but significantly lower mRNA stability and greater translational efficiency than proximal isoforms on average. The diversity of alternative 3′ UTRs also enabled inference and evaluation of both positively and negatively acting cis-regulatory elements. The 3′ UTR elements with the greatest implied influence were microRNA complementary sites, which were associated with repression of 32% and 4% at the stability and translational levels, respectively. Nonetheless, both the decay and translation rates were highly correlated for proximal and distal 3′ UTR isoforms from the same genes, implying that in 3T3 cells, alternative 3′ UTR sequences play a surprisingly small regulatory role compared to other mRNA regions.
The sequence features of an mRNA constitute a platform for trans factors to bind and exert regulatory control over its localization, stability, and translational efficiency. The specific combination of cis-regulatory sequences present in an mRNA is determined through processing of the pre-mRNA into a mature mRNA composed of three DNA-encoded regions: the 5′ untranslated region (5′ UTR), the open reading frame (ORF), and the 3′ UTR.
At least three attributes of 3′ UTRs favor their utility for hosting binding sites for trans-regulatory factors. First, 3′ UTRs have fewer sequence and length constraints and, therefore, provide a more fertile context for the evolutionary emergence and maintenance of regulatory sites. Second, sites within 3′ UTRs are out of the path of the scanning/translating ribosome, which can displace regulatory factors before they exert their effects (Grimson et al. 2007). Third, because of mRNA circularization, sites within 3′ UTRs are near key points of regulation of both mRNA degradation and translation initiation. Hence, 3′ UTR–bound trans factors can affect the amount of mRNA translated via several mechanisms, including (1) mRNA deadenylation, decapping, and degradation, (2) endonucleolytic cleavage and degradation (e.g., by SMG6 in nonsense-mediated mRNA decay [Huntzinger et al. 2008; Eberle et al. 2009]), (3) partial deadenylation that reduces the amount of PABP bound to the poly(A) tail, which might decrease translation-promoting circularization (Weill et al. 2012), (4) disruption of cap-bound initiation factors (Jackson et al. 2010), and (5) changes in subcellular localization that sequester mRNAs away from ribosomes (Sonenberg and Hinnebusch 2009). For example, the effects of the AU-rich element (ARE)-binding protein ELAVL1 (formerly known as HuR) on mRNA stability are mediated through binding to 3′ UTRs (Lebedeva et al. 2011; Mukherjee et al. 2011). Most known binding sites of the PUF family of regulatory proteins are also in the 3′ UTR (Quenault et al. 2011), and cytoplasmic mRNA localization is most frequently mediated through cis-regulatory elements found in the 3′ UTR (Andreassi and Riccio 2009).
With these attributes, 3′ UTRs can be the major drivers of tissue-specific expression. This power of 3′ UTRs to regulate gene expression is illustrated in the Caenorhabditis elegans germline, in which reporter mRNAs controlled by 3′ UTR sequence, but not promoter sequence, recapitulate the cell-type–specific expression patterns of 24 different genes (Merritt et al. 2008). In murine fibroblasts, transcriptional regulation contributes substantially to differences in gene expression, but translational regulation is another major determinant, while mRNA and protein degradation rates are less important (Schwanhäusser et al. 2011). The degree to which this translational regulation is mediated through 3′ UTR elements is unknown.
Alternative cleavage and polyadenylation of transcripts from the same gene enables cell-type– or condition-specific expression of 3′ UTR isoforms, thereby adding another layer of post-transcriptional regulation. Analysis of expressed sequence tags (ESTs) suggests that a majority of mammalian genes contain multiple cleavage and polyadenylation sites (Tian et al. 2005), either in “tandem UTRs,” for which choice of an earlier site by the cleavage and polyadenylation machinery precludes use of later sites, or in alternative last exons, for which the proximal alternative site falls within an intron and thus can be spliced out of the primary transcript, allowing cleavage at the distal alternative site. Moreover, 3′ UTR isoform choice is itself regulated. A coordinated shift toward shorter tandem UTR isoforms is observed upon cell proliferation, and this pattern appears to generalize across tissues: the more highly proliferative a tissue, the shorter the average genome-wide 3′ UTR length (Sandberg et al. 2008), with the longest average 3′ UTR lengths occurring in the brain (Ramskold et al. 2009). The correlation between 3′ UTR length and cell proliferation is also observed across mouse embryonic development (Ji et al. 2009). Additionally, oncogenic transformation can sometimes favor 3′ UTR shortening (Mayr and Bartel 2009), although subsequent genome-wide analyses have suggested that UTR shortening is associated with transformation in only some cell lines (Shepard et al. 2011). A progressive lengthening of 3′ UTRs is also observed during zebrafish development, with particularly strong preference for longer isoforms in the brain (Li et al. 2012; Ulitsky et al. 2012).
The functional relevance of tandem UTR shortening remains unclear. An early genome-wide study suggested that genes with particularly long 3′ UTRs (>1 kb) had relatively short mRNA half-lives (Yang et al. 2003), but a subsequent higher-resolution study did not observe a significant relationship between 3′ UTR length and stability (Sharova et al. 2009). However, these genomic surveys relied on published genome annotations of 3′ UTRs and compared stabilities for mRNAs of different genes rather than stabilities of different mRNA isoforms from the same genes, leaving open the possibility that the observed effects (or lack thereof) were caused by attributes merely correlated with 3′ UTR length. Reporter genes have supported the destabilizing effect of longer UTRs. For a handful of genes, luciferase reporters fused to longer tandem 3′ UTR isoforms had decreased protein production compared with those fused to the shorter 3′ UTR isoforms (Sandberg et al. 2008; Mayr and Bartel 2009). Some of this down-regulation is due to mRNA destabilization of the longer isoform, as assayed by half-life measurements performed on three genes across several cell lines (Mayr and Bartel 2009). Whether shortened 3′ UTRs generally lead to increased mRNA stability and translational efficiency is still an open question.
Multiple methods have been developed to annotate or quantify cleavage and polyadenylation events globally using high-throughput sequencing (Ozsolak et al. 2010; Fu et al. 2011; Jan et al. 2011; Shepard et al. 2011; Hoque et al. 2012; Martin et al. 2012), most of which involve oligo(dT) priming off the poly(A) tail. Oligo(dT) priming does well at identifying the most distal poly(A) sites but for proximal sites yields some false positives resulting from internal priming off of A-rich regions coded within the mRNAs. The 3P-seq (poly[A]-position profiling by sequencing) method does not use oligo(dT) priming and thus lacks these internal-priming artifacts and the consequent false-positive annotation of proximal sites (Jan et al. 2011). At their 3′ end, 3P-seq tags typically include residues corresponding to a 3- to 7-nt fragment of the poly(A) tail (Fig. 1A). These untemplated adenosines provide an unambiguous marker of a true poly(A) site, thereby enabling confident annotation.
Figure 1.

Experimental methods used to assay alternative cleavage and polyadenylation. (A) 3P-seq (Jan et al. 2011). This method captures 3′ end mRNA fragments without using oligo(dT) priming and, therefore, is highly specific for true poly(A) sites. A bridge oligo (black) is used to ligate a biotinylated oligo (blue); reverse transcription primer (red); captured mRNA 3′ end fragment (gray); Illumina sequencing adapters (purple and green). (B) Poly(A)-primed sequencing, or 2P-seq. This simplified method is useful for quantifying usage of known poly(A) sites. (C) Reproducibility of 2P-seq measurements of isoform abundance across two biological replicates. Only isoforms with at least a single read in either replicate were considered, and a single pseudocount was added prior to taking the log of each value. (D) Correspondence between mRNA quantified using 2P-seq and RNA-seq data sets from 3T3 cells. A minimum of two tags or 0.1 RPKM was required for 2P-seq and RNA-seq, respectively, and pseudocounts of 1 and 0.1, respectively, were added prior to taking the log of each value.
To explore the role of alternative cleavage and polyadenylation on mRNA stability and translation, we first used 3P-seq to produce high-quality annotations of poly(A) sites in mouse fibroblasts. We then used a simpler poly(A)-tail-primed sequencing method called 2P-seq to quantify the mRNA half-life and translational efficiency of each expressed, annotated 3′ UTR isoform. mRNAs with shorter tandem UTR isoforms tended to be somewhat more stable than those with longer isoforms, and we identified known and novel regulatory motifs associated with stability differences. However, the differences in stability between short and long isoforms were small, with 3′ UTR sequences playing a surprisingly small role in modulating mRNA half-life when compared to attributes of the mRNA coding region. Likewise, differences between isoform-level translation rates were also generally small, with mRNA-specific translational regulation occurring primarily through features of the 5′ UTR and coding sequence.
Results
3P-seq annotation of murine poly(A) sites
To generate a reference set of poly(A) sites in mouse cells, we prepared 3P-seq libraries using RNA isolated from NIH 3T3 cells and embryonic stem cells (Fig. 1A). Of the 49 million sequencing reads, 20 million (41%) mapped uniquely to the mouse genome. These reads had an average of approximately four A's at their 3′ ends, as expected from incomplete RNase H digestion of the poly(A) tail (Jan et al. 2011). To the extent that these 3′-terminal A's did not match the genome, they provided a key indicator that the read originated from a true cleavage and polyadenylation site and was not a degradation product from within the body of the mRNA. The 10 million reads that contained at least two 3′-terminal A's were used to annotate a total of 33,590 poly(A) sites genome wide (Supplemental Fig. S1; Methods). The fraction of informative reads (51%) was comparable to that achieved using a modified 3P-seq protocol, despite the claim that the modification improves this fraction (Hoque et al. 2012). These included at least one site for 12,759 of 17,946 (71%) RefSeq genes (Pruitt et al. 2009) (using a strict definition of nonoverlapping genes). These 3P-seq–annotated poly(A) sites typically mapped near 3′ ends of RefSeq-annotated transcripts, and of those that were distant, more were upstream of RefSeq-annotated sites than downstream. The mean 3′ UTR length was 1209 nt, with a median of 775 nt. We used these 3P-seq–annotated poly(A) sites for all subsequent analyses.
2P-seq quantifies alternative cleavage and polyadenylation
Although 3P-seq has clear advantages for annotating alternative poly(A) sites (Jan et al. 2011) and can also be used to quantify the usage of these alternative sites (Ulitsky et al. 2012), the method is cumbersome compared to other methods. Therefore, we developed a more streamlined method, called poly(A)-primed sequencing, or 2P-seq, to more conveniently quantify the alternative usage of known 3′ UTR isoforms (Fig. 1B). For 2P-seq, poly(A)-selected mRNA is first fragmented by partial digestion with RNase T1, which does not cleave within the poly(A) tail. mRNA fragments are next reverse transcribed using a primer ending in a stretch of 20 T's that were followed by a VN anchor (N represents a fully degenerate base, and V is any base except for T), and the resulting cDNAs were circularized, PCR-amplified, and sequenced. The Illumina sequencing used a custom primer that ended with 20 T's, and thus sequencing began at the next non-A nucleotide of the template. Hence, for cDNAs primed at the poly(A) tail, the first sequenced base corresponded to the 3′ most non-A nucleotide of the mRNA.
Because 2P-seq uses oligo(dT) priming, it is susceptible to mispriming at genomically encoded A-rich regions of mRNAs. Indeed, genomic nucleotides immediately downstream from mapped 2P tags were significantly enriched for adenosine. However, this enrichment was not detected downstream from 2P tags mapping near to 3P-seq–annotated poly(A) sites (Supplemental Fig. S2). Therefore, all subsequent analyses were restricted to 2P tags mapping within 20 nt of 3P-seq–annotated poly(A) sites, a range that allowed for the considerable microheterogeneity observed at cleavage and poly(A) sites (Hart et al. 1985; Jan et al. 2011).
In mouse 3T3 cells, 2P-seq results for poly(A)-site usage correlated very highly when comparing two biological replicates (Pearson rp = 0.94, Spearman rs = 0.96) (Fig. 1C; Supplemental Fig. S3). 2P-seq results were also highly correlated with 3P-seq results (rs = 0.83) (Supplemental Fig. S3). Combining 2P tags from all poly(A) sites of each gene generated an mRNA expression profile that correlated well with the results of mRNA-seq (rp = 0.74, rs = 0.78) (Fig. 1D; Supplemental Fig. S3), which further validated the highly quantitative nature of 2P-seq. Finally, isoform quantification using a Northern blot agreed closely with that of 2P-seq, while demonstrating the increased resolution of 2P-seq for distinguishing isoforms (Supplemental Fig. S4).
ORF exon-junction density is most important for predicting mRNA stability
To explore the genome-wide effects of alternative cleavage and polyadenylation on mRNA stability, we treated 3T3 cells with actinomycin D to block transcription and prepared 2P-seq libraries over an 8-h time course, which provided a measure of the amount of each 3′ UTR isoform remaining at each time point, as illustrated for mRNAs from the Cstf1 gene (Fig. 2A,B). Half-lives for each 3′ UTR isoform were generated by fitting these data.
Figure 2.

mRNA and isoform half-life values, and modeling of associated mRNA features. (A) Schematic of approach used to measure half-lives of tandem 3′ UTR isoforms. Distal and proximal isoforms are illustrated for cleavage stimulation factor subunit 1 (Cstf1). For each time point after transcriptional arrest with actinomycin D, 2P tags are plotted for each isoform as well as a gene-level aggregate, showing the corresponding half-life estimates below. Tags for the 0 h (steady-state) replicates were averaged. (B) Fitted exponential degradation curves, shown for the same isoforms as in A. An additional normalization factor not considered here was used to correct for the slight decrease in total mRNA at later time points, resulting in slightly lower final half-lives of 0.52 h (proximal) and 1.20 h (distal). (C) Correlation between gene-based half-lives determined in 3T3 cells (2P-seq) with previous results in mouse embryonic stem cells (Sharova et al. 2009). (D) Correspondence between our measured half-lives and mRNA half-lives predicted using a model trained on a separate 50% of our data. For each gene with multiple tandem 3′ UTRs, only the most proximal isoform was used to quantitate gene half-life, although results were similar when averaging over all isoforms or using only the most distal isoform. (E) The correspondence between mRNA half-life and ORF exon-junction density. A maximum of 0.02 exons/bp was used for ORF exon-junction density, affecting seven of the 7028 genes. (F) Sequence features used to predict half-lives, arranged in approximate order of importance in the model. Adding more features resulted in diminishing returns, with a model including only the top three to six features performing nearly as well as any of the models with more features. Because of significant multicollinearity, features correlating well were grouped together (e.g., ORF length and ORF exon-junction density) with their pairwise correlation indicated (Pearson r). Also reported are each individual R2 with mRNA stability, and each cumulative R2 calculated for a linear model including all previous features. Predicted targeting for the top 10 miRNAs or for individual miRNAs was quantified using context scores (Grimson et al. 2007; Garcia et al. 2011). Error bars show 95% confidence intervals.
Two tests supported the ability of our 2P-seq experiment to measure half-lives accurately. First, the data set was split into two independent replicates, each with three time points (each derived from a different plate of cells), and isoform-specific half-lives calculated from these two biological replicates were compared and found to be in good agreement (rs = 0.76, n = 3192 isoforms) (Supplemental Fig. S5). Second, half-lives were calculated for each gene after combining tags mapping to all of its poly(A) sites, and these gene-level half-lives correlated well with stability values previously measured in 3T3 cells using metabolic labeling followed by sequencing (rp = 0.62) (Supplemental Table S1; Schwanhäusser et al. 2011). These values also correlated well with half-lives measured using actinomycin D and microarrays in mouse ES cells (rp = 0.68) (Fig. 2C; Sharova et al. 2009) and mouse neuroblastoma cells (rp = 0.70) (Clark et al. 2012). Indeed, these were among the best pairwise correlations observed between re-analyzed half-life data sets from different cell types (Supplemental Table S1), although biases of the actinomycin D and metabolic labeling approaches presumably influence these comparisons (Dolken et al. 2008).
In agreement with previous studies (Yang et al. 2003; Sharova et al. 2009), we found that the top gene ontology categories associated with high mRNA stability (top quartile of half-lives) were related to cellular metabolism and translation (Supplemental Table S2). The top gene ontology categories associated with low mRNA stability (bottom quartile) were related to transcription factors, cellular signaling pathways, and regulation of various cellular processes. These enrichments were significant even after controlling for the different steady-state mRNA levels observed between high- and low-stability gene sets.
To provide context for our novel intragenic analysis, in which we measured the influence of the different 3′ UTR isoforms of the same gene, we began with a more conventional inter-genic analysis, considering sequence determinants common to all tandem 3′ UTR isoforms, including in the 5′ UTR, ORF, and proximal 3′ UTR. A multivariate model of mRNA stability, which considered many potential sequence features, was trained on a subset of the data and then tested on the remaining data (Fig. 2D). Our model explained over one-third of the variance in half-lives between mRNAs of different genes (R2 = 0.39 using MARS, 95% confidence interval [0.389,0.409]; linear R2 = 0.35, 95% confidence interval [0.329,0.369]), a substantial improvement over the previously reported R2 value of 0.20 (Sharova et al. 2009). Our model still outperformed the previous model when trained and tested on previously published data sets (R2 range, 0.23–0.28) and when trained on our 3T3 data and tested on other data sets (R2 range, 0.21–0.23), which confirmed its generality. For comparison, the correlations between previously published half-life data sets ranged from R2 = 0.16 to R2 = 0.49 (Supplemental Table S1), suggesting that our model encompasses a large proportion of the universal, cell-type–independent determinants of message stability.
A single feature dominated our model of mRNA stability: The number of exon junctions per kilobase of coding sequence was positively associated with and accounted for 28% of the variation in mRNA half-lives (Fig. 2E). Exon-junction density has been previously observed to be correlated with stability, but the magnitude of this relationship (R2 = 0.11 [Sharova et al. 2009] and R2 = 0.15 [Clark et al. 2012]) was substantially lower than that observed using our data. Perhaps exon-junction density plays a larger role in determining half-life in 3T3 cells compared to either mES or neuroblastoma cells. Alternatively, the increased correlation might indicate reduced experimental noise in measuring half-lives or more stringent filtering for high-quality stability measurements.
Another important predictive feature was 3′ UTR length. As reported by an early study (Yang et al. 2003) but not a subsequent higher-resolution study (Sharova et al. 2009), 3′ UTR length was negatively associated with stability. It explained 9% of the variation in mRNA half-lives when tested individually, and increased the R2 from 0.28 to 0.32 when included in the full model (Fig. 2F). Some other sequence features correlated with mRNA stability individually but contributed only slightly to the predictive accuracy of the full model in the context of the other features (Fig. 2F). Sequence features such as target sites for a specific highly expressed microRNA (miRNA) occur more sparsely throughout the expressed mRNAs, which helps explain their limited predictive capabilities when considering all mRNAs.
Shorter 3′ UTR isoforms are slightly more stable
For three human genes examined over a number of cell lines, proximal 3′ UTR isoforms are substantially more stable than distal isoforms, typically by 1.5- to 6.6-fold (Mayr and Bartel 2009). To explore the extent to which these findings generalize to other genes in other species, we compared the half-lives of all tandem 3′ UTR isoforms detected in mouse 3T3 cells. A significantly larger number of genes had more stable proximal isoforms (n = 318) compared with the number of genes with more stable distal isoforms (n = 215, P = 9.4 × 10−6 binomial test) (Fig. 3A). Despite this detectable shift toward more stable proximal isoforms, the half-lives of the proximal and distal tandem UTR isoforms of individual genes correlated very well overall (rs = 0.74, rp = 0.72), and the correlation increased (to rs = 0.80, rp = 0.79) when restricting the analysis to the more highly expressed genes (top 50%), for which half-life estimates were the most robust. The high correlation between half-lives of tandem 3′ UTR isoforms persisted when splitting actinomycin D time points (each derived from a separate plate of cells) into independent biological replicates (rs = 0.66, rs = 0.67), as did the significant shift toward more stable proximal isoforms (P = 0.01, P = 6 × 10−4, binomial test) (Supplemental Fig. S5).
Figure 3.

Isoform-specific quantification of mRNA stability. (A) Relationship between half-lives of the adjacent proximal and distal tandem 3′ UTR isoforms' half-lives, highlighting the statistically significant differences (P < 0.05, colored points) and their tallies (colored numbers). (B) Cumulative distribution function of data shown in A. Plotted is the fraction of genes with half-life exceeding the value of the x-axis. The very slight shift toward more stable proximal tandem UTR isoforms is statistically significant (P = 4.0 × 10−3, Mann-Whitney test, n = 3463 tandem isoform pairs).
These results indicating that tandem 3′ UTR choice plays a relatively subtle role in regulating mRNA abundance under the conditions studied supported the previous finding that the primary determinant of mRNA stability is exon-junction density in the coding sequence (Fig. 2E; Sharova et al. 2009; Clark et al. 2012), which should be identical across all tandem 3′ UTR isoforms of each gene. Overall, the median proximal half-life was just 7.5% greater than the median distal half-life (Fig. 3B). This observation and our detection of more than 200 genes with more stable distal than proximal 3′ UTR isoforms suggest that stabilizing regulatory elements in 3′ UTRs (which could offset the effects of destabilizing elements) play a more prominent role than currently appreciated.
As an initial assessment of the biological roles of genes whose stability is regulated by alternative cleavage and polyadenylation, we performed gene ontology analysis, comparing genes with and without significant differences in stability between tandem UTR isoforms. Genes with destabilized distal UTR isoforms were enriched for cellular localization-related categories, including membrane-, adhesion-, and extracellular-associated GO terms (Supplemental Table S3). The mRNAs encoding proteins with these specific subcellular localizations are themselves often localized, with a distinctive distribution in subcellular fractions consistent with rough ER or plasma membrane location (Wang et al. 2012), raising the possibility that alternative isoforms might often contribute to mRNA localization.
Motifs associated with increased or decreased stability of alternative isoforms
Because tandem 3′ UTR isoforms typically share 5′ UTR and coding sequences, differences in stability should depend largely on the sequence unique to the distal 3′ UTR isoform. With this in mind, we searched the distal regions for motifs associated with increased or decreased stability of the distal isoform. To reduce the sequence space for this de novo motif search, all 8-nt sequences (allowing up to one degenerate position) were first ranked by their excess conservation in 3′ UTRs compared to control motifs. For the 901 most-conserved motifs (top 0.5%), all distal 3′ UTRs containing each k-mer were identified. Because requiring the presence of a specific motif selects for longer 3′ UTRs, non-motif-containing distal 3′ UTRs were sampled to match distal UTR lengths of the motif-containing UTRs. Using these length-matched controls, we tested whether the median differences in half-life between the proximal and distal isoforms were significantly greater or less for the motif-containing compared to the non-motif-containing sets.
The three motifs most significantly associated with destabilized distal isoforms perfectly matched (1) the relatively common consensus binding motif for the PUF family factors (Fig. 4A; White et al. 2001; Hafner et al. 2010); (2) a canonical UAUUUAUU AU-rich element (Barreau et al. 2005); and (3) a C-rich motif (Table 1; Neff et al. 2012). The fifth and tenth motifs on this list contained UG repeats, matching the preferred binding motif of CELF proteins, which are known to bind and destabilize mRNAs containing such motifs (Vlasova-St. Louis and Bohjanen 2011). Also in the top 20 were matches to let-7 (canonical 8-nt site) (Fig. 4B) and miR-29 (canonical 8-nt site with one N), the two most highly expressed miRNAs in 3T3 cells (Rissland et al. 2011), although these miRNA regulatory motifs were identified after the analysis had reached false discovery rates (Q) of 0.20 and 0.47, respectively (Table 1). Perhaps most intriguing were the motifs associated with stabilization, including the second-most-significantly associated motif, CNUAAUAA (Table 1), as stabilizing elements within 3′ UTRs have been far less characterized. A recent report identified several motifs associated with stabilization in mammalian cells as measured by metabolic labeling (Goodarzi et al. 2012). Neither of the top two motifs identified in their analysis was correlated with mRNA stabilization in our data set whether analyzed at the level of genes (P = 0.47 and 0.28 for sRSM1 and sRSM2, respectively) or isoforms (P = 0.21 and 0.75).
Figure 4.

Relationships between isoform half-life and either motifs or isoform accumulation. (A) Half-life differences for proximal versus distal tandem UTR isoforms containing a PUF-binding site in the distal region (red curve) or those for length-matched control tandem UTRs without the site (blue curve). Fold changes >0 indicate a more stable proximal isoform, and those <0 indicate a more stable distal isoform. The P-value reports a statistically significant difference between the two cumulative distributions (Mann-Whitney test). (B) Half-life differences for proximal versus distal tandem UTR isoforms with and without an 8-nt site for the let-7 miRNA in the distal region. Otherwise, as in A. (C) Half-life differences for tandem UTR isoforms with and without the top candidate stabilizing motif in the distal region. Otherwise, as in A. (D) Relationship between half-life and steady-state mRNA accumulation, calculated as the average over all 3′ UTR isoforms. mRNA expression levels were the average of two biological replicates. (E) Differences in proximal and distal tandem UTR half-lives compared to differences in their accumulation. On average, distal isoforms were slightly more highly expressed than were proximal isoforms, as indicated by the vertical line, but proximal isoforms were on average slightly more stable than were distal isoforms, as indicated by the horizontal line.
Table 1.
Top 20 motifs associated with stabilization or destabilization of tandem 3′ UTRs
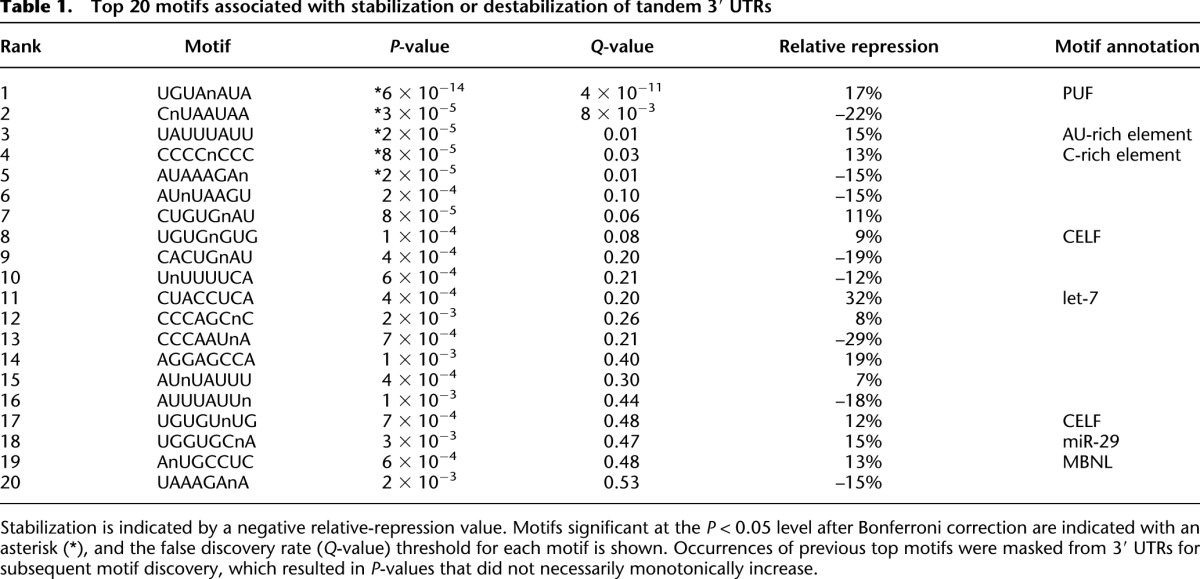
Although our unbiased motif search identified some known regulatory sequences and some interesting candidate elements, the magnitude of repression or derepression associated with each of these was modest, with the let-7 motif associated with the strongest regulation (32% repression) (Table 1). Encouraged by our success in modeling mRNA stability differences between genes (Fig. 2F), we attempted to model the differences between tandem UTR isoforms of the same gene, incorporating 3′ UTR sequence determinants (including PUF sites, miRNA sites, and AU-rich elements), 3′ UTR length (as well as changes in % length), and sequence conservation. The resulting model performed poorly (R2 < 0.01) (data not shown), which reinforced the notion that these elements play subtle roles in regulating transcriptome-wide mRNA stability.
mRNA steady-state levels are governed by the rates of both transcription and degradation, with transcription rates reported to be the more influential determinant in 3T3 cells (Schwanhäusser et al. 2011). Likewise, when comparing different genes, we found a significant but small positive correlation between mRNA half-life and steady-state levels, with an R2 = 0.06 (Fig. 4D), very similar to the previous estimate (Schwanhäusser et al. 2011). With respect to the tandem 3′ UTR isoforms for the same genes, if differential stability played a major role in determining their relative levels, we would have expected a strong positive correlation between the differences in mRNA levels and half-lives for isoform pairs. In fact, we found the opposite—a slight but statistically significant negative correlation (rp = –0.06, P = 2.3 × 10−4) (Fig. 4E). This correlation remained negative, albeit no longer statistically significant, after removing all of the most distal 3′ UTR isoforms, which tend to be the most highly expressed. Taken together, these results show that differential cleavage and polyadenylation are responsible for most of the differences in the steady state levels of tandem 3′ UTR isoforms, and that differential mRNA stability is only a minor contributor.
Proximal and distal tandem 3′ UTR isoforms are often translated similarly
After finding surprisingly little systematic difference with respect to mRNA stability (Fig. 3), we combined global polysome profiling (Arava et al. 2003) with 2P-seq to assess the contribution of isoform choice to translational efficiency. After arresting translating ribosomes with cycloheximide, cells were harvested, and cytoplasmic lysate was fractionated on a sucrose gradient (Fig. 5A). RNA from each of the six gradient fractions was then analyzed by 2P-seq. From these data the average number of ribosomes per mRNA isoform was calculated (Fig. 5B), normalizing to tag counts for yeast RNA that we had spiked into each fraction as it came off the gradient. As expected, isoforms of mRNAs with longer ORFs were generally bound by more ribosomes (rs = 0.44). Further evidence that our polysome fractionation accurately separated mRNAs based on relative translation came from the comparison of our translational efficiency values with those obtained by other studies. To calculate the translational efficiency for each gene, we combined the data for its different isoforms, and normalized for ORF length. These values correlated well with previous estimates derived from high-throughput proteomics (rs = 0.47) (Schwanhäusser et al. 2011) and correlated even better with those derived from ribosome footprint profiling (rs = 0.56) (Supplemental Fig. S6).
Figure 5.

Isoform-specific quantification of mRNA translational efficiency. (A) Polysome profile used to separate mRNAs into six fractions based on the number of bound ribosomes. Fractions are labeled by the average number of ribosomes bound per message. The two peaks in fraction 0 are the 60S and 80S ribosomal subunits. (B) Schematic of polysome-gradient sedimentation, illustrating calculation of translation rates for individual mRNA isoforms. In this example, the distal isoform has a greater average number of ribosomes bound (1.71 ribosomes/mRNA) compared to the proximal isoform (1.43 ribosomes/mRNA). Because isoforms share the same ORF, we did not normalize by ORF length when comparing values for tandem isoforms. (C) Polysome profiles for the DNA methyltransferase Dnmt1 mRNA, which has two tandem UTR isoforms colored red (proximal), and blue (distal). 2P-seq read counts were normalized to known amounts of yeast spike-in RNA. Translational efficiencies, calculated as the average number of ribosomes bound, are shown as vertical lines. (D) Comparison of translation rates for proximal and distal tandem 3′ UTR isoforms. Results for the 851 most highly expressed tandem 3′ UTR isoforms are shown in red, with results of the remainder shown in gray. Because tandem UTR isoforms presumably share the same ORF, translation rates were not divided by ORF length; doing so increased the correlation between proximal and distal isoforms but did not affect the tendency toward increased translation of the distal isoforms. (E) Translational efficiency differences for proximal versus distal tandem UTR isoforms with and without a 7-nt site for the let-7 miRNA in the distal region. Otherwise, as in Figure 4A.
Recent genome-wide studies of translation using either proteomics or ribosome footprint profiling provide relative translation rates averaged over all copies of an mRNA present in the cell. As with global polysome profiling, our complementary approach separated individual mRNAs based on the number of bound ribosomes. This enabled detection of cases in which a gene's mRNAs spanned distinct subpopulations, some highly translated, others less efficiently translated or sequestered away from ribosomes. For example, Dnmt1 mRNAs had a bimodal distribution across the gradient, with peaks at 2–3 and approximately 12 ribosomes per message, suggestive of just such a partitioning of its cellular mRNAs (Fig. 5C). In principle, our approach would also detect differences in elongation rates, with slower elongation resulting in more ribosomes bound per message and faster elongation resulting in fewer. However, with the exception of certain stress conditions (Liu et al. 2013; Shalgi et al. 2013), translation is not generally thought to be regulated at the elongation step. Therefore, we interpreted the isoform-specific differences in bound ribosomes as differences in translational efficiency, with fewer bound ribosomes presumably reflecting less translational initiation. This interpretation is analogous to those made previously in studies that use global ribosome profiling or ribosome footprint profiling to compare translational efficiency at the level of genes rather than isoforms (Arava et al. 2003; Hendrickson et al. 2009; Ingolia et al. 2009). Very few mRNA isoforms peaked in the premonosomal fraction, perhaps because ribosomes can continue to associate at start codons even after cycloheximide blocks translocation, but in any event, this phenomenon would increase the detected number of bound ribosomes by no more than one per mRNA.
Measuring the number of ribosomes at the mRNA level rather than at the gene level, when combined with 2P-seq, enabled comparison of the relative translational efficiencies for isoforms of the same gene. The plots for different tandem 3′ UTR isoforms of the same gene typically had the same shape and indicated strikingly similar translational efficiencies (Fig. 5C; Supplemental Fig. S7). When considering all genes for which we had translation data for tandem 3′ UTR isoforms, translational efficiencies of proximal and distal isoforms correlated highly (R2 = 0.58, n = 4298 tandem UTRs) (Fig. 5D). Moreover, this correlation improved markedly when restricting analysis to the most highly expressed genes (R2 = 0.81, n = 851) (Fig. 5D), suggesting that measurement imprecision or biological noise was responsible for much of the increased variation observed in the more inclusive analysis. We conclude that most translational regulation occurs through regions of the mRNA that are in common between the tandem isoforms, with relatively little variation in translational efficiency (<20%) remaining to be explained by a combination of measurement imprecision, noise, and elements unique to the distal isoforms.
In contrast to our results related to the stability of tandem 3′ UTR isoforms, we found a small but significant genome-wide bias toward higher translational efficiencies for longer tandem 3′ UTRs using the fullest set of tandem UTR isoforms (P < 10−8, n = 4298, binomial test) (Fig. 5D) or just the most highly expressed isoforms (P = 1 × 10−5, n = 851) (Fig. 5D). Median translation rates for proximal and distal isoforms differed by <3%. The increased translational efficiency of the distal isoform might be due to recruitment of trans factors to cis-regulatory motifs in the distal isoform, although a de novo motif search uncovered few candidate positive regulatory motifs (see below). Alternatively, increased 3′ UTR length might itself lead to an increase in translational efficiency.
Motifs associated with translational repression
We next searched for motifs associated with translational regulation, applying the same approach used to identify potentially stabilizing and destabilizing elements. Unlike the motifs associated with differential mRNA stability, the top motifs identified (12/12) were all associated with repression (Table 2). These included motifs that matched the let-7 (Q = 10−4) (Fig. 5E) and miR-29 sites, which were the first and the fifth most enriched motifs from this search, respectively. An AC-rich motif matching hnRNP L binding sites was the third most significant repressive motif, and hnRNP L has been reported to play a role in regulating translation of some mRNAs (Ray et al. 2009). Two 8-mers containing one or two occurrences of UGCU, the canonical binding motif of MBNL proteins (Goers et al. 2008), occurred lower down on the list, albeit with Q-values of 0.4 and 0.7. MBNL proteins regulate pre-mRNA splicing, but are also expressed cytoplasmically and have been implicated in control of mRNA localization and translation (Wang et al. 2012). The PUF motif was not found to be associated with differential translation (P = 0.52).
Table 2.
Top 20 motifs associated with increased or decreased translational efficiency
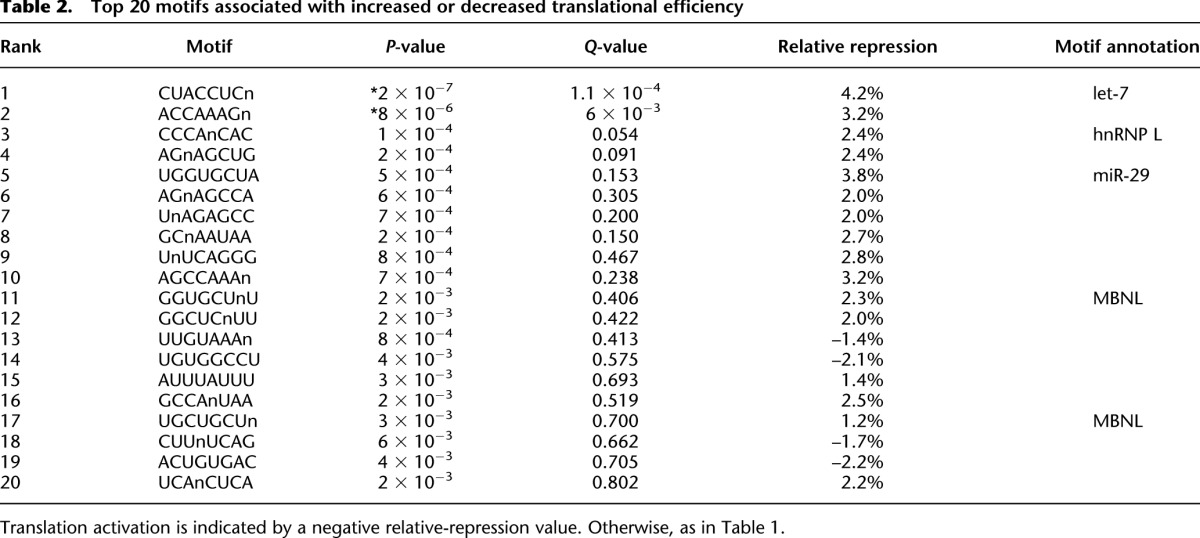
Although several motifs were highly significant in our de novo search, the magnitude of the change potentially mediated by the presence of each motif was even smaller than that for the motifs potentially regulating stability. For example, the implied relative translational repression averaged only 4% for the let-7 and miR-29 sites, even when focusing on only those UTRs with perfect 8-nt sites. Most prior measurements of global miRNA repression required comparison of miRNA-transfected or knockout cells to control cells. When evaluated at steady state, which allows the miRNAs to exert their full effects, those previous comparisons show that miRNAs predominantly act to destabilize mRNA targets, with smaller, albeit detectable, effects on translation (Baek et al. 2008; Guo et al. 2010). Our measurements of 4% translational repression and 30% mRNA repression, obtained without experimental perturbation of miRNAs, agree with these previous results. Altogether, our results suggest that 3′ UTRs play a minor role in regulating translational efficiency in 3T3 cells.
Discussion
Our large-scale analyses found that the choice of 3′ UTR isoform caused surprisingly modest changes in mRNA half-lives and translation rates in 3T3 cells. This result tempers the inferences that our labs had made previously when considering potential consequences of the preferential use of shorter isoforms in rapidly dividing or transformed cells (Sandberg et al. 2008; Mayr and Bartel 2009). Our previous studies observed relatively large differences in stability (Mayr and Bartel 2009) and translation (Sandberg et al. 2008; Mayr and Bartel 2009) when using luciferase assays or Northern blots to examine a handful of tandem 3′ UTR isoforms. However, the alternative isoforms previously chosen for experimental follow-up were selected based on their biological function and/or the differential presence of multiple sites to coexpressed miRNAs. For this reason and because the majority of distal UTR regions do not contain target sites for expressed miRNAs, these previously published results were not representative of all alternative isoforms.
When inspecting our current results, proximal isoforms were more highly translated for the two genes studied in Mayr and Bartel (2009) for which mouse homologs were evident. This observation is in qualitative agreement with this previous human study, although only one of these two genes showed increased stability of the proximal isoform. With respect to genes studied in Sandberg et al. (2008), we found poor agreement in overall expression differences between proximal and distal isoforms for four out of the five genes with similar isoforms expressed between that study and the current one. Differences between what we observed now and previously may be attributed to large differences in the cell types studied—3T3 fibroblasts in this study compared with mouse primary lymphocytes in Sandberg et al. (2008)—and to potential regulatory divergence of orthologs in human (Mayr and Bartel 2009) compared to mouse (this study). In summary, the previous experiments illustrate the potentially large consequences that 3′ UTR isoform choice can exert for selected genes in certain contexts. Our results concur, highlighting several hundred genes, or ∼15% of tandem UTR-containing genes, with significantly different stabilities (Fig. 3). However, we find that these large consequences are not the norm in 3T3 cells.
Although our major finding from the current large-scale study might be considered “negative” in the sense that, when comparing isoforms from the same gene, we usually did not observe the large differences in stability and translation that might have been expected, we emphasize that this negative finding is not subject to the caveats normally associated with a negative experimental result. This is because our negative finding was the outcome of positive experimental results, i.e., the strikingly strong and significant correlations that we observed between the short and long isoforms, in terms of both their stability (Fig. 3A) and their translation (Fig. 5D). To the extent that our methods were not perfect, they underreported the correlation between the short and long isoforms and thus, if anything, overestimated (not underestimated) the impact of alternative 3′ UTR isoforms on translation efficiency/mRNA stability. This being said, the strong correlations observed demonstrate that 2P-seq was in fact quite accurate for measuring isoform abundance. Because the ends for different mRNA isoforms have different sequences and reside on different molecules, 2P-seq provided essentially independent measurements for each isoform. Thus, for each gene with tandem UTR isoforms, it independently measured the distal isoform and independently measured each of the proximal isoforms. The striking correlations that we observe between these independent measurements for the long and short isoforms, in terms of both their half-lives (Fig. 3A) and their translation efficiencies (Fig. 5D), proved that 2P-seq was accurate, since any error in these measurements would tend to reduce these correlations.
It is formally possible that unknown systematic biases in the 2P-seq method somehow increase the similarity in expression measured for isoforms from the same gene. However, given the high correspondence in gene quantification compared to RNA-seq, the high correspondence in isoform quantification between 2P-seq and 3P-seq, the good agreement with isoform quantification by Northern blot, and the high correspondence in measured half-lives compared to previous data sets, the 2P-seq method appears to be both highly precise as well as accurate in quantifying 3′ UTR isoforms.
Less than 35% of all 3′ UTR sequence is common across all tandem UTR isoforms expressed in 3T3 cells. With no reason to suspect that regulatory elements more densely populate the regions of the UTRs shared between isoforms, the surprisingly small amount of stability and translational regulation imparted by distal isoforms can be extrapolated to the remainder of the 3′ UTRs to imply that most regulation of message stability and translation in 3T3 cells occurs through features of other mRNA regions, i.e., either the 5′ UTRs or ORFs. This conclusion is surprising when considering that 3′ UTRs comprise nearly one-third of all mRNA sequence and, as outlined in our introduction, have features expected to provide a highly favorable location for post-transcriptional regulatory elements.
Our quantitative modeling also indicated a secondary role for 3′ UTRs in regulating mRNA stability in 3T3 cells, with ORF exon-junction density acting as the primary predictive factor. Exon junction complexes (EJCs) are deposited on mRNAs during cotranscriptional splicing and serve to identify premature stop codons, triggering nonsense-mediated decay (Schoenberg and Maquat 2012), and can also function in mRNA localization (Ghosh et al. 2012). Perhaps the stabilizing effect of exon junctions is also mediated through EJCs or through other protein factors such as SR proteins that are deposited near exon junctions in the nucleus (Singh et al. 2012). This stabilization might also involve translation, because the association is strongest when using exon-junction density in the ORF rather than over the entire mRNA. However, it is difficult to envision how this stabilizing effect could be retained after the pioneer round of translation, during which these factors would be displaced from the mRNA by the translating ribosome.
Four models could explain the abundance and sequence conservation of alternative 3′ UTR isoforms despite the apparently subtle differences in stability and translational regulation imparted by these isoforms in 3T3 cells. (1) Perhaps most of these regulatory differences are biologically important despite their small magnitudes. (2) Perhaps most of these differences are not important, but a small subset (perhaps preferentially those with greatest magnitudes) are biologically important. In this scenario, emergence of a tandem 3′ UTR isoform would typically lead to little change in protein output and essentially no change in evolutionary fitness. Such a scenario would allow the widespread acquisition of many tandem isoforms per gene but would not explain their sequence conservation. (3) Perhaps the regulatory differences imparted by tandem UTR isoforms in 3T3 fibroblasts are magnified in other cell types or conditions. We suspect that our results will generalize to most mammalian cells in culture and perhaps to cells of most adult tissues, for which robust transcriptional regulation might dominate, thereby providing less need for 3′ UTR-mediated differences in post-transcriptional gene regulation. For other cells, however, regulation occurs in the absence of robust transcription (e.g., in the early embryo, in red blood cells, or under certain stress conditions) or far from transcription (e.g., at neuronal synapses); these cells might rely more heavily on differences in stability or translation of alternate 3′ UTR isoforms. (4) Perhaps alternative 3′ UTR isoforms primarily serve to regulate other aspects of mRNA function such as mRNA localization. Localized translation of mRNAs is important in a wide variety of cell types, and the cis-regulatory elements implicated in mRNA localization have most often been found in 3′ UTRs (Andreassi and Riccio 2009). To distinguish between these models and to evaluate the spatial and temporal conservation of gene regulatory processes operating through alternative 3′ UTR isoforms and their magnitudes in other contexts, global quantitative approaches like that described here could be performed in other cell types and conditions, perhaps augmented by fractionation approaches to detect differences in subcellular localization (Wang et al. 2012).
By globally comparing stability and translation of 3′ UTR isoforms, we were able to estimate the relative contributions of these two post-transcriptional regulatory modes and simultaneously quantify the amount of repression or derepression associated with many conserved cis-regulatory elements. Compared to other global approaches, ours more conveniently and potentially more accurately measures both the absolute and relative amount of regulation associated with each potential element. The convenience and accuracy came from our ability to simultaneously evaluate all types of regulatory elements, without requiring knockdown, knockout, or overexpression of the trans factors and the associated indirect effects that can result. An advantage of the alternative approach involving trans-factor perturbation is that factors can more confidently be assigned to their corresponding regulatory elements, whereas our approach has the advantage of identifying and evaluating the relative importance of candidate elements for trans factors that have yet to be characterized. Both our approach and trans-factor perturbation provide associations that speak to the function of the elements, whereas complementary biochemical approaches, such as those involving crosslinking, provide important information regarding factor occupancy.
Our de novo searches revealed multiple motifs significantly associated with mRNA destabilization and/or translational repression. The accuracy of our results was illustrated by our ability to identify expected cis-regulatory motifs, including target sites for Pumilio and the two most highly expressed miRNAs in mouse fibroblasts. We also discovered several novel motifs associated with stabilization of distal 3′ UTR isoforms. Competitive binding to otherwise destabilizing cis motifs, such as miRNA sites (Kedde et al. 2007) and AU-rich elements (Barreau et al. 2005), has been reported. However, little is known about cis motifs that act independently to stabilize mRNAs—an exception being motifs that mediate cytoplasmic polyadenylation, which occurs during oocyte maturation and early embryonic development and in some other settings (Villalba et al. 2011). Although further experiments are required to validate our candidate mRNA stabilizing elements, their presence could help counteract the effects of known destabilizing elements and thereby explain the small average difference in stability of tandem 3′ UTR isoforms.
Methods
Cell culture
Mouse NIH 3T3 cells were plated at a density of 1 to 2 × 106 cells per 15-cm plate and allowed to grow for ∼48 h before being used for downstream experiments. These conditions allowed a substantial number of cells to be harvested from each plate while preventing large-scale contact inhibition. 3P-seq experiments were performed as previously published (Jan et al. 2011) using 60 μg of total 3T3 RNA and 16 cycles of PCR amplification.
Annotation of murine polyadenylation sites
Mouse 3T3 3P-seq data were used to annotate cleavage and polyadenylation sites as previously described in Jan et al. (2011), with the following modifications: (1) Because of the larger 3′ UTR size and lower gene density in mammals, RNA-seq coverage was not required to connect potential poly(A) sites to annotated gene regions; (2) tags within a ±20-nt window were consolidated into a single poly(A)-site annotation; (3) to define an alternative isoform, tags supporting the poly(A) site were required to constitute ≥10% of all the tags with 3′-terminal adenosines that mapped to the genic region; (4) in order to ensure that non-genome-matching A's were not the result of sequencing errors, each cluster was required to include (1) at least one tag with greater than or equal to four 3′-terminal A's, (2) two unique tags with non-genome-matching 3′-terminal A's (NGAs), and (3) a minimum percentage (10%) of the total NGA tags mapping to the genic region, or three unique NGA tags, at least one of which contained multiple NGAs. Python scripts used to annotate poly(A) sites from 3P-seq data are available online at http://bitbucket.org/nspies/buildpolyaclusters.
Stability measurements and 2P-seq
Actinomycin D was added to plates at a final concentration of 10 μg/mL, and cells were harvested by trypsinization and TRIzol extraction. Approximately 15 μg of total RNA for each sample were prepared for sequencing using 2P-seq. Briefly, poly(A)+ RNA was selected using oligo dT25 beads (Dynal) and eluted directly into 25 μl of RNase T1 sequence buffer. RNase digestion was performed for 20 min at 22°C using 0.5 U RNase T1 (biochemistry grade; Ambion) and 1 μl glycoblue (15 mg/mL). The reaction was quenched and precipitated using the T1 Precipitation/Inactivation Buffer. Partially digested RNA was reverse transcribed (20-μl reaction, 30 min at 48°C, with 250 nM 2P-RT primer, 200 units SuperScript III Reverse Transcriptase; Invitrogen), then base hydrolyzed. Single-stranded cDNA was purified on a denaturing polyacrylamide gel, then circularized (20-μl reaction, 2 h at 60°C, with 100 units CircLigase II; EpiBio). CircLigase was heat-inactivated for 10 min at 80°C. Circularized cDNA was amplified by high-fidelity PCR for 12–15 cycles using barcoded PCR primers, then gel-purified and sequenced using a custom primer (2P-seq-PE1.1). A detailed, step-by-step protocol is included in the Supplemental Material.
Half-life estimation was performed by fitting a nonlinear exponential curve to the library-size-normalized read counts for tactD = 0,0,1,2,4,8 (two biological replicates at t = 0) using SciPy (http://www.scipy.org/). Significance of the differences in half-lives between isoforms was assessed using a t-test comparing values and variances of the exponential parameters; we found this standard approach closely matched the bootstrap P-values and was far more computationally efficient.
Translation measurements
Cells were incubated in the presence of 10 μg/mL cycloheximide for 5–10 min, and all subsequent solutions included cycloheximide at this concentration. Cells were harvested by trypsinization, then quickly washed and lysed using cold solutions. Lysate was spun on a 10-mL, 10%–50% sucrose gradient for 2 h, then separated using a gradient master (Biocomp). Fractions were manually defined according to the A280 peaks indicative of ribosomal subunits or mono- and polyribosomes. Approximately 200 ng Saccharomyces castellii total RNA was added to each fraction immediately following fractionation, and then RNA was prepared by GuHCl extraction. Approximately 5–20 μg/sample was then prepared for sequencing using the 2P-seq protocol and 12–16 PCR cycles.
Stability models
Multiple linear regression was performed using the core R packages, and nonlinear regression was performed using Multivariate Adaptive Regression Splines (MARS), using the earth package for R (Milborrow 2011). Both dependent and independent variables were log-transformed prior to regression analysis. miRNA targeting was quantitatively predicted for each mRNA using TargetScan context scores (Grimson et al. 2007). Because context scores are negative, and a lower negative number indicates greater predicted repression, the inverse of the context score was used to reflect the predicted repressive effects of miRNA targeting. For miRNAs with multiple expressed family members, the most highly expressed member was chosen to calculate supplemental pairing. AU-rich elements were scored by simple GC content as well as using AREScore (Spasic et al. 2012). The pumilio binding motif was defined as UGUAnAUA (Hafner et al. 2010).
Motif analysis
To reduce the search space for this analysis, enabling the statistical analysis of 7- and 8-nt motifs, we ranked all k-mers by their excess conservation in 3′ UTRs. For a given k-mer at a given position in the genome, conservation was defined as the total branch-length across all species containing the exact motif in the full-genome MULTIZ 30-way vertebrate alignment (Blanchette et al. 2004; Kheradpour et al. 2007). Evolutionary conservation above background was calculated by a hypergeometric test of the conserved and nonconserved occurrences of a given motif, relative to a cohort of shuffled motifs, keeping the number of CpG dinucleotides constant. The top 901 (0.5%) most highly conserved motifs were used as candidates for subsequent analysis.
Stability- and translation-associated motifs were identified by comparing the half-lives or translation rates of tandem 3′ UTRs containing a candidate motif to control tandem 3′ UTRs. Non-motif-containing tandem UTRs were sampled with replacement to closely match the 3′ UTR length distribution of motif-containing UTRs. A Mann-Whitney U-test was used to assess the significance of the difference in delta-stability (or delta-translation) between the motif-containing and control tandem UTRs. To increase the number of tandem UTRs, all adjacent pairs of tandem UTRs were used from genes containing more than 2 UTR isoforms (that is, individual comparisons of isoforms 1 and 2, 2 and 3, 3 and 4, etc., were performed) after applying a tag threshold of 10 reads in each 0-h (actinomycin D control) sample for each isoform to ensure reliable measurements.
The relative repression of a motif was calculated as the amount of repression for motif-containing tandem UTRs compared to the repression in control tandem UTRs:
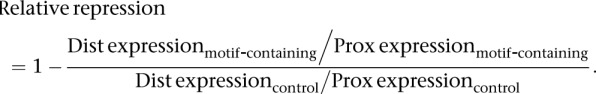 |
For example, control distal isoforms are 94% as stable as control proximal isoforms, compared to let-7–containing distal isoforms, which are 64% as stable as the paired proximal isoforms, yielding a relative repression of 1 – (64%/94%) = 32%. The effects of miRNA targeting were evaluated by using either the experimentally defined motifs (see Tables 1, 2) or the full 8-nt seed-matched sites, which are generally most responsive to the miRNA (Grimson et al. 2007; Baek et al. 2008).
Gene ontology analysis
A custom GO analysis was performed using annotations from the Gene Ontology Consortium (Ashburner et al. 2000). P-values were calculated using a hypergeometric test.
Data access
Raw 2P-seq and 3P-seq sequencing data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlh.nih.gov/geo/) under accession number GSE44698. Murine poly(A) site annotations and isoform-level stability and translation data are available in Supplemental Table S4.
Acknowledgments
We thank W. Gilbert and P. Vaidyanathan for technical assistance with polysome fractionation, C. Jan for mouse embryonic stem cell 3P-seq data, V. Butty for use of RNA-seq data, S. Eichhorn for ribosome profiling data, J. Stefano for experimental assistance, and A. Subtelny, J. Arribere, E. Wang, V. Agarwal, and other members of the Burge and Bartel laboratories for comments and suggestions. This work was supported by NIH grant GM067031 (D.P.B.) and GM085319 (C.B.B.). D.P.B. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.156919.113.
References
- Andreassi C, Riccio A 2009. To localize or not to localize: mRNA fate is in 3′UTR ends. Trends Cell Biol 19: 465–474 [DOI] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D 2003. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci 100: 3889–3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP 2008. The impact of microRNAs on protein output. Nature 455: 64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreau C, Paillard L, Osborne HB 2005. AU-rich elements and associated factors: Are there unifying principles? Nucleic Acids Res 33: 7138–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MB, Johnston RL, Inostroza-Ponta M, Fox AH, Fortini E, Moscato P, Dinger ME, Mattick JS 2012. Genome-wide analysis of long noncoding RNA stability. Genome Res 22: 885–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolken L, Ruzsics Z, Radle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, et al. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14: 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH 2009. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16: 49–55 [DOI] [PubMed] [Google Scholar]
- Fu Y, Sun Y, Li Y, Li J, Rao X, Chen C, Xu A 2011. Differential genome-wide profiling of tandem 3′ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res 21: 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP 2011. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol 18: 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Marchand V, Gaspar I, Ephrussi A 2012. Control of RNP motility and localization by a splicing-dependent structure in oskar mRNA. Nat Struct Mol Biol 19: 441–449 [DOI] [PubMed] [Google Scholar]
- Goers ES, Voelker RB, Gates DP, Berglund JA 2008. RNA binding specificity of Drosophila muscleblind. Biochemistry 47: 7284–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Najafabadi HS, Oikonomou P, Greco TM, Fish L, Salavati R, Cristea IM, Tavazoie S 2012. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature 485: 264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP 2007. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol Cell 27: 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart RP, McDevitt MA, Ali H, Nevins JR 1985. Definition of essential sequences and functional equivalence of elements downstream of the adenovirus E2A and the early simian virus 40 polyadenylation sites. Mol Cell Biol 5: 2975–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, Brown PO 2009. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol 7: e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, Tian B 2012. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat Methods 10: 133–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E 2008. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14: 2609–2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Friedman RC, Ruby JG, Bartel DP 2011. Formation, regulation and evolution of Caenorhabditis elegans 3′ UTRs. Nature 469: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Lee JY, Pan Z, Jiang B, Tian B 2009. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc Natl Acad Sci 106: 7028–7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JA, Slanchev K, le Sage C, Nagel R, Voorhoeve PM, van Duijse J, Orom UA, et al. 2007. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131: 1273–1286 [DOI] [PubMed] [Google Scholar]
- Kheradpour P, Stark A, Roy S, Kellis M 2007. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res 17: 1919–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedeva S, Jens M, Theil K, Schwanhausser B, Selbach M, Landthaler M, Rajewsky N 2011. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell 43: 340–352 [DOI] [PubMed] [Google Scholar]
- Li Y, Sun Y, Fu Y, Li M, Huang G, Zhang C, Liang J, Huang S, Shen G, Yuan S, et al. 2012. Dynamic landscape of tandem 3′ UTRs during zebrafish development. Genome Res 22: 1899–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Han Y, Qian SB 2013. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell 49: 453–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, Zavolan M 2012. Genome-wide analysis of pre-mRNA 3′ end processing reveals a decisive role of human cleavage factor I in the regulation of 3′ UTR length. Cell Rep 1: 753–763 [DOI] [PubMed] [Google Scholar]
- Mayr C, Bartel DP 2009. Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt C, Rasoloson D, Ko D, Seydoux G 2008. 3′ UTRs are the primary regulators of gene expression in the C. elegans germline. Curr Biol 18: 1476–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milborrow S 2011. Multivariate adaptive regression spline models. http://www.milbo.users.sonic.net/earth [Google Scholar]
- Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr, Tuschl T, Ohler U, Keene JD 2011. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell 43: 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff AT, Lee JY, Wilusz J, Tian B, Wilusz CJ 2012. Global analysis reveals multiple pathways for unique regulation of mRNA decay in induced pluripotent stem cells. Genome Res 22: 1457–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM 2010. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143: 1018–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Klimke W, Maglott DR 2009. NCBI reference sequences: Current status, policy and new initiatives. Nucleic Acids Res 37: D32–D36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quenault T, Lithgow T, Traven A 2011. PUF proteins: Repression, activation and mRNA localization. Trends Cell Biol 21: 104–112 [DOI] [PubMed] [Google Scholar]
- Ramskold D, Wang ET, Burge CB, Sandberg R 2009. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput Biol 5: e1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL 2009. A stress-responsive RNA switch regulates VEGFA expression. Nature 457: 915–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Hong SJ, Bartel DP 2011. MicroRNA destabilization enables dynamic regulation of the miR-16 family in response to cell-cycle changes. Mol Cell 43: 993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Neilson JR, Sarma A, Sharp PA, Burge CB 2008. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg DR, Maquat LE 2012. Regulation of cytoplasmic mRNA decay. Nat Rev Genet 13: 246–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M 2011. Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Krykbaeva I, Taipale M, Lindquist S, Burge CB 2013. Widespread regulation of translation by elongation pausing in heat shock. Mol Cell 49: 439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MSH 2009. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res 16: 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi E-A, Lu J, Flanagan LA, Hertel KJ, Shi Y 2011. Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17: 761–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ 2012. The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151: 750–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG 2009. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 136: 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spasic M, Friedel CC, Schott J, Kreth J, Leppek K, Hofmann S, Ozgur S, Stoecklin G 2012. Genome-wide assessment of AU-rich elements by the AREScore algorithm. PLoS Genet 8: e1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Hu J, Zhang H, Lutz CS 2005. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res 33: 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Shkumatava A, Jan CH, Subtelny AO, Koppstein D, Bell GW, Sive H, Bartel DP 2012. Extensive alternative polyadenylation during zebrafish development. Genome Res 22: 2054–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba A, Coll O, Gebauer F 2011. Cytoplasmic polyadenylation and translational control. Curr Opin Genet Dev 21: 452–457 [DOI] [PubMed] [Google Scholar]
- Vlasova-St Louis I, Bohjanen PR 2011. Coordinate regulation of mRNA decay networks by GU-rich elements and CELF1. Curr Opin Genet Dev 21: 444–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. 2012. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 150: 710–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill L, Belloc E, Bava F-A, Mendez R 2012. Translational control by changes in poly(A) tail length: Recycling mRNAs. Nat Struct Mol Biol 19: 577–585 [DOI] [PubMed] [Google Scholar]
- White EK, Moore-Jarrett T, Ruley HE 2001. PUM2, a novel murine puf protein, and its consensus RNA-binding site. RNA 7: 1855–1866 [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE 2003. Decay rates of human mRNAs: Correlation with functional characteristics and sequence attributes. Genome Res 13: 1863–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]