

Abstract
While landmark studies have shown that microbiota activate and educate host immunity, how immune systems shape microbiomes and contribute to disease is incompletely characterized. Primary immunodeficiency (PID) patients suffer recurrent microbial infections, providing a unique opportunity to address this issue. To investigate the potential influence of host immunity on the skin microbiome, we examined skin microbiomes in patients with rare monogenic PIDs: hyper-IgE (STAT3-deficient), Wiskott-Aldrich, and dedicator of cytokinesis 8 syndromes. While specific immunologic defects differ, a shared hallmark is atopic dermatitis (AD)–like eczema. We compared bacterial and fungal skin microbiomes (41 PID, 13 AD, 49 healthy controls) at four clinically relevant sites representing the major skin microenvironments. PID skin displayed increased ecological permissiveness with altered population structures, decreased site specificity and temporal stability, and colonization with microbial species not observed in controls, including Clostridium species and Serratia marcescens. Elevated fungal diversity and increased representation of opportunistic fungi (Candida, Aspergillus) supported increased PID skin permissiveness, suggesting that skin may serve as a reservoir for the recurrent fungal infections observed in these patients. The overarching theme of increased ecological permissiveness in PID skin was counterbalanced by the maintenance of a phylum barrier in which colonization remained restricted to typical human-associated phyla. Clinical parameters, including markers of disease severity, were positively correlated with prevalence of Staphylococcus, Corynebacterium, and other less abundant taxa. This study examines differences in microbial colonization and community stability in PID skin and informs our understanding of host–microbiome interactions, suggesting a bidirectional dialogue between skin commensals and the host organism.
Although the relationships between microbiota, host, and disease are complex, investigators have long relied upon Koch's postulates to impute causation between microorganism and disease. However, the requirement that disease-causing microorganisms uniformly recapitulate disease in healthy individuals was compromised when Koch discovered asymptomatic carriers of Vibrio cholera and Salmonella typhi, documenting the important distinction between apparent colonization and infection. Potentially pathogenic bacteria that colonize humans are influenced not only by host factors, but also by their native microbial community. This paradigm requires that diseases of microbial origin must be investigated within the context of their microbial community, host factors, and immunity.
Genome-wide surveys of the microbes inhabiting the different microenvironments of the human body (e.g., gut, skin, oral) (The Human Microbiome Project Consortium 2012), model organisms (Chung et al. 2012), and geographic regions (Yatsunenko et al. 2012) have suggested the presence of strong selective control on the microbial communities that can colonize and persist in particular environments. The identity and relative representation of microbes at a given site, the overall microbial density (bioburden) and diversity are all components that can be constrained by extrinsic or intrinsic factors, such as nutrient accessibility, temperature, oxygen, or products produced by other microbes.
Innovations in sequencing technology have enabled explorations of the complexity of the vast human-associated microbiota. Recent reports have examined shifts in microbial species and communities associated with diseases as varied as atopic dermatitis, inflammatory bowel disease, colorectal cancer, obesity, and metabolic syndrome (Balter 2012). Microbes also promote human health by performing vital functions such as aiding in digestion and educating the immune system (Naik et al. 2012; Upadhyay et al. 2012). Less well understood is the role that human immunity plays in shaping resident microbial communities. Some environmental microbial communities are extremely simple, containing just a few species (e.g., acid mine drainage biofilms) (Denef et al. 2010), while others are enormously complex including hundreds of phyla, including the Guerrero Negro hypersaline mats (Harris et al. 2013), soil, or ocean. Animal-associated microbial symbioses range from monospecific association of Vibrio fischeri inhabiting the light organ of Euprymna scolopes (squid) to the modestly complex communities inhabiting the human body (Dethlefsen et al. 2007). Even at its most complex, the human system appears to be colonized by approximately 10 phyla, an order of magnitude fewer than many environmental ecosystems. An open question is the extent to which symbiotic bacteria are selected for mutualistic interactions with the human host based on the limited ecological niches of human tissues versus a more active modulation of microbial communities by the immune system.
The microbiome of healthy human skin shows significant topographical variation between sebaceous, dry, and moist microenvironments. Likewise, the temporal stability of bacterial community membership and structure varies by skin site (Costello et al. 2009; Grice et al. 2009; The Human Microbiome Project Consortium 2012). These attributes differ significantly in disease states such as atopic dermatitis. Skin disease severity in atopic dermatitis patients was closely associated with marked shifts in the composition, diversity, and interpersonal and temporal variation of the resident microbial communities (Kong et al. 2012).
Studying primary immunodeficiency (PID) patients provides an opportunity to evaluate the degree to which altered immunity may influence the human microbiome and how, in turn, microbiota may interact with the host to cause disease. As cutaneous immunity has been shown to be distinct from that of other organs (Jiang et al. 2012; Naik et al. 2012), we surveyed the skin microbiomes of three different groups of PID patients whose genetically defined diseases are characterized by AD-like skin disease, diminished memory T and B cells, and variable eosinophilia and elevated IgE. Importantly, despite this broad phenotypic overlap, each of these diseases has its own distinct monogenic mutations and other extracutaneous clinical manifestations: Signal transducer and activator of transcription 3 (STAT3) mutations cause autosomal dominant hyper-IgE syndrome (STAT3-HIES; Job syndrome, OMIM 243700); mutations of dedicator of cytokinesis 8 (DOCK8) cause autosomal recessive hyper IgE syndrome (DOCK8 deficiency, OMIM 611432); and mutations in WAS cause X-linked recessive Wiskott-Aldrich syndrome (WAS; OMIM 301000).
STAT3-HIES arises from dominant-negative STAT3 mutations that prevent Th17 cell differentiation, abrogating CD4+ interleukin (IL)-17 production, which is important in epithelial fungal and bacterial infections as well as defensin expression (Milner et al. 2010). Clinical manifestations include recurrent staphylococcal infections, eczema, sinopulmonary infections, mucocutaneous candidiasis, and abnormalities in connective tissue, vessels, teeth, and bones, as well as an increased rate of lymphoma (Holland et al. 2007; Minegishi et al. 2007). DOCK8 deficiency more generally impairs T-cell differentiation, including Th17 cells (Engelhardt et al. 2009; Zhang et al. 2009), and is postulated to result from cytoskeletal abnormalities that may inhibit proper migration and differentiation of immune cells. In addition to recurrent sinopulmonary infections and staphylococcal skin infections, DOCK8 deficiency is associated with eczema, persistent viral infections of the skin, and a predisposition to malignancies (Renner et al. 2004). WAS results from X-linked recessive mutations in WAS. WAS has cytoskeletal abnormalities that lead to defects in T- and B-lymphocyte function via deficiencies in locomotion, signaling, and immune synapse formation (Ochs and Thrasher 2006). Clinical manifestations of WAS include eczema, thrombocytopenia, neutropenia, and a predisposition to autoimmunity and malignancy.
PID patients suffer from recurrent infections caused by readily isolated common bacteria and fungi (e.g., Staphylococcus aureus and Candida albicans) as well as uncommon opportunistic pathogens. Diagnosis typically relies on culture-based analyses; however, cultivation methods are limited in their ability to investigate complex microbial communities or to detect fastidious microbiota. Culture-independent metagenomic approaches, e.g., 16S ribosomal RNA (rRNA) sequencing, greatly increase the range of detectable microbes and allow assessment of the relative abundance of members of the microbial community. The 16S rRNA gene is present in all bacteria/archaea and contains both variable regions, which enable taxonomic classification, and conserved regions, which serve as binding sites for PCR primers.
Here, we present the results of a culture-independent comparative analysis, using 16S rRNA sequencing, of the skin microbiota of three groups of PID patients compared with classical AD patients and healthy controls. Our goal was to examine alterations in skin microbial communities in human immunodeficiencies as a basis for understanding the forces exerted by both the ecological niche and immune selection.
Results
Data generated for microbiome analysis
To investigate the extent to which the skin microbiome is altered in PID patients compared with controls, we analyzed a total of 170,167 full-length 16S rRNA Sanger sequences (mean 608 reads/sample; greater than 199 reads minimum, 280 samples), 6,850,352 V1-3 (average 11,650 reads/sample; greater than 1000 reads minimum, 595 samples), and 1,496,770 internal transcribed sequence regions 1 (ITS1; 13,607 mean reads/sample; greater than 1051 reads minimum, 90 samples) pyrosequences for four skin/nares sites in 49 healthy controls, 13 classical AD, 25 STAT3-HIES, 10 WAS, and six DOCK8 patients (Supplemental Table S1). Patient characteristics are described in Table 1. In general, the correlations of relative abundances of 17 major skin taxa obtained via Sanger compared with 454 sequencing were robust (partial Spearman correlation adjusting for multiple measurements 0.42 < ρ < 0.81; 2.6 × 10−9 < P < 9.9 × 10−117). Our findings justified use of sequences from the two platforms interchangeably for taxonomy-based analysis, while noting that full-length Sanger sequencing differentially recovered sequences for Enterobacteriaceae (Supplemental Fig. S1; Supplemental Table S2). For analyses based on operational taxonomic units (OTU; 97% similarity cutoff), such as alpha and beta diversity metrics, only 454 data were used. Rarefaction curves indicated that sampling for both Sanger and 454 sequencing provided sufficient coverage to analyze the dominant members of the bacterial communities (Supplemental Fig. S2; Supplemental Table S3).
Table 1.
Summary of patient characteristics
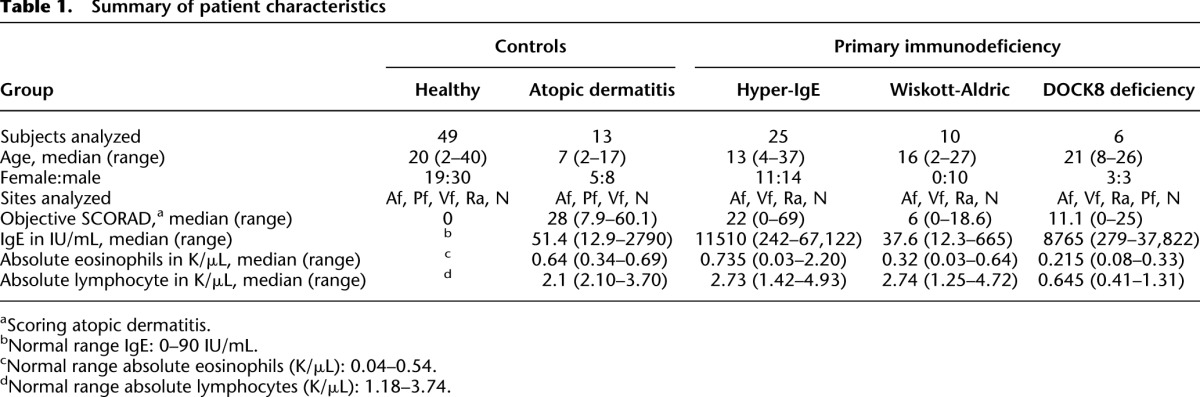
Since PID patients often use systemic and/or topical medications to prevent and/or treat infections, we included both healthy volunteers and AD patients undergoing treatment as controls (Supplemental Table S4). For each patient cohort, we examined the use of antimicrobial agents and immunosuppressants for possible associations with changes in the skin and mucosal bacterial communities. Bacterial community diversity was measured by the Shannon diversity index, an ecological measure of microbial communities that considers the following: (1) richness, or the total number of bacterial types, and (2) evenness, or the relative proportion of these bacterial types. Within each PID group, patients who had received systemic treatment within 30 d (antimicrobials, steroids) and/or topical treatment within 7 d (antimicrobials, steroids) did not harbor generalized changes in bacterial community diversity when compared to PID patients not receiving recent treatment (Supplemental Fig. S3; Supplemental Table S4). We did not observe an obvious shift in the bacterial community diversity related to recent treatment, distinct from findings identified in AD patients in which use of AD treatments was associated with an increase in bacterial community diversity compared with an untreated state of AD disease exacerbation (Kong et al. 2012). Interestingly, a recent study demonstrated that the skin microbiome in mice did not change after oral antibiotic treatment, in contrast to the gut microbiome (Naik et al. 2012). Finally, as skin microbiota differ based on age (Oh et al. 2012), patients and healthy controls were matched based on the two major groupings of prepubertal/early-pubertal versus late adolescent/adult (Supplemental Table S6), with the exception of the retroauricular crease (Ra), for which no prepubertal/early-pubertal data existed.
Given the site specificity observed in the skin microbiome (Costello et al. 2009; Grice et al. 2009; The Human Microbiome Project Consortium 2012) and the tendency for dermatoses to present in characteristic sites of predilection, we examined multiple skin sites in our patients: the antecubital fossae (Af) and the Ra as sites of disease predilection; volar forearm (Vf) as an adjacent control skin site that is not characteristically affected; and the nares (N), a potential reservoir for pathogens such as S. aureus and Streptococcus pneumoniae (von Eiff et al. 2001). For AD, STAT3-HIES, WAS, and DOCK8 patients, skin disease severity was assessed quantitatively with objective SCORAD (scoring AD), a well-validated clinical assessment tool (Williams et al. 1994, 1996; Kunz et al. 1997; Oranje et al. 2007). Laboratory studies in PID patients included serum IgE and a complete blood count (Supplemental Table S4). Representative images comparing skin disease manifestations in each patient group are shown in Figure 1, A through D. Because our largest PID patient cohort was the STAT3-HIES group, with numerous recurring visits, we focused the bulk of our analyses on comparisons between STAT3-HIES patients and healthy controls, with additional analyses including the other PID/AD cohorts to broaden insights into the microbiomes of these disorders.
Figure 1.

Representative clinical images of disease severity in the different patient groups. (A) Non-PID atopic dermatitis, (B) Hyper IgE syndrome, (C) Wiskott-Aldrich, and (D) DOCK8 deficiency.
Altered permissiveness to microbial colonization in PID patients
We sought to determine whether underlying immunodeficiency altered the bacterial communities present on human skin, specifically to include non-native taxa or unique community structures on PID skin. We classified the sequence data to (1) determine if unique bacteria colonized the skin of PID patients and (2) assess the taxonomic differences between PID patients and healthy controls (Fig. 2A,B; Supplemental Fig. S4A–D; Supplemental Table S9). We analyzed sites of disease predilection (Af, Ra) and observed that the environmental microbe and opportunistic pathogen Proteobacteria Serratia marcescens was increased at both sites in STAT3-HIES patients, as well as the adjacent arm site (Af, Vf: adjusted P ≤ 0.05; Ra: N.S.) (Fig. 2A). Although S. marcescens is ubiquitous in the environment and is commonly found in households, this bacterium has not previously been identified as a component of the skin microbiome of healthy individuals or AD patients, independent of treatment with antimicrobials. S. marcescens is a nosocomial pathogen associated with wounds, urinary tract infections, and bacteremia, but it is not known to cause clinical disease in STAT3-HIES patients. Moreover, S. marcescens was observed infrequently in the other two PID groups (Supplemental Fig. S4) but can cause disease in some patient populations, e.g., chronic granulomatous disease.
Figure 2.

Bacterial taxonomic classifications show colonization with unique taxa and altered representation of diverse taxa. Relative abundances of 14 major phyla-family taxonomies in the antecubital fossa (Af) are shown in A and retroauricular crease (Ra) in B. Shown are seven representative healthy controls and all Sanger-sequenced STAT3-HIES patients. Full versions of all skin and nares sites are in Supplemental Figure S4. (C) Inlaid pie charts compare the mean relative abundances of major skin genera across patient groups. Mean ± SEM for C are shown in Supplemental Figure S5 and Supplemental Tables S9 and S10. Patient identifiers are in Supplemental Table S13.
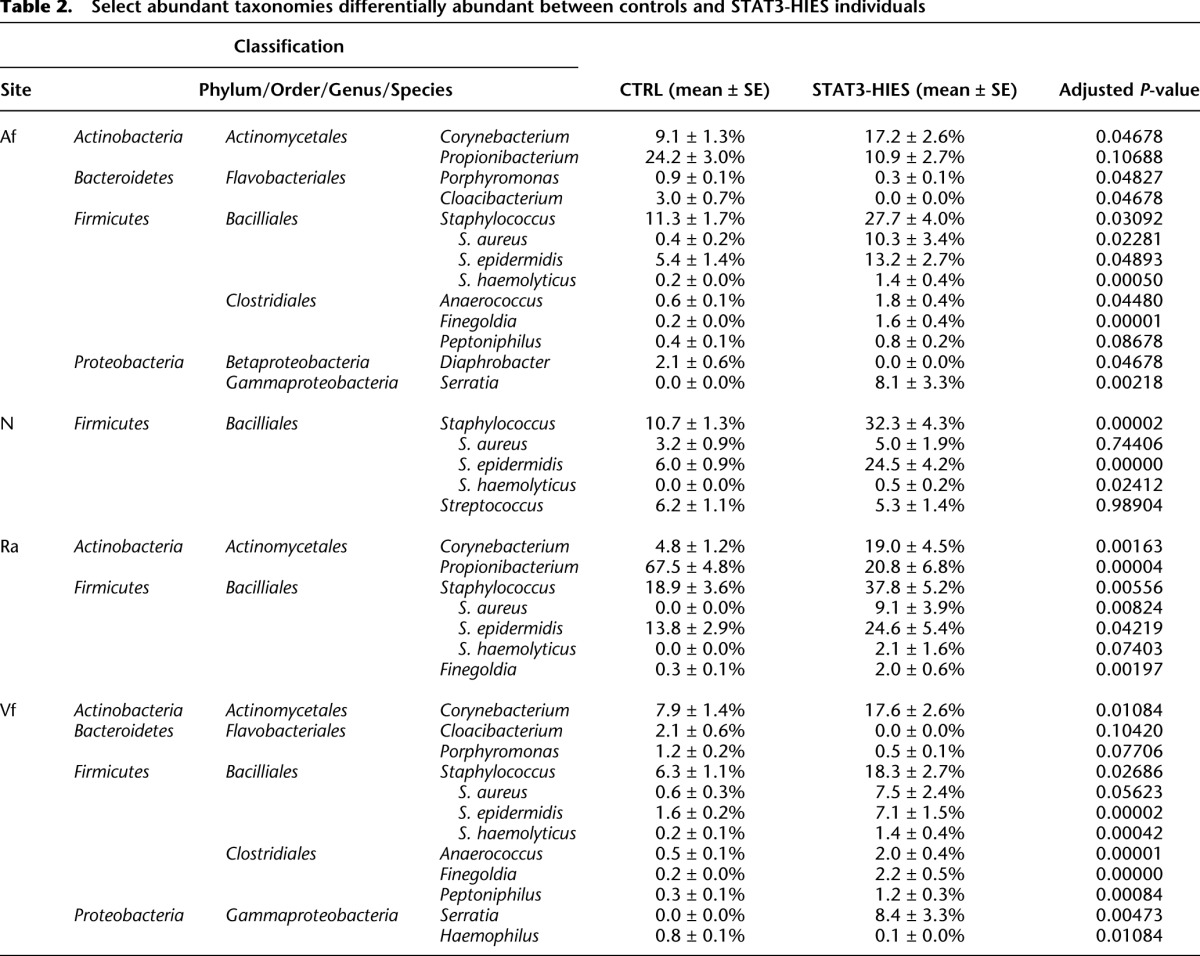
The Af (Fig. 2A) and Vf (Supplemental Fig. S5) are typified by intermediate bacterial diversity with moderate amounts of Propionibacterium, Staphylococcus, and Corynebacterium, while the sebaceous Ra (Fig. 2B), which presents a different cutaneous environment, is characterized in healthy individuals by low bacteria diversity with a predominance of Propionibacterium. Further analyses of the skin sites (Table 2; Supplemental Table S9) showed a significant overrepresentation of Firmicutes in the STAT3-HIES skin, particularly of the Clostridiales class (Anaerococcus, Finegoldia, and/or Peptoniphilus, FDR-adjusted P ≤ 0.1). The common skin bacteria Corynebacterium and Staphylococcus were highly overrepresented on STAT3-HIES skin compared with healthy controls (adjusted P < 0.05) (Fig. 2C). Bacteroidetes (particularly Porphyromonas and/or Cloacibacterium, adjusted P ≤ 0.1 Af, Vf) and the abundant skin commensal Propionibacterium (adjusted P = 0.1) were significantly depleted at sites of disease predilection (Af, Ra) in the STAT3-HIES individuals. With the finding of a distinct species (Serratia marcescens) colonizing STAT3-HIES skin, a possible hypothesis is that a deficiency in the host immune system may increase the skin's ecological permissiveness, e.g., allowing atypical microbiota to colonize the skin. However, we did not identify a generalized increase in the number of different taxa on PID skin, even in patients who had not received antimicrobials. Although we observed a unique colonization by S. marcescens, the bacterial phylum that includes S. marcescens, Proteobacteria, also includes other species common to the human skin. Thus, the ability of human skin to host certain bacterial phyla remained restricted to those previously identified.
Table 2.
Select abundant taxonomies differentially abundant between controls and STAT3-HIES individuals
To assess which taxa contributed to the variation observed between samples, we performed principal coordinates analysis (PCoA) using the Yue-Clayton theta (θ) similarity coefficient (Clayton and Yue 2005), which takes into consideration the number of bacterial species present and their relative abundances in the two communities being compared. Spearman correlation of the relative abundances of different taxa to the axes, shown by biplot lines, identifies taxa that most significantly contribute to axis variation. At sites of disease predilection, the Af and Ra (Fig. 3; for additional skin and N sites, see Supplemental Fig. S6), the predominance of Staphylococcus, Propionibacterium, Corynebacterium, and members of the Clostridiales family appeared to be the primary drivers of variation between individuals and groups. In particular, Staphylococcus, and to a lesser extent, Corynebacterium, contributed most heavily to the axis variation associated with PID and AD patients, while Propionibacterium was heavily associated with healthy controls. To quantify the level of similarity of bacterial community structures in our patient cohort and healthy controls, we calculated and compared interpersonal variation: (1) between individuals of a patient group (“within-group”), (2) between different patient groups and controls (“vs. controls”), and (3) between different PID groups (“between PID”) (Supplemental Fig. S7). Interpersonal variation between the microbial communities of the STAT3-HIES individuals was significantly lower than in healthy controls and other PID groups at all sites (multiple comparison test after Kruskal-Wallis, P < 0.05) (Supplemental Table S8) except the Ra, which displayed low interpersonal variation irrespective of patient group. Interpersonal variation within STAT3-HIES, DOCK8, and WAS groups was generally similar (P < 0.05), again with the exception of the Ra. At sites of eczematous disease predilection (Af and Ra), STAT3-HIES and DOCK8 individuals were significantly more similar to each other than to WAS individuals (P < 0.05, Af), while WAS individuals were generally significantly more similar to controls (P < 0.05 for Af, Ra, Vf) than other PID individuals.
Figure 3.

Taxonomies associated with variation within and between individuals. Principal coordinates analysis (PCoA) of the Yue-Clayton theta coefficient, which calculates the similarity between two samples based on (1) number of species in common between two samples, and (2) their relative abundances. Samples that have similar principal coordinates appear closer together, i.e., are more similar. Biplot lines indicate the most significant unique consensus taxonomies contributing to variation along axis 1; Spearman correlations (ρ) are with axis 1. Length of biplot lines reflects the contribution of that taxa to the top three axes. Associated P-values < 2.2 × 10−16. Percentage variation attributed to an axis is indicated. Antecubital fossa (Af) and retroauricular crease (Ra) are shown in A and B, respectively; nares and volar forearm are shown in Supplemental Figure S6.
In general, consistent with the beta diversity metrics showing more similarity between the WAS and control groups, there were few genera whose mean relative abundances were significantly different between the WAS patients and controls, irrespective of site. While the DOCK8 patient population was too small to demonstrate statistically significant differences between groups, several of the trends observed in the STAT3-HIES population, such as elevated Clostridiales and Staphylococcus epidermidis levels, were also observed in this population (Supplemental Fig. S6).
Finally, to identify whether there were key indicator taxa that could be used to classify an individual as belonging to the STAT3-HIES immunodeficient patient class, we used Random Forests, a supervised machine learning technique (Knights et al. 2011). To determine those most differentiating taxa for each site, we ranked taxa by its mean decrease in accuracy, a measure of its differentiating power calculated by estimating the increase in classification error caused by removing that taxa from the set of predictors. The error rate for each site ranged from 10% (Vf) to 18% (Ra; Af: 16%; N: 13%). Consistent with our over- and underrepresentation analysis, taxa of the Clostridiales order, P. acnes, S. aureus, Serratia, and Cloacibacterium were key indicator taxa differentiating the two groups consistently across skin sites, with key differentiating taxa in the N S. epidermidis, Enterococcus, and unclassified Actinomycetales (Supplemental Table S14).
Ecological differences in microbial communities of PID patients
To further explore microbial community changes, we compared bacterial community diversities in the skin of the PID cohort and controls using Shannon diversity, an ecological measure of community richness and evenness. At the Af, diversity in STAT3-HIES individuals trended toward being lower compared with healthy controls (P = 0.08) (Supplemental Table S7), with levels more comparable to those of AD patients (P > 0.05) (Fig. 4A). At characteristically unaffected sites such as the Vf and N, diversity was comparable between all PID patient groups, AD, and healthy controls (Kruskal-Wallis test, P > 0.05; STAT3-HIES N excepted P = 0.003). While the Af, Vf, and N communities are moderately complex, the sebaceous Ra is characterized by markedly low bacterial diversity with a predominance of Propionibacterium. Notably, we observed a loss of this homogeneity with the bacterial diversity at the Ra being significantly elevated in all PID cohorts (Kruskal-Wallis test, P = 0.01). This ecological measure of community diversity mirrored results obtained by taxonomic classification of the PID skin in which expansion in the number of phyla able to colonize the PID skin was not observed. However, dysbiosis, in terms of bacterial community diversity, existed in a site-specific manner, i.e., sites of disease predilection.
Figure 4.

Community-wide metrics suggest altered permissivity in niche specificity in colonization in PID individuals. Site codes: (Af) antecubital fossa; (N) nares; (Ra) retroauricular crease; (Vf) volar forearm. (A) Mean ± SEM for Shannon diversity is plotted for each patient group (colored as indicated) at all sites. (B) Site specificity is measured by the Yue-Clayton theta coefficient as in Figure 3, calculated between moist (antecubital fossa), dry (volar forearm), and sebaceous (retroauricular crease) skin sites and nares. (C) Longitudinal stability was assessed by calculating the theta coefficient between two and four timepoints for controls (light blue) and STAT3-HIES patients (red).
While overall community diversity was not significantly increased in PID skin, we hypothesized that PID could potentially increase permissiveness of microbial colonization in the skin, such that community composition at different sites is dysregulated. To compare the similarities of bacterial community structures between different sites, we calculated the mean theta index across three diverse microenvironments of the skin (Af, Ra, and Vf: moist, sebaceous, and dry, respectively) and the N. The skin microbiomes in healthy individuals are often strikingly dissimilar from site to site (Costello et al. 2009; Grice et al. 2009). However, we observed significant blunting of this site-to-site variation, or decreased site-specificity, in the bacterial community structure of the DOCK8 and STAT3-HIES microbiomes (P = 0.02 and P = 0.0001, respectively) and a similar trend in WAS (P = 0.07) (Fig. 4B; Supplemental Table S8).
Another trend that might reflect increased permissiveness of PID skin would be aberrant stability of the microbial communities over time. In healthy controls, longitudinal stability of microbial communities is highly site-specific, associated most closely with microenvironment characteristics, including moist or sebaceous skin. Since immunodeficiency may alter temporal stability of skin microbial communities, we compared the longitudinal stability between healthy controls and STAT3-HIES patients, calculating the mean theta index across two to four timepoints for STAT3-HIES patients (Fig. 4C; Supplemental Table S8). Across multiple sites, microbiomes of STAT3-HIES patients were significantly less stable over time (Af: P = 0.19; N: P = 0.02; Ra: P = 0.002; Vf: P = 0.02) compared with healthy controls. Alterations in skin bacterial diversity, blunting of site-to-site variability, and increase in temporal instability, taken together with the ability of unique taxa to associate with PID skin, suggest that immunodeficiency may result in dysregulation of the typical host-specific constraints on community composition.
Association of the microbiome with clinical disease manifestations and severity
Since skin disease in PID patients is often associated with S. aureus, we classified Staphylococcus to the species level (Fig. 5A; Supplemental Fig. S4; Supplemental Table S10). While staphylococci were generally more abundant in the AD group than in the STAT3-HIES Af and Vf (adjusted P = 0.03, 0.06, respectively), we observed significant differences in the species composition in the STAT3-HIES and DOCK8 groups. In contrast to AD patients, whose staphylococci are primarily S. aureus (Fig. 5A; Supplemental Figs. S4, S5), at all sites S. epidermidis was significantly enriched in the STAT3-HIES group (adjusted P ≤ 0.05), and Staphylococcus haemolyticus, an opportunistic pathogen, was less abundant but also significantly enriched (adjusted P ≤ 0.07). While S. aureus was also significantly enriched at skin sites (adjusted P ≤ 0.06; equally abundant in the N), surprisingly, it comprised a much smaller proportion of staphylococci in the STAT3-HIES and DOCK8 groups as compared to AD. The decreased S. aureus proportion may relate to the antibiotic prophylaxis given to reduce S. aureus skin and lung infections, and these antibiotics frequently are ineffective against other staphylococcal species.
Figure 5.

Taxonomic association with clinical features. (A) Pie charts comparing the mean relative representation of major staphylococcal species across patient groups at all sites. Site codes: (Af) antecubital fossa; (N) nares; (Ra) retroauricular crease; (Vf) volar forearm. (B) Correlation of taxonomy with clinical markers of disease severity for the antecubital fossa. Unsupervised hierarchical clustering of Spearman correlation coefficients for the 34 taxa whose mean abundance was >0.25% across STAT3-HIES patients. Correlations are calculated against blood levels of lactate dehydrogenase, lymphocytes, IgE, and eosinophils, and a score of skin disease severity (scoring atopic dermatitis [SCORAD]). Hemoglobin was a control. Reds indicate correlation; blues indicate anti-correlation.
Frequent infections are common in PID patients, and the increased frequency or severity of infections is a marker of worsening disease. Therefore, we examined laboratory studies and a skin disease severity score in the STAT3-HIES patients to identify taxa or groups of taxa that correlated with disease severity. We calculated Spearman correlation coefficients for the correlation between taxonomic relative abundance at the Af, a site of disease predilection, to the following: SCORAD, serum IgE, serum lymphocytes, lactate dehydrogenase, and serum eosinophils. Hemoglobin was included as a laboratory study that was not expected to correlate with disease severity.
Propionibacterium acnes abundance was significantly inversely correlated with skin disease severity (P = 3.7 × 10−05) (Supplemental Table S11), while S. aureus was significantly correlated (P = 0.002). From unsupervised clustering of the correlation coefficients obtained for the 34 most abundant taxa (Fig. 5B), we identified two major clusters. The first, smaller cluster contained taxa in addition to S. aureus, whose relative abundances were positively correlated with disease severity. The second, larger cluster contained potential markers of a healthier skin microbiome in our patient population; their relative abundances were inversely correlated with markers of disease severity. In addition to P. acnes, this group included the Firmicutes Lactobacillus (P = 0.05) and several Proteobacteria, including Stenotrophomonas (P = 0.01), Novosphingobium (P = 0.04), and Burkholderia (P = 0.01). Interestingly, while the majority of staphylococci in the STAT3-HIES individuals were not S. aureus, these non-S. aureus staphylococci were not correlated with disease severity.
Fungal characterization of PID
In addition to bacterial infections, patients with STAT3-HIES often develop fungal infections, including candidiasis and Aspergillus pneumonias. Thus, we performed ITS1 sequencing in a pilot study of STAT3-HIES patients. At nondisease sites, the Vf and N, we observed a trend toward increased fungal richness in the STAT3-HIES individuals (Fig. 6A; Supplemental Table S12), which would further support the hypothesis of a more permissive microbial niche in the PID skin. This increase in diversity appeared to involve a reduction in the relative abundance of the common skin fungi Malassezia (P < 0.05, all sites) (Fig. 6B; Supplemental Table S12). Moreover, we observed a significant increase in the relative abundance of Aspergillus (P < 0.06) and Candida (P < 0.05) at all sites in the STAT3-HIES patients (Supplemental Table S12; Supplemental Fig. S8). Since antifungal medications are commonly administered to STAT3-HIES patients, we examined their medication histories for use of antifungals. We observed Aspergillus even in the STAT3-HIES patients who had not previously received any antifungal treatment. These findings further suggest that the altered immunity that modifies the potential bacterial microbiome niche in PID patients also applies to fungal skin/mucosal communities and may contribute to the clinical observation of increased fungal infections in this patient population.
Figure 6.

Characterization of fungal skin communities suggest increased permissivity and elevated levels of opportunistic pathogenic fungi. (A) Boxplots showing fungal richness (species observed) of STAT3-HIES versus controls at all sites; black bars indicate median. (B) Select ITS1 fungal taxonomic classifications for the antecubital fossa, grouped by patient category. Full versions of all skin and nares sites are shown in Supplemental Figure S8. Patient identifiers are in Supplemental Table S13.
Fungal–bacterial associations in PID
To evaluate major fungal–bacterial associations, we performed a strain-by-strain co-occurrence analysis based on a Spearman correlation of fungal and bacterial taxonomic relative abundances at each site. Generally, patterns of correlation/anti-correlation of relative abundances differed between controls and STAT3-HIES heatmaps (Supplemental Fig. S9). For example, in the Ra, Malassezia globosa was generally correlated with most genera of Firmicutes and Actinobacteria in both controls and STAT3-HIES patients and Malassezia restricta was anti-correlated. In the STAT3-HIES group, streptococci, coryneforms, and S. aureus were uniquely correlated with the presence of Candida, Aspergillus, Cryptococcus, and others. In the Af and Vf, streptococci, staphylococci, and Corynebacterium were significantly correlated with non-Malassezia fungi in STAT3-HIES individuals, while controls exhibited lesser interactions between these taxa.
To assess co-occurrence and community assembly on a population level, including both fungal and bacterial taxa, we calculated the C-score for each site, which reflects exclusion patterns of different taxa within a niche, i.e., the tendency of each taxa to exclude one another from a given niche (Stone and Roberts 1990). To impute significance to the score, we compared it against the mean score obtained from a null distribution of 10,000 permutations drawn from the same control or STAT3-HIES matrix. Based on statistical significance of C-scores, the microbiomes at each site in the STAT3-HIES and control groups were not composed of interacting bacterial and fungal consortia (P > 0.05, STAT3-HIES Vf [P = 0.01]), suggesting no significant pattern to community-wide co-occurrence in the skin. While these results suggest on a broader scale that communities of the skin do not have obvious assembly rules, co-occurrence of specific fungal–bacterial partners begin to suggest some level of interaction between members in the two kingdoms. Further investigation is required to determine the extent of such an interaction, which may occur at a local level that is not reflected by these broader microbial surveys.
Discussion
Pathogens are typically considered as discrete causative agents in virulence studies, yet they exist within a rich milieu of other microbial species that can influence pathogenicity. Disease could derive from dysbiosis of the microbial community without an invading pathogen dominating the community. To begin to investigate the complex interrelationships between host, microbe, and disease, we characterized the bacterial and fungal microbiota of several groups of PID patients compared with classical AD patients and healthy controls. Our study offers a unique opportunity to study potential alterations in the human microbiome in the setting of PID. We focused on rare inherited disorders that shared the cutaneous phenotype of AD-like skin disease, so that we could identify differences in the microbiota that may reflect the specific immunological and genetic features of these distinct populations. Our analyses demonstrate an increased permissivity in the bacterial and fungal colonization of the skin of PID patients, dysbiosis in community diversity, association of bacterial communities with skin disease manifestations and severity, and unique bacterial–fungal taxonomic co-occurrences.
Based on our observations of decreased site specificity and longitudinal microbiome stability, as well as colonization by unique taxa, we hypothesize that PID increases the host's ability to be colonized by atypical microbiota. Interestingly, this increased permissiveness of skin did not result in significant increases in microbial diversity or colonization by novel phyla. This suggests that host-specific constraints on the taxonomic diversity of skin microbiota, e.g., site physiology, persist despite defective host immunity in both the recognition and the clearance of cutaneous taxa. While the issue of bioburden, which may or may not be significantly altered in these patient populations, is important and remains a challenge, our study focused on taxonomic diversity.
We found that presence of skin disease had a dominant signature that appeared to be strongly associated with community composition. In AD, the presence of skin disease was very strongly associated with the presence of S. aureus. PID patients are frequently treated with antimicrobial therapeutics to manage or prevent chronic or recurrent skin and systemic infections, yet the impact of systemic antibiotics on intact skin is incompletely understood. We determined that while S. aureus was most significantly correlated with disease severity in these patients, other taxa such as Clostridiales and Corynebacterium were correlated as well. In contrast to AD patients, the primary staphylococcal species in the PID patients were S. epidermidis and S. haemolyticus rather than S. aureus, in spite of the presence of disease. In these PID patients, antibiotics, including penicillins and trimethoprim-sulfamethoxazole, are administered to reduce S. aureus infections and appear to effectively reduce S. aureus relative abundance. Whether the non-S. aureus staphylococci significantly contribute to skin disease is currently unknown; in our previous study of atopic dermatitis, we observed a significant increase in the relative abundance of S. epidermidis concurrent with increases in S. aureus (Kong et al. 2012). A possibility is that these other staphylococci may share a mutualistic or commensal relationship with S. aureus by enabling greater resistance to antimicrobial peptides (Peschel et al. 2001; Sieprawska-Lupa et al. 2004; Lai et al. 2007) or antibiotics (Wielders et al. 2001; Berglund and Söderquist 2008) or by potentiating the S. aureus toxic response as with C. albicans (Peters and Noverr 2013). Alternately, these species could take advantage of inflamed skin conditions and co-colonize with S. aureus (Nilsson et al. 1998; McCrea et al. 2000; Cho et al. 2001; Williams et al. 2002).
While PID skin microbiomes shared the reduced diversity seen in AD, we observed microbial traits unique to the skin disease of these PID patients. We compared PID patients with AD patients who also were undergoing treatment for their recurring skin disease. While treatment, particularly with antibiotics, may skew the relative representation of different taxa, our results reflect the day-to-day state in which these patients remain at risk for infections despite prophylactic antibiotic treatment. Of note, our cohort of WAS patients had less skin disease and were less likely to be on systemic antimicrobials compared with the other PID groups in our study. This may have contributed to the observed similarities in skin bacterial community diversity (except the Ra) between WAS patients and healthy controls. However, the increased bacterial community diversity in the Ra of the untreated and treated WAS patients compared with healthy controls suggests that host factors, i.e., immunodeficiency, are likely important causes of the observed skin microbiome differences in our PID patients, even when use of antimicrobials is taken into account.
Interactions with skin commensals are key in developing cutaneous immunity (Naik et al. 2012). One possibility is that the skin microbes in our patient cohort may alter the activation of cutaneous immunity. The markedly elevated IgE levels in these patients may be related to allergy or atopy, or may reflect an aberrant response to a colonizing microbe. Our data support the hypothesis that the skin in PID patients exhibit different selective pressures leading to permissiveness for different organisms such that an environmental microbe, S. marcescens, uniquely colonized PID skin. Its longitudinal fluctuations, as well as its ability to colonize both characteristically affected and unaffected sites (Af, Ra, and Vf), suggest that S. marcescens may be a transient taking advantage of the ecological permissiveness of the immunodeficient skin, as opposed to being pathogenic. Notably, S. marcescens infections in chronic granulomatous disease patients are typically treated with the antibiotics, e.g., trimethoprim-sulfamethoxazole, which were used in our PID patient populations. In addition, several members of the Clostridiales family also significantly colonized PID skin, whereas in controls, their representation was very low.
Since STAT3-HIES patients suffer from oral and pulmonary fungal infections and DOCK8 patients experience multiple recalcitrant viral infections, investigating nonbacterial microbiota is of importance. The observation of increased Candida and Aspergillus species on the skin of STAT3-HIES patients may correlate with their risk of pulmonary aspergillosis and oral candidiasis with the skin serving as a possible reservoir for infection, much as the N is a reservoir for S. aureus and Streptococcus pneumoniae infections (von Eiff et al. 2001). The presence of Aspergillus did not differ in patients on antifungal medications with activity against Aspergillus compared with patients without a history of antifungal agents with activity against Aspergillus, again highlighting the significant skin microbial differences observable in PID patients in the absence of use of antimicrobial agents. In addition, coinfection of certain C. albicans isolates with S. aureus can result in infectious synergism with enhanced toxicity and host inflammation (Peters and Noverr 2013).
From a genetic and mechanistic perspective, WAS and DOCK8 deficiency syndromes likely share many similarities. WAS acts downstream from DOCK8 as an effector of CDC42 signaling (McGhee and Chatila 2010). WAS patients have cytoskeletal dysfunction leading to immune deficiency via defective migration or immune synapse formation by a wide range of immune cells, and DOCK8-deficient patients are also thought to have cytoskeletal abnormalities based on known functions of other DOCK family members. In contrast, defects in STAT3 signaling, a key regulator of many immunologic pathways, impair defensin expression and neutrophil recruitment and production, in part due to defects in Th17 differentiation. A distinction among the PID groups was that severe skin disease was not observed in these WAS patients. Interestingly, the overall skin microbiomes in DOCK8-deficient and STAT3-HIES patients were more similar to each other, perhaps reflecting their phenotypic rather than mechanistic convergence. Concurrent host/microbiome transcriptional profiling will further our understanding of the PID immunologic response to shifts in their resident microbiome, and functional metabolomics will examine the microclimate of PID skin and how the microbiota respond to, or in turn, affect the host.
While PIDs are rare monogenic disorders, periods of immune suppression are frequent in cancer patients and transplant recipients. Future studies should investigate whether loss of immunity similarly alters microbial community in these states. Many of these patients receive prophylactic antibiotic treatment to decrease bacterial load, preventing both infection and microbial translocation. Further characterization of dysbiosis in immune-compromised individuals, as well as the antibiotic resistance reservoir in these chronically treated patients, might direct both usage of antibiotic prophylaxis and infection control. Future studies may also explore a role for prebiotics/probiotics to maintain microbial diversity and health as preventative approaches.
By Koch's postulates, the relationship between pathogen and disease denotes characteristics of necessity and sufficiency. However, it is increasingly clear that microbial pathogens, such as S. aureus, which can asymptomatically colonize human hosts, may only be disease-associated in the context of their microbial communities and host factors. A corollary to this assertion is that dysbiosis of the microbial community can itself function as an etiological agent. General instability and dysbiosis of the skin microbiome, as observed in PID patients, may be an underlying contributor to their recurrent skin disease. If therapies target not only pathogenic agents but also microbial homeostasis, the risks of developing antibiotic-resistant bacteria might be reduced. In summary, the increased instability and permissiveness of the bacterial PID skin microbiome lends insight into host–microbiome interactions and its potential contribution to a distinct PID-associated microbiome.
Methods
Subjects
All samples for this study were obtained according to a protocol approved by the Institutional Review Board of NHGRI, NIH. Healthy and AD subjects were recruited from the greater Washington, DC, metropolitan region. PID patients were recruited from patient cohorts evaluated at the Clinical Center, NIH. All subjects gave informed consent. Demographic and clinical data were collected for each subject. All subjects provided medical and medication history and underwent dermatologic examination at all clinic visits. The diagnoses of PID patients were based on clinical history and genetic testing. Assessment of extent of skin disease involvement was based on the objective scoring atopic dermatitis (SCORAD) (Kunz et al. 1997). Bloodwork included serum IgE (IU/mL; normal range: 0–90), hemoglobin (g/dL; normal range: males 13.7–17.5, females 11.2–15.7), absolute lymphocyte count (K/μL; normal range: 1.18–3.74), absolute eosinophil count (K/μL; normal range: 0.04–0.36), and lactate dehydrogenase (U/L; normal range: 113–226). A summary of subject characteristics are shown in Table 1, with detailed information about sampling, sites analyzed, and date sampled in Supplemental Table S1. For Sanger data, data for healthy controls and AD patients were obtained from previous studies (Grice et al. 2009; Kong et al. 2012).
Sample collection and processing
All samples were collected by H.H.K. as described in Grice et al. (2009) and Kong et al. (2012). Briefly, skin preparation instructions included avoiding bathing and avoiding emollients or antimicrobial soaps or shampoos for 24 h prior to all sampling. Sampling sites included the nares (N), retroauricular crease (Ra), antecubital fossa (Af), and volar forearm (Vf). From a 4-cm2 area, bacterial swabs (via Epicentre swabs) and scrapes (via sterile disposable surgical blade) were obtained and incubated in enzymatic lysis buffer and lysozyme for 30 min at 37°C. For the nares, only swab samples were taken. Scrapes and swabs were obtained from the other sites as available. Correlation analysis of log proportions of genera identified in scrape versus swab sampling confirmed the similarity of techniques (R = 0.87, P < 2.2 × 10−16; data not shown).
Preparation of samples for 16S Sanger sequencing
Samples were processed as described in Oh et al. (2012). Briefly, skin swabs were incubated in enzymatic lysis buffer and lysozyme (20 mg/mL) for 30 min at 37°C. Two 5-mm stainless steel beads (Qiagen) were added to the solution, placed in a TissueLyser (Qiagen), and processed for 2 min at 30 Hz. The standard protocol for the PureLink Genomic DNA kit (Invitrogen) was followed for all subsequent steps. Near full-length 16S rRNA genes (16S) were amplified from purified genomic DNA using primers 8F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1391R (5′-GACGGGCGGTGWGTRCA-3′) and were sequenced at the National Institutes of Health Intramural Sequencing Center (NISC). Three hundred to 400 unique sequences were obtained from 16S rRNA amplicons of each sample.
Preparation of samples for 454 sequencing
Genomic DNA extractions were performed as for the Sanger sequencing. 16S rRNA V1-V3 amplicon libraries were prepared from sample DNA using AccuPrime HF Taq (Invitrogen, 12346-086) and universal primers flanking variable regions V1 (27F, 5′-AGAGTTTGATCCTGGCTCAG-3′) and V3 (534R, 5′-ATTACCGCGGCTGCTGG-3′). For each sample, the universal primers were tagged with unique sequences (“barcodes”) to allow for multiplexing/demultiplexing (Lennon et al. 2010). For ITS1 amplicon library preparation, universal primers flanking the ITS1 region—adapter + 18SF (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAGGTAAAAGTCGTAACAAGGTTTC-3′) and 5.8S-1R + barcode (5′-GTTCAAAGAYTCGATGATTCAC-3′)—were used to amplify a select number of genomic DNA extractions. PCR products were then purified using the Agencourt AMPure XP Kit (A63880) and quantitated using the Quant-iT dsDNA high-sensitivity assay kit (Invitrogen, Q33120). Approximately equivalent amounts of each PCR product were then pooled and purified with a Qiagen MinElute column (Qiagen, 28004) into 30 μL TE prior to sequencing at the NIH Intramural Sequencing Center. Amplicon libraries were sequenced on a 454 GS FLX (Roche) instrument using titanium chemistry.
16S rRNA amplicon sequence analysis pipeline
Supplemental Table S1 shows samples for which analyzable data were obtained, as a certain number of samples failed during the sequencing and quality control pipeline. Mothur version 1.26.0 (Schloss et al. 2009) was used for sequence processing, definition of operational taxonomic units (OTUs), and downstream analyses. For full-length 16S rRNA amplicon processing, sequence assembly, filtering, and alignment were performed as described in Grice et al. (2009) and Kong et al. (2012). Briefly, sequences matching the human genome were removed (E-value < 0.1) and then aligned using the Greengenes NAST (DeSantis et al. 2006) aligner. For V1-V3 region 16S rRNA amplicons, 454 flowgram data were denoised using the mothur implementation of PyroNoise (Quince et al. 2011). The resulting sequences were trimmed of primers and barcodes (tolerant of primer errors ≤1 and barcode errors ≤2) and filtered for homopolymers ≤8 and length ≥200 bp. Sequences were aligned in mothur using a SILVA reference database (Pruesse et al. 2007).
Chimera removal was performed with the mothur implementation of UCHIME (Edgar et al. 2011) and classified using a ribosomal database project naïve Bayesian classifier (Wang et al. 2007). Full-length and V1-V3 16S-rRNA sequences were processed in parallel, and to ensure that generally results from either method resulted in equivalent data, we calculated the partial Spearman correlation coefficient between the relative abundances of the dominant taxa (Supplemental Fig. S1), adjusting for multiple patient timepoint measurements.
Following sequence quality control processing, all uncorrected pairwise distances were calculated and operational taxonomic units (OTUs) defined at 97% similarity using average neighbor joining. No lane masking was applied. To estimate sampling saturation, rarefaction curves were generated for each site for each sequencing method used (Supplemental Fig. S2A,B). While OTUs did not achieve saturation, the rarefaction curves confirmed that our sampling gave reasonable coverage to analyze the dominant members of the bacterial communities. Alpha diversity (community evenness and richness: Shannon diversity index, “diversity”) and beta diversity (shared community structure/membership: Theta, θ “similarity” index) were calculated at a 97% similarity cutoff in mothur.
Species-level classification for Staphylococcus and Streptococcus was performed using the pplacer algorithm (v1.1.alpha13-0-g1ec7786, (32)) in which each 16S rRNA gene classifying to either of these genera are compared with a curated set of reference packages created as described in Matsen et al. (2010). Briefly, a high-quality 16S rRNA reference database was built from rRNA records extracted from RefSeq genomes (as of April 2012) and RDP type species sequences (release 10, update 24), and sequences for each genus were then extracted from this trusted set and placed on a phylogenetic tree using the taxtastic suite of tools (taxit v0.3.1) (Conlan et al. 2012). Sequences classified as Staphylococcus or Streptococcus by the RDP naïve Bayesian classifier were then placed on a phylogenetic reference tree with “–keep-at-most 1000–max-pitches 1000” and taxonomic classifications were generated using the guppy program in pplacer, with likelihood cutoffs set to 0.60.
ITS1 amplicon sequence analysis pipeline
Sequences were denoised, trimmed of barcodes and primers, and chimeras removed with a pipeline as previously described for skin fungal community analyses (Findley et al. 2013). Sequences were then classified to the genus level using this database and the k-nearest neighbor (knn) implementation in mothur and then assigned to phylotypes. Sequences of Malassezia species were identified using a custom curated Malassezia reference library as described in Findley et al. (2013).
Statistics
All data are represented as mean ± SEM unless otherwise indicated. Unless otherwise indicated, P-values were adjusted for multiple comparisons greater than six using the p.adjust function in R using method = “fdr” (Benjamini and Hochberg 1995). Statistical significance was ascribed to an alpha level of the adjusted P-values ≤ 0.1. Each site was treated as a separate data set based on spatial physiological differences between different body niches (Grice et al. 2009). All per-sample calculations are available upon request as a separate data set. For Ra, statistical comparisons were calculated for both age-adjusted and non-age-adjusted values.
For subjects with multiple measurements (e.g., symmetric sites or multiple sampling timepoints), samples were processed independently and symmetric sites were combined. Multiple sampling timepoints were adjusted for using a mixed effects model with a random intercept for multiple timepoints. Processed sequences were then subsampled to 1000 sequences per sample (454 only), and diversity statistics and proportions were calculated from the subsampled data. For alpha and beta diversity statistics, pools were subsampled 100× and resultant values averaged. More than five subsamplings yielded a 99.8% correlation with results from one subsampling, and five to 100 subsamplings were up to 99.99% similar. For longitudinal analyses based on the theta coefficient, we calculated the average similarity between two and four timepoints within an individual and then averaged the mean longitudinal stability of all individuals for either STAT3-HIES or control patients. For spatial (intersite) diversity, we calculated the mean theta coefficient between moist (antecubital fossa), dry (volar forearm), and sebaceous (retroauricular crease) skin sites and the nares for each individual, and then calculated the mean for all individuals within a category. For mean theta indices, multiple timepoints were averaged prior to calculating a between-category average. Finally, the nonparametric Kruskal-Wallis test was used with θ to determine statistically significant differences between microbial populations. To identify significant intercategory comparisons, we used a post-hoc multiple comparison test, implemented by the kruskalmc test in the pgirmess package in R. For ITS1-derived measurements, symmetric site and multiple timepoint measurements were averaged prior to statistical testing.
For taxonomy-based analyses, we did not subsample typical filtering strategies (e.g., taxa must occur in >25% of individuals within a patient category) or remove low-abundance/low-incidence taxa that generally result from unequal sampling. Where they existed, we used relative abundances obtained from full-length Sanger sequencing in place of the relative abundances obtained from 454 sequencing. To identify taxa that were differentially abundant between controls and PID patients, we employed a mixed effects model using category as a factor variable and taxa relative abundance as the dependent variable, including a random intercept for multiple patient measurements. P-values were generated from the mixed effects model using the glht test (category = “Tukey”) in the multcomp package in R. Random Forests analysis was implemented by the randomForest package in R on the filtered data. C-scores were calculated using a null hypothesis of random community assembly calculated against 10,000 random matrices through the vegan and bipartite R packages. For correlations with patient clinical metadata, we selected taxa whose mean relative abundance across the STAT3-HIES patient group was >0.25% and calculated a Spearman correlation coefficient with clinical blood markers IgE, eosinophil, lymphocytes, lactate dehydrogenase, and hemoglobin levels, and SCORAD. For heatmaps, we visualized those taxa with mean relative abundance >0.25%.
Data access
The sequence data from this study have been submitted to NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number 46333. Patient and sample metadata have been submitted to dbGAp (http://www.ncbi.nlm.nih.gov/gap) under accession number phs000266.v3.p1.
Acknowledgments
We thank members of the Segre laboratory, Helen C. Su, and Mark C. Udey for their helpful discussions and, in particular, Sean Conlan, Clayton Deming, Jennifer Meyer, Effie Nomicos, Deborah Schoenfeld, and Elizabeth Garabedian for their underlying efforts. This work was supported by NIH NHGRI and NCI Intramural Research Programs and in part by 1K99AR059222 (H.H.K.). Sequencing was funded by grants from the National Institutes of Health (1UH2AR057504-01 and 4UH3AR057504-02).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.159467.113.
References
- Balter M 2012. Taking stock of the human microbiome and disease. Science 336: 1246–1247 [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300 [Google Scholar]
- Berglund C, Söderquist B 2008. The origin of a methicillin-resistant Staphylococcus aureus isolate at a neonatal ward in Sweden: Possible horizontal transfer of a staphylococcal cassette chromosome mec between methicillin-resistant Staphylococcus haemolyticus and Staphylococcus aureus. Clin Microbiol Infect 14: 1048–1056 [DOI] [PubMed] [Google Scholar]
- Cho S-H, Strickland I, Boguniewicz M, Leung DYM 2001. Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol 108: 269–274 [DOI] [PubMed] [Google Scholar]
- Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. 2012. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149: 1578–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton MK, Yue JC 2005. A similarity measure based on species proportions. Commun Statist-Theor Method 34: 2123–2131 [Google Scholar]
- Conlan S, Kong HH, Segre JA 2012. Species-level analysis of DNA sequence data from the NIH Human Microbiome Project. PLoS ONE 7: e47075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R 2009. Bacterial community variation in human body habitats across space and time. Science 326: 1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denef VJ, Mueller RS, Banfield JF 2010. AMD biofilms: Using model communities to study microbial evolution and ecological complexity in nature. ISME J 4: 599–610 [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL 2006. NAST: A multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34: W394–W399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, McFall-Ngai M, Relman DA 2007. An ecological and evolutionary perspective on human–microbe mutualism and disease. Nature 449: 811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, Chen A, Kim HS, Lloret MG, Schulze I, et al. 2009. Large deletions and point mutations involving DOCK8 in the autosomal recessive form of the hyper-IgE syndrome. J Allergy Clin Immunol 124: 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D, Nomicos E, Park M, NIH Intramural Sequencing Center Comparative Sequencing Program, et al. 2013. Topographic diversity of fungal and bacterial communities in human skin. Nature 498: 367–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, NISC Comparative Sequencing Program, Bouffard GG, Blakesley RW, Murray PR, et al. 2009. Topographical and temporal diversity of the human skin microbiome. Science 324: 1190–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JK, Caporaso JG, Walker JJ, Spear JR, Gold NJ, Robertson CE, Hugenholtz P, Goodrich J, McDonald D, Knights D, et al. 2013. Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J 7: 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, et al. 2007. STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357: 1608–1619 [DOI] [PubMed] [Google Scholar]
- The Human Microbiome Project Consortium 2012. Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS 2012. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nature 483: 227–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D, Costello EK, Knight R 2011. Supervised classification of human microbiota. FEMS Microbiol Rev 35: 343–359 [DOI] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, et al. 2012. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22: 850–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz B, Oranje AP, Labrèze L, Stalder JF, Ring J, Taïeb A 1997. Clinical validation and guidelines for the SCORAD index: Consensus report of the European Task Force on Atopic Dermatitis. Dermatology (Basel) 195: 10–19 [DOI] [PubMed] [Google Scholar]
- Lai Y, Villaruz AE, Li M, Cha DJ, Sturdevant DE, Otto M 2007. The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Mol Microbiol 63: 497–506 [DOI] [PubMed] [Google Scholar]
- Lennon NJ, Lintner RE, Anderson S, Alvarez P, Barry A, Brockman W, Daza R, Erlich RL, Giannoukos G, Green L, et al. 2010. A scalable, fully automated process for construction of sequence-ready barcoded libraries for 454. Genome Biol 11: R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsen F, Kodner R, Armbrust EV 2010. pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics 11: 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea KW, Hartford O, Davis S, Eidhin DN, Lina G, Speziale P, Foster TJ, Höök M 2000. The serine-aspartate repeat (Sdr) protein family in Staphylococcus epidermidis. Microbiology 146: 1535–1546 [DOI] [PubMed] [Google Scholar]
- McGhee SA, Chatila TA 2010. DOCK8 immune deficiency as a model for primary cytoskeletal dysfunction. Dis Markers 29: 151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JD, Sandler NG, Douek DC 2010. Th17 cells, Job's syndrome and HIV: Opportunities for bacterial and fungal infections. Curr Opin HIV AIDS 5: 179–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. 2007. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448: 1058–1062 [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, et al. 2012. Compartmentalized control of skin immunity by resident commensals. Science 337: 1115–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Frykberg L, Flock J-I, Pei L, Lindberg M, Guss B 1998. A Fibrinogen-binding protein of Staphylococcus epidermidis. Infect Immun 66: 2666–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs HD, Thrasher AJ 2006. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol 117: 725–738 [DOI] [PubMed] [Google Scholar]
- Oh J, Conlan S, Polley EC, Segre JA, Kong HH 2012. Shifts in human skin and nares microbiota of healthy children and adults. Genome Medicine 4: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oranje AP, Glazenburg EJ, Wolkerstorfer A, de Waard-van der Spek FB 2007. Practical issues on interpretation of scoring atopic dermatitis: The SCORAD index, objective SCORAD and the three-item severity score. Br J Dermatol 157: 645–648 [DOI] [PubMed] [Google Scholar]
- Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, et al. 2001. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor Mprf is based on modification of membrane lipids with l-lysine. J Exp Med 193: 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters BM, Noverr MC 2013. Candida albicans-Staphylococcus aureus polymicrobial peritonitis modulates host innate immunity. Infect Immun 81: 2178–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO 2007. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quince C, Lanzen A, Davenport R, Turnbaugh P 2011. Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner ED, Puck JM, Holland SM, Schmitt M, Weiss M, Frosch M, Bergmann M, Davis J, Belohradsky BH, Grimbacher B 2004. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J Pediatr 144: 93–99 [DOI] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, et al. 2004. Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother 48: 4673–4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone L, Roberts A 1990. The checkerboard score and species distributions. Oecologia 85: 74–79 [DOI] [PubMed] [Google Scholar]
- Upadhyay V, Poroyko V, Kim T, Devkota S, Fu S, Liu D, Tumanov AV, Koroleva EP, Deng L, Nagler C, et al. 2012. Lymphotoxin regulates commensal responses to enable diet-induced obesity. Nat Immunol 13: 947–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Eiff C, Becker K, Machka K, Stammer H, Peters G 2001. Nasal carriage as a source of Staphylococcus aureus bacteremia. N Engl J Med 344: 11–16 [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielders C, Vriens M, Brisse S, de Graaf-Miltenburg L, Troelstra A, Fleer A, Schmitz F, Verhoef J, Fluit A 2001. Evidence for in-vivo transfer of mecA DNA between strains of Staphylococcus aureus. Lancet 357: 1674–1675 [DOI] [PubMed] [Google Scholar]
- Williams HC, Burney PG, Pembroke AC, Hay RJ 1994. The U.K. Working Party's diagnostic criteria for atopic dermatitis. III. Independent hospital validation. Br J Dermatol 131: 406–416 [DOI] [PubMed] [Google Scholar]
- Williams HC, Burney PG, Pembroke AC, Hay RJ 1996. Validation of the U.K. diagnostic criteria for atopic dermatitis in a population setting. U.K. Diagnostic Criteria for Atopic Dermatitis Working Party. Br J Dermatol 135: 12–17 [PubMed] [Google Scholar]
- Williams RJ, Henderson B, Sharp LJ, Nair SP 2002. Identification of a fibronectin-binding protein from Staphylococcus epidermidis. Infect Immun 70: 6805–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. 2012. Human gut microbiome viewed across age and geography. Nature 486: 222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, Matthews HF, Davis J, Turner ML, Uzel G, et al. 2009. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med 361: 2046–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]