

Abstract
The development of cancer is a dynamic evolutionary process in which intraclonal, genetic diversity provides a substrate for clonal selection and a source of therapeutic escape. The complexity and topography of intraclonal genetic architectures have major implications for biopsy-based prognosis and for targeted therapy. High-depth, next-generation sequencing (NGS) efficiently captures the mutational load of individual tumors or biopsies. But, being a snapshot portrait of total DNA, it disguises the fundamental features of subclonal variegation of genetic lesions and of clonal phylogeny. Single-cell genetic profiling provides a potential resolution to this problem, but methods developed to date all have limitations. We present a novel solution to this challenge using leukemic cells with known mutational spectra as a tractable model. DNA from flow-sorted single cells is screened using multiplex targeted Q-PCR within a microfluidic platform allowing unbiased single-cell selection, high-throughput, and comprehensive analysis for all main varieties of genetic abnormalities: chimeric gene fusions, copy number alterations, and single-nucleotide variants. We show, in this proof-of-principle study, that the method has a low error rate and can provide detailed subclonal genetic architectures and phylogenies.
Cancer clones evolve by a dynamic Darwinian process of mutational diversification under selective pressures exerted by tissue ecosystems, the immune system, and therapy (Nowell 1976; Merlo et al. 2006; Greaves and Maley 2012). Not only do cancers of a similar type differ in their genomic landscapes—indeed, each is unique—but intraclonal genetic and phenotypic diversity is an inherent feature of this disease (Marusyk et al. 2012). Progression of disease (Merlo et al. 2010; Park et al. 2010) and possibly the general intransigence of advanced disease may be attributable to the genetic diversity within the cells of a tumor and/or within the propagating or stem cells (Greaves 2013). The current vogue for personalized medicine and targeted therapy could well be thwarted or achieve only transient benefit if the targets are themselves subclonally segregated (Greaves and Maley 2012; Swanton 2012).
Second- or next-generation sequencing (NGS) allows whole exome or whole genome sequencing of bulk cancer cells (Stephens et al. 2009; Yates and Campbell 2012), and with adequate depth of parallel sequence reads, it is apparent that many acquired mutations in the cancer genome are subclonally distributed (Nik-Zainal et al. 2012). Subclonal genetic diversity of genome-wide copy number changes has also been demonstrated in a variety of cancers (Klein and Stoecklein 2009; Navin et al. 2010). Cancer cell genetic heterogeneity at the single-cell level has long been recognized by chromosome karyotyping (Wolman 1986) and by fluorescence in situ hybridization (FISH) of tissue sections (Clark et al. 2008). The use of multiplex targeted FISH with three or four colored probes to interrogate the copy number of specific DNA targets facilitates a deeper interrogation of clonal architecture in cancer cell populations from which evolutionary relationships of subclones at diagnosis and relapse can be derived (Anderson et al. 2011). Whole-genome amplification of single cells allowing both sequence and copy number analysis at the single-cell level is now providing even more subclonal information (Navin et al. 2011; Baslan et al. 2012; Zong et al. 2012). These methods combined provide a striking portrait of cancer cell diversity and clonal evolution through the construction of phylogenetic trees characterized by a nonlinear, branching architecture (Anderson et al. 2011; Navin et al. 2011; Gerlinger et al. 2012; Yates and Campbell 2012). But the type of mutation that can be interrogated restricts this approach, and the complexity of clonal architectures is underestimated.
Ideally, what is required for a comprehensive interrogation of the complex genomics of cancer cells is a methodology for single-cell analysis that has the following attributes: (1) an unbiased cell sample from the cancer; (2) highly efficient single-cell sorting; (3) a relatively high-throughput analysis of at least a few hundred cells; and (4) a method that allows the simultaneous detection of multiple genetic alterations of different types, e.g., chimeric fusion genes, copy number alterations (CNA), and single-nucleotide variants (SNVs) in a single cell. We here provide proof-of-principle data using single acute lymphoblastic leukemic (ALL) cells to demonstrate that this is feasible using multiplex targeted DNA amplification from flow-sorted single cells followed by high-throughput Q-PCR using the BioMark HD microfluidic platform (Fluidigm). The versatility of Q-PCR allows simultaneous analysis of a variety of DNA alterations, and coupling this technology with a microfluidic platform capitalizes on a high-throughput system with small reaction volumes that lends itself to single-cell analysis. Q-PCR analysis combined with the BioMark HD has been used for single-cell gene expression analysis (Citri et al. 2012; Pina et al. 2012) and single nucleotide polymorphism (SNP) allelic discrimination analysis with bulk DNA (Wang et al. 2009) but not, to date, for single-cell mutational screening.
Results
Development of the method using the REH leukemia-derived cell line
The established B-cell precursor cell line REH was first used to develop and refine this single-cell analysis method. Our previous FISH (Horsley et al. 2008) and more recent SNP array analysis (data not shown) has characterized this cell line as ETV6-RUNX1 fusion gene positive with multiple CNAs including CDKN2A and MX1 and single nucleotide polymorphisms. In this study, the EPOR SNP (SNP rs318720) was adopted as a surrogate acquired heterozygous SNV anticipated to be present in every cell.
Briefly, single carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled REH and cord blood cells (normal diploid control) were sorted into individual wells of a 96-well plate, lysed, and DNA target amplification completed for regions encompassing the clonotypic ETV6-RUNX1 fusion genomic sequence, CDKN2A, MX1, and the SNP rs318720. The B2M locus, located in a diploid region of the genome, was used as a control. The resulting reaction mix was then diluted and Q-PCR-completed using the 96.96 dynamic array and the BioMark HD. The workflow for this method is shown in Figure 1. A cell was deemed to be positive for a SNP (or SNV) if the Q-PCR cycle threshold (CT) value was below 28. The presence or absence of the signal from the probe complementary to the wild-type sequence determined heterozygous or homozygous mutations, respectively, but the copy number of each allele cannot be inferred (Fig. 2A). The ΔΔCT method (Applied Biosystems, Life Technologies Ltd.) with modifications to incorporate the results from multiple Taqman assays targeting the same region was used to determine the relative copy number for each locus; the use of multiple assays to target one region increased the accuracy of attributed DNA CNAs (Fig. 2B).
Figure 1.

Schematic workflow of the multiplex targeted Q-PCR approach for the simultaneous detection of gene fusions, DNA copy number alterations, and mutations in single cells. Initially, CFSE-labeled single cells are sorted into each well of a 96-well plate and then lysed. Multiplex DNA specific target amplification is then completed to amplify a DNA region of interest (fusion gene, copy number alteration, or SNV) from the two copies found in a single cell to an amount that can be detected by Q-PCR using the BioMark HD. Amplified samples and assays are then loaded into a 96.96 dynamic array that utilizes a valve-controlled capillary network to bring these two mixes together at nanoliter volumes (completed in the IFC controller) for the Q-PCR reaction to take place. This final Q-PCR step determines the gene fusion, mutation, or copy number alteration status for each single cell.
Figure 2.

Single-cell genetic analysis of leukemic cells using the multiplex targeted Q-PCR approach and the BioMark HD platform. (A) Heatmap depicting an example of raw Q-PCR data from the BioMark HD. The rows represent single cells including six cord blood cells and seven REH cells. The columns represent assays, each completed in quadruplicate including B2M (one of three assays), MX1 (two of three assays), CDKN2A (two of three assays), the ETV6-RUNX1 fusion gene assay, and the EPOR SNP (rs318720) assay containing two Taqman probes, one complementary to the wild-type sequence (labeled with FAM) and the other complementary to the SNP sequence (labeled with VIC). The colored boxes at the junction of a row and column indicate the raw CT value (according to the key on the right) obtained for a Q-PCR reaction involving the indicated cell and assay. Assays targeting a mutation or the fusion gene provide a definitive positive or negative result indicating the presence or absence, respectively, of an alteration. The DNA copy number assays provide a raw CT value that requires further analysis (standard ΔΔCT method, Applied Biosystems) to attribute a DNA copy number to the target gene of interest for a single cell; an example can be found in B (refer to the Single-Cell Analysis section in the Methods, and Supplemental Material). (Green arrow) The Q-PCR amplification curves generated from each copy number assay for a single cord blood cell. (Black cross in a colored box) An inadequate amplification curve. (B) Graph depicting the estimated DNA copy number of MX1 attributed reliably to 89 single cells given the assay results from B2M (assay 3) and MX1 (assay 3); one of the nine estimated copy number results used to confidently attribute a DNA copy number to the gene of interest for a single cell. The height of the bar indicates the estimated DNA copy number, and the color of the bar indicates the integer; (light blue) one copy; (dark blue) two copies; (green) three copies; and (yellow) four copies. (CB) Cord blood. (C) Subclonal genetic architecture of the REH cell line inferred by multiplex targeted Q-PCR and confirmed by FISH analysis (126 and 100 cells, respectively); the percentages in parentheses are those obtained by FISH analysis. All cells harbored the ETV6-RUNX1 fusion (F) and the EPOR SNP compared with cord blood cells. DNA copy number is indicated for each gene and subclonal population. Representative FISH images are shown next to their respective subclone. (D) Subclonal genetic architecture of leukemic cells from a child with Down’s syndrome and acute lymphoblastic leukemia (DS-ALL) generated by multiplex targeted Q-PCR and FISH analysis (115 and 100 cells, respectively); 98% of cells harbored the P2RY8-CRLF2 fusion and the IL7R mutation (IL7Rm) by multiplex targeted Q-PCR. Of these, the majority were heterozygous mutations (IL7Rm hete). A minor subclone (2%) had a homozygous IL7R mutation (IL7Rm homo). Loss of CDKN2A was subclonal to the IL7R mutation and proceeded to homozygous loss in 11% of cells. FISH for the P2RY8-CRLF2 fusion and CDKN2A copy number confirmed these results (percentages in parentheses); the IL7R mutation cannot be detected by FISH.
Several approaches were adopted during this experiment to optimize and confirm the presence of a single cell and ensure that all assays performed efficiently under these experimental conditions (details can be found in the Methods). Efficient FACS sorting of single cells was initially confirmed by microscopy. The fluorescent cells were sorted onto a glass slide with the aim of collecting 48 independent single cells; this established the efficiency of the BD FACSAria I (SORP) instrument (BD). In our hands, the failure rate is 2%–4% (one to two occasions per 48 attempts), the majority of which are a failure to sort a cell compared with two cells found in 0.002% of occasions (one in every 528 attempts to sort a single cell). As it is not possible to visualize single cells in a 96-well plate, we sought to quantify the DNA in each well to identify those with high amounts (of the B2M gene) indicating that two or more cells may have inadvertently been collected. These data were consequently removed from the phylogenetic analysis but only constituted a maximum of two wells per plate.
To estimate the error rate of each assay (gene fusion, CNA and SNV assays), a control experiment consisting of 48 cord blood cells was completed for each patient-specific multiplex experiment. Gene fusion and SNV assay false-positive error rates can be found in Supplemental Table 2. Only two assays (SNVs in EZH2 and BAZ2A) generated false-positive results in 2% (1/48) of cord blood cells. CNA assays had an error rate ranging from 4.3% to 7.1% (Supplemental Table 3). False-negative errors rates could not be directly determined as each assay is patient-specific and no positive single-cell control sample is available. However, when using allelic discrimination assays to detect SNVs, each assay generates a signal either for the wild-type sequence, the mutant sequence, or both (most common here). Each single cord blood control cell produced a signal for the wild-type sequence confirming the assay efficiency in the multiplex system. The SNP rs318720 analyzed in the REH cell line produced a signal for the wild-type and polymorphic sequence in each cell interrogated. The assay error rates were used to define a bona fide minor subclonal population (Methods, Defining Subclonal Populations at Low Frequencies section). We were also careful to compare the mutation allele burdens generated by each approach used in this study, including exome sequencing, 454 pyrosequencing, allelic discrimination by digital PCR using bulk DNA, and genotyping of each single cell (Table 1).
Table 1.
Whole-exome sequencing results for two ETV6-RUNX1 positive acute lymphoblastic leukemia cases
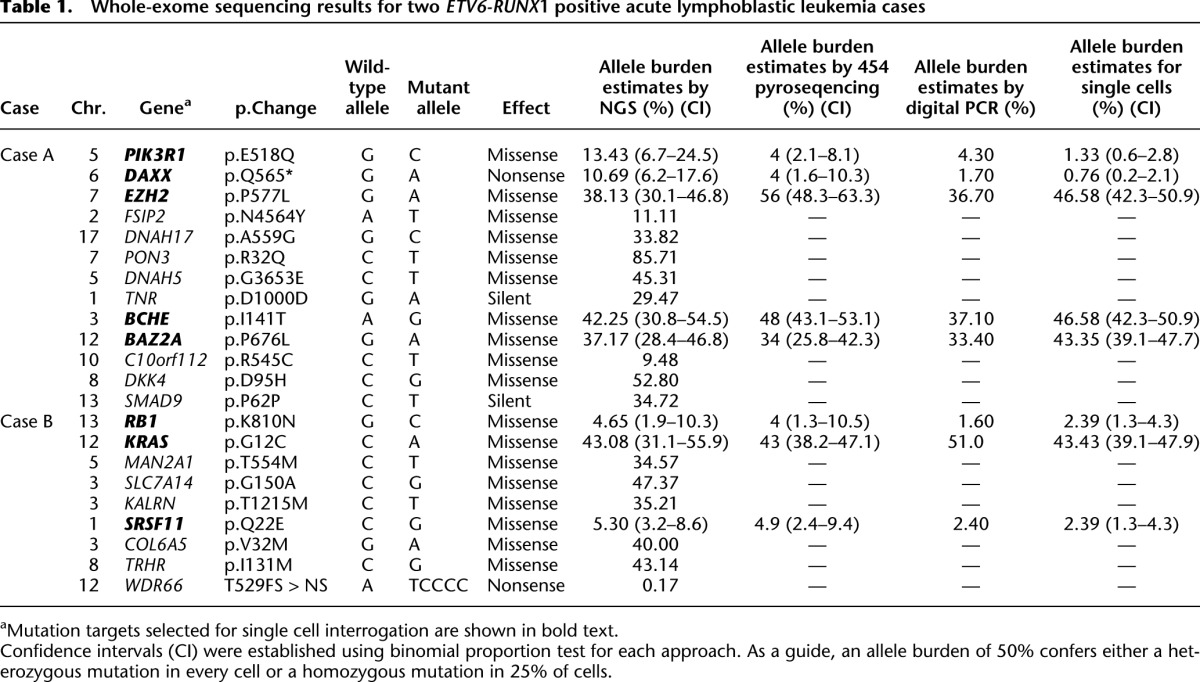
Data from single cells that were removed from the Q-PCR analysis included those wells that showed no data (no cell), those wells in which all B2M assays did not have a strong signal (<28 CT), and wells in which all CNA assays for a target region of interest did not produce CT results within one CT. Data from suggested minor subclonal populations that did not exceed assay error rates were also removed. On average, 75% of interrogated single cells generated complete comprehensive results. A detailed breakdown of the single-cell experiment data with explanations as to why data were removed can be found in Supplemental Table 4.
All REH cells (n = 126 − single cells considered for phylogenetic analysis) scored positive for the ETV6-RUNX1 fusion gene and the EPOR SNP rs318720. Major subclonal populations had varying copies of the MX1 gene; 49% of cells retained two copies of MX1 compared with 42% of cells that had gained either one or two copies of this gene (Fig. 2C). The majority of cells showed loss of both CDKN2A copies, but minor subclones were characterized by either loss of MX1 and/or loss of only one copy of CDKN2A. These DNA copy number data were independently confirmed by FISH using BAC probes complementary to the ETV6-RUNX1 fusion gene, CDKN2A, and MX1. The subclonal structure and percentages obtained by FISH correlated well with the results obtained by single-cell genetic analysis using multiplex targeted Q-PCR and the BioMark HD (Fig. 2C).
Single-cell genetic analysis of a Down’s syndrome ALL case
We replicated this approach using leukemic cells from a child with Down's syndrome and acute lymphoblastic leukemia (DS-ALL) for which we had previously characterized the genomic alterations using SNP analysis and targeted Sanger Sequencing. This case was expected to have a simple but informative clonal architecture involving a P2RY8-CRLF2 fusion gene, loss of CDKN2A, and an IL7R mutation involving the deletion of 8 base pairs and the insertion of 10 nonconsensus base pairs in exon 6.
The subclonal genetic architecture of this DS-ALL case was more complex than suggested by SNP analysis. Using the single-cell multiplex targeted Q-PCR approach, 115 single cells were analyzed and compared with a further 100 cells using FISH (Fig. 2D). Cells that did not harbor any alterations were present at 4% and 2% by FISH and multiplex targeted Q-PCR experiments, respectively, reflecting the 90% BLAST count/low nonleukemic cell mix in this diagnostic sample. The P2RY8-CRLF2 fusion and the IL7R mutation were present in 98% of cells by multiplex targeted Q-PCR. A minor subclone (2%) had lost the IL7R wild-type allele but retained the mutant allele. Loss of CDKN2A was subclonal to the IL7R mutation and proceeded to homozygous loss in 11% of cells. FISH for the P2RY8-CRLF2 fusion and CDKN2A copy number confirmed these results with respect to these loci. The loss of the wild-type IL7R allele in a minor subclone was not anticipated. Subclonal segregation of a homozygous mutation would be very difficult to clarify, except using single-cell analysis.
Single-cell genetic analysis of ETV6-RUNX1 positive ALL cases with exome sequencing data
To expand our approach and confirm that it could be applied to more complex genetic data sets, we selected two ETV6-RUNX1 positive ALL diagnostic bone marrow samples that had been subjected to both whole-exome sequencing to identify SNVs (Table 1) and SNP arrays for CNA (confirmed by exome analysis) (Table 2). Variations in mutation allelic burden suggested the presence of subclonal populations in both cases and was confirmed using Digital PCR (Table 1). Each case also harbored varied chromosomal alterations in both size and location, and known recurrent secondary CNAs in ALL (Mullighan et al. 2007) were identified including loss of PAX5 and CDKN2A.
Table 2.
DNA copy number data for two ETV6-RUNX1 positive acute lymphoblastic leukemia cases
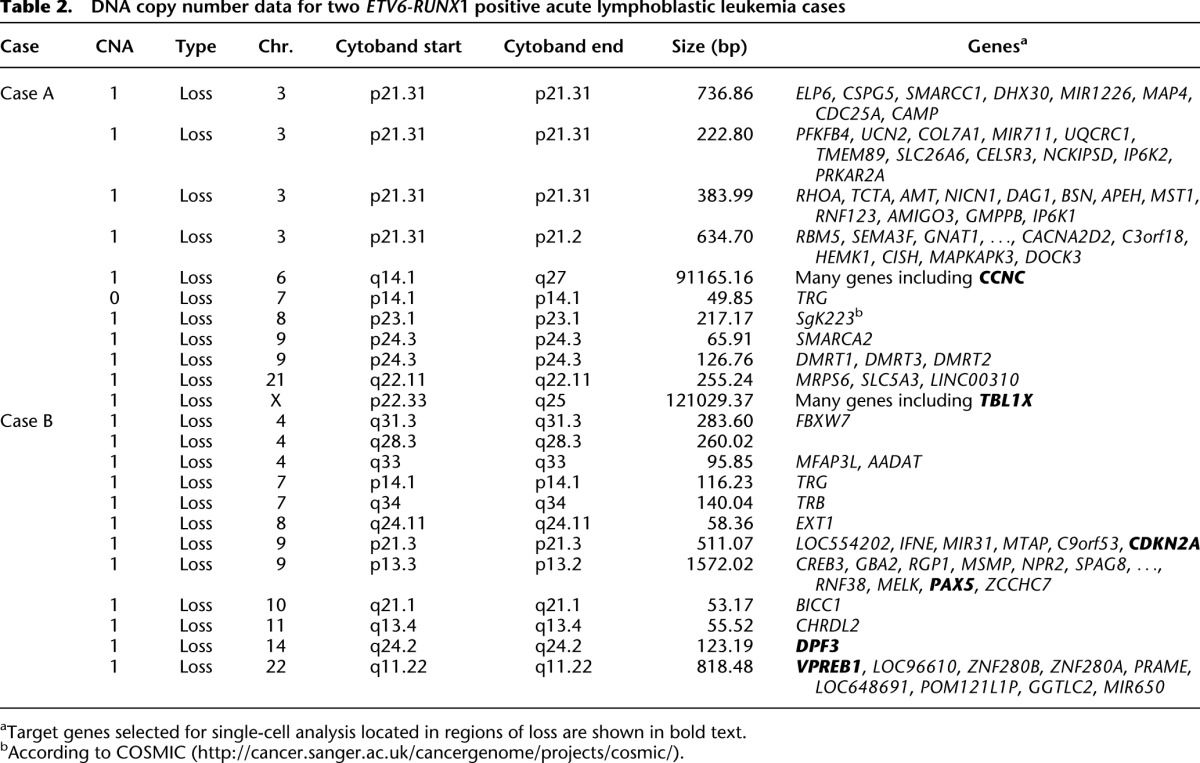
Genomic targets for Case A included SNVs in BCHE (exome SNV read depth 71×, variant reads 30), EZH2 (exome SNV read depth 139×, variant reads 53), PIK3R1 (exome SNV read depth 67×, variant reads 9), DAXX (exome SNV read depth 131×, variant reads 14), and BAZ2A (exome SNV read depth 113×, variant reads 42) and CNAs in CCNC and TBL1X. Targets for Case B included SNVs in KRAS (exome SNV read depth 65×, variant reads 28), RB1 (exome SNV read depth 129×, variant reads 6), and SRSF11 (exome SNV read depth 302×, variant reads 16) and CNAs in VPREB1, PAX5, CDKN2A, and DPF3 (Tables 1, 2). SNVs were chosen based on allelic burden encompassing both high, low, and intermediate frequencies; not all SNVs were included. However, allelic discrimination assays designed for DNAH17, MAN21, and SLC7A14 did not show sufficient target specificity to wild-type and mutant sequence in the multiplex Q-PCR reaction, and the resulting data were inconclusive. Only one attempt was made to design assays for each chosen SNV. The patient-specific ETV6-RUNX1 genomic fusion gene sequence was obtained by long-distance PCR (Wiemels and Greaves 1999) guided by fusion break-point coordinates from whole-exome sequencing. Comprehensive data were collected from 261 single cells for Case A and 254 from Case B.
Derivation of phylogenetic trees using maximum parsimony
To uncover the clonal phylogeny from these single-cell data, we used the maximum parsimony (MP) method (Page and Holmes 1998). The most parsimonious phylogenetic tree is the tree describing the best estimated phylogenetic relationships given the included taxa (group of related cells or clones). Tree branch lengths are directly proportional to the number of evolutionary changes inferred, and the points at which the branches diverge (nodes) represent the ancestor state of a clonal clade (a monophyletic group that includes all descendants of the ancestor). A phylogeny inferred using this criterion shows how the clonal expansion has evolved from a common ancestor toward the observed states. In the event that two (or more) trees hold equal parsimony, all trees are accepted (Page and Holmes 1998). Detailed method explanations of this approach can be found in the Methods and the Supplemental Material.
Maximum parsimony searches for Case A resulted in one maximum parsimonious tree (Fig. 3A). The phylogenetic architecture of the tree shows a quasi-linear structure with a direction toward subclone A4 representing 87.4% of the clonal population; the subclonal heterogeneity in this case is therefore modest, given the number of genomic alterations investigated. The most recent common ancestor (MRCA) of this tree, which is the most recent ancestor from which all clones of the group directly descend, is subclone A3. This clone harbors the ETV6-RUNX1 fusion in addition to BCHE and EZH2 mutations, suggesting that these alterations were relatively early mutational changes in the pathogenesis of this individual ALL. The major clone A4 may have a selective fitness advantage over all other subclones associated with the acquisition of CCNC deletions and a BAZ2A SNV. But, as this is a single time point snapshot of a dynamic process, we cannot exclude the possibility that subclone A4 was spawned before subclones A5 and A2, which are characterized by heterozygous mutations in PIK3R1 and DAXX, respectively. The maximum parsimony algorithm shows the inferred MRCA of the A4–A5 clonal clade (Fig. 3A, gray box), a group of cells that has died out or been outcompeted or if still present, exists at too low a frequency to detect.
Figure 3.

Phylogenetic analysis results for Cases A and B. In each case the observed clone is indicated by a circle. Yellow circles indicate tumor clones, and black circles indicate the normal cell population. The alterations are listed below each subclone; excluding ETV6-RUNX1, those without a number indicate the presence of a mutation and those with a number indicate DNA copies accordingly. The boxed subclone in gray is inferred; a group of cells that has died out or been outcompeted, or if still present, exists at a low frequency that cannot be reliably detected by this approach. The number in italics at each node indicates the jackknifing value. The distance unit is indicated. (A) One parsimonious tree was found for Case A consisting of four subclones with modest heterogeneity. The major clone (A4) represents 87.4% of the population. The size of each circle is proportional to the number of single cells included in the subclone except A4, which has been reduced by a third in this tree. (B) In Case B, there are two equally parsimonious trees composed of seven subclones. These two trees differ by the position of subclones B4 and B5, which are equal parsimonious ancestors to subclones B3, B7, and B8. This case shows increased heterogeneity with the major clone representing 54.9% of the population (B3). The major clone B3 is reduced by half in this tree.
Maximum parsimony searches for Case B produced two equally parsimonious trees with identical topological structures (Fig. 3B). Both trees show complex branching structures with a marked subclonal heterogeneity of seven clones that differ only for the position of subclones B4 and B5. The MRCA of the clonal expansion is represented by subclone B2, which shows a homozygous deletion of VPREB1 in addition to the ETV6-RUNX1 fusion. This subclone is the biggest clonal group after clone B3 (at diagnosis) despite the genetic diversity of the progressive subclonal populations, suggesting that this subclone may be quiescent or reside in a niche environment. The acquisition of the KRAS mutation presumably provides selective advantage as a secondary driver event. A minor subclone was also identified in which the wild-type KRAS allele was lost, suggesting the acquisition of homozygosity for the KRAS mutation. The number of mutant gene copies (one or two) in each single cell cannot be determined using the Taqman KRAS SNV assay used here, but the increased allele burden characterized by whole-exome sequencing, 454 pyrosequencing, and digital PCR validates this conclusion.
The position of subclones B4 or B5 indicates that these populations are equally parsimonious ancestors of the clonal clade constituted by subclones B3, B7, and B8, representing ∼65% of all examined cells. This represents the reiteration of either PAX5 or CDKN2A loss independently in two different branches. Reiterated CNA of the same gene is a feature of the subclonal genetic architecture in pediatric ALL (Anderson et al. 2011; Waanders et al. 2012), suggesting that these deletions may not only provide selective advantage but may be a target for DNA level breakage via, for example, RAGS (Kitagawa et al. 2002; Waanders et al. 2012) or AID (Longerich et al. 2006; Klemm et al. 2009).
Discussion
Cancers have complex patterns of acquired mutations, and patients with closely related subtypes of cancer have unique or clone-specific mutation patterns. Additional complexity clearly exists within each individual cancer, as mutations are acquired serially and with a variegated pattern of distribution in subclones. As mutational profiles are increasingly used for differential diagnosis, prognostication, and therapeutic targeting, this multidimensional complexity is of some consequence.
Systematically interrogating subclonal genetic complexity poses a considerable technical challenge. A clonal architecture or phylogeny can be inferred bioinformatically by analysis of high-depth NGS data (Nik-Zainal et al. 2012; Yates and Campbell 2012). Nik-Zainal et al. (2012) reconstructed clonal phylogenies through the development of novel algorithms that rely on whole-genome sequencing data (200× coverage) to phase mutations with germline polymorphisms and define clonal and subclonal phylogenies. This is in marked contrast to the exome sequencing data used in this study, which do not offer the opportunity of continuous genomic information that would allow one to reconstruct such phylogenies with confidence. Additionally, exome data in leukemias show low total mutation burden (on average 10), which renders them further limited to differentiate clonal phylogenies. It is, in fact, this limitation of exome sequencing data and to a lesser extent whole-genome sequencing that the present study is precisely addressing where, as shown in the example of Case B, two SNVs of close allele burden estimates RB1 and SRSF11 (4.65 and 5.30, respectively) could be in the same (or separate) subclone with equal probability. Without single-cell data, this level of phylogenetic resolution is not possible. We conclude that single-cell genetic analysis is required for a robust and definitive designation of the segregation pattern of mutations within cancer clones. Theoretically, the best approach might be to sequence the genomes of individual cells. Significant progress has recently been made in this regard (Baslan et al. 2012; Zong et al. 2012), but error rates and low cell throughput are, currently, significant limitations. We adopted the alternative strategy of analyzing single cells from cancers whose genomic, mutational profile at the cell population level was already established by high-resolution SNP arrays and whole-exome sequencing.
Using the strategy outlined here, we were able to detect all three categories of mutational change—fusion gene, CNA, and SNV—in a single cell. Our method is amenable to high-throughput analysis: In each case, we were able to analyze 200–300 leukemic cells. We assume that the profiles that emerge from these numbers of cells are representative of the patients' leukemias. This would be more demanding with adult carcinomas where the topographical segregation of distinct subclones could result in selective sampling, but clearly multiple small biopsies could be assessed (Park et al. 2010; Gerlinger et al. 2012). The screening of many thousands of single cells is possible by the method we report but is restrained at present by cost.
The phylogenetic trees inferred from the cellular distribution of SNVs in larger numbers of cells provide additional evidence that cancers evolve within a branching architecture of subclones. These detailed and complex subclonal architectures would not be detected by other genetic techniques. Prior studies on ALL suggested that the latter is generated and sustained by genetically diverse cancer propagating or stem cells (Anderson et al. 2011; Notta et al. 2011), and it will be important to confirm this using the current microfluidic platform.
This proof-of-principle study using leukemic cells illustrates that it is possible to assess single cells simultaneously for different types of genetic lesions—fusion genes, copy number alterations, and single nucleotide variants—and from these data to construct a clonal, evolutionary phylogeny. Leukemias are likely to be significantly less complex in this respect than carcinomas (Vogelstein et al. 2013), but nevertheless, the subclonal architectures we illustrate here for ALL are likely to be a significant underestimation of complexity. Higher definition of clonal structure, including minor clones at <1%, is however achievable by both increasing the depth of initial sequencing and by screening larger numbers of single cells. Also, informative though these clonal analyses are, they remain single time point snapshots of a very dynamic evolutionary process. Ideally, single-cell genetics and phylogenetic tree construction should be applied to serial samples from individual patients, i.e., diagnosis versus relapse, primary versus metastases, and pre- versus post-chemotherapy. It is known that these major transitions can involve selective sweeps or stringent clonal selection (Mullighan et al. 2008; Liu et al. 2009; Anderson et al. 2011; Diaz et al. 2012).
Dissecting the detailed clonal architecture of cancer has significant clinical implications. The extent of intraclonal genetic diversity may be predictive of progression of disease (Maley et al. 2006) or clinical outcome (Mroz et al. 2013). Cancer genomics holds the promise of personalized medicine with therapy targeted at products of recurrent “driver” mutations (Chin et al. 2011). Subclonal segregation would not be a desirable credential of any candidate target. Targeting even a major branch of the phylogenetic tree rather than the founder lesion would be predicted to have only transient benefit and, moreover, provide selective pressure for the emergence of previously minor clones (Greaves and Maley 2012; Swanton 2012).
Methods
Samples
The precursor B-cell leukemic cell line, REH, was purchased from American Type Culture Collection (ATCC, Virginia, USA) and cultured in RPMI-1640 medium, 10% FCS.
The patient samples studied in this investigation were collected from Italian or UK hospitals, with local ethical review committee approval (CCR 2285, Royal Marsden Hospital NHS Foundation Trust). Bone marrow aspirates or peripheral blood samples underwent lymphoprep separation and were viably frozen in 10% DMSO, 90% FCS and stored in liquid nitrogen (10% DMSO, 90% FCS).
Bulk sample analysis
Single nucleotide polymorphism and DNA copy number array analysis for Case A and Case B
To define DNA copy number alterations and SNVs for these cases, we used the Affymetrix Cytogenetics Whole Genome 2.7M Array (Affymetrix). Briefly, 100 ng of genomic DNA from both diagnostic and remission samples was whole-genome amplified (WGA) using the Affymetrix Cytogenetics Reagent Kit and the Affymetrix assay protocol according to the manufacturer's instructions. Samples were then fragmented to generate small products (<300 bp), which were subsequently biotin-labeled, denatured, and loaded into the arrays. After hybridization, the chips were washed, stained (streptavidin-PE), and scanned using the GeneChip Scanner 3000. CEL files were generated using Affymetrix GeneChip Command Console (AGCC) v3.1 and analyzed by Chromosome Analysis Suite (Affymetrix) software, version 1.2.2. Quality-control metrics for each case can be found in Supplemental Table 1A. SNP and DNA copy number analysis for REH and the DS-ALL sample was completed using the Affymetrix Genome-Wide Human SNP Array 6.0. The method details can be found in the Supplemental Material.
ETV6-RUNX1 fusion breakpoints
Genomic sequences for the ETV6-RUNX1 positive samples (REH cell line and patient Cases A and B) were cloned using long-distance inverse PCR using DNA from bulk cells as described before (Wiemels and Greaves 1999). P2RY8-CRLF2 fusions were sequenced according to Mullighan et al. (2009).
Whole-exome sequencing
Matched genomic DNA (3–5 μg) from leukemic and complete remission samples from the two cases with childhood acute lymphoblastic leukemia was prepared for Illumina paired-end sequencing (Illumina). Exome enrichment was performed using the Agilent SureSelectXT Human All Exon 50Mb kit (Agilent Technologies Ltd.) as per the manufacturer's guidelines but without the pre-enrichment PCR amplification ENREF_1. Solid phase reversible immobilization (SPRI) bead cleanup was used to purify products in preparation for sequencing (Agencourt AMPure XP beads, Beckman Coulter). Flow-cell preparation, cluster generation, and paired-end sequencing (75 bp reads) were performed according to the Illumina protocol guidelines on an Illumina GAII Genome Analyzer. The target coverage per sample was for 70% of the captured regions at a minimum depth of 30× sequencing coverage.
Sequencing reads were aligned to the human genome (NCBI build 37) using the BWA algorithm on default settings (Li and Durbin 2010). Duplicate reads derived by PCR were removed using Picard, and reads mapping outside the targeted region of the genome were excluded from the analysis. Standard internal quality-control evaluation of sequencing data including the percent of uniquely mapped reads, the percent of target region covered, the percent of unmapped reads, sequence quality metrics, and total sequencing output (in GB) was performed for all samples. The remaining uniquely mapping reads (∼60%) provided 60%–80% coverage over the targeted exons at a minimum depth of 30×. Leukemic sample identity relative to the matched remission sample was controlled by digital genotyping of 100 genome-wide SNP markers prior to variant calling.
In house-variant caller CaVEMan (cancer variants through expectation maximization) was used to call single nucleotide substitutions (Varela et al. 2011). To call insertions and deletions, split-read mapping was implemented as a modification of the Pindel algorithm (Ye et al. 2009). Copy number and loss of heterozygosity (LOH) analysis was performed using ASCAT (Van Loo et al. 2010). Further details of these steps can be found in the Supplemental Material. For validation, all putative somatic indels were confirmed by capillary sequencing via 454 pyrosequencing (Roche) of both tumor and remission samples from each patient. Mutant allele burden estimates were derived from the fraction of reads reporting the mutant allele over the total read depth at each genomic location, and confidence intervals were derived using the binomial distribution.
Commercial and custom primers for Q-PCR and digital PCR
Primer Express Software (Applied Biosystems) was used to design custom genotyping Taqman Q-PCR assays for an SNV that could distinguish the mutant allele from its wild-type counterpart. Each SNV assay contained allele-specific minor-groove binder (MGB) probes for the wild-type allele (FAM-labeled) and the mutant allele (VIC-labeled) (Supplemental Table 2). These assays were tested to ensure specificity and reliability (refer to the Assay Validation section in the Supplemental Material). Assays to detect a fusion breakpoint were designed using a similar approach with an FAM-labeled MGB probe straddling the fusion break point. DNA copy number Taqman assays were purchased from Applied Biosystems as these have been designed for uniform amplification efficiency and have been commercially validated. Three CNA assays were chosen within each DNA target region of interest and the diploid reference region encompassing B2M.
Digital PCR
Digital PCR was used to quantify target sequences and estimate mutant allele burdens within bulk DNA from each patient at diagnosis and in cord blood. This was completed using the 12.765 digital array (Fluidigm) and the BioMark HD. This digital array contains 12 panels each with 765 individual microfluidic chambers (6 nL volume per chamber). Six targets were simultaneously interrogated according to the manufacturer's instructions; 5 ng of DNA per panel. The number of target molecules per panel was determined using BioMark HD Digital PCR software; SNV frequencies were calculated by dividing the number of mutant allele copies by the total number of copies for the wild-type and mutant alleles.
Single-cell analysis
Single-cell labeling and flow sorting
Single-cell sorting was performed on a BD FACSAria I (SORP) instrument (BD) equipped with an automated cell deposition unit using the following settings: 100-μm nozzle, 1.4 bar sheath pressure, 32.6 kHz head drive, and a flow rate that gave one to 200 events per second. Details of viable cell thawing, single-cell CFSE staining (according to the manufacturer's instructions), cell sorting parameter explanations, and the assessment of single-cell sorting efficiencies can be found in the Supplemental Material. Two 96-well plates of single cells were collected for REH and the DS-ALL Case, and four plates were collected for the two ALL Cases. The plates were composed of a no template control (NTC), 11 control cord blood cells, and 84 target cells (REH or patient cells).
Single-cell multiplex targeted pre-amplification and Q-PCR
Labeled single cells were sorted into 2.5 μL of lysis buffer composed of 1 mg/mL proteinase K (Qiagen) and 0.5% Tween 20 in HEPES-buffered saline (Sigma-Aldrich). Lysis was carried out for 50 min at 60°C followed by 10 min at 98°C. Specific (DNA) targeted amplification (STA) was then performed prior to Q-PCR. This multiplex STA reaction was composed of 5 μL of pre-amplification master mix (Life Technologies) and 2.5 μL of 1:40 primer mix (containing all primers for the gene targets of interest). Denaturation was completed for 15 min at 95°C, followed by 24 cycles of amplification for 15 sec at 95°C and for 4 min at 60°C. The STA product was then diluted 1:6 using DNA suspension buffer (Teknova). Finally, 2.7 μL of the single-cell target amplified DNA was interrogated by Q-PCR for each DNA target of interest using the 96.96 dynamic microfluidic array and the BioMark HD as recommended by the manufacturer; thermal phase for 1800 sec at 70°C, for 60 sec at 25°C, followed by a hot start phase of 60 sec at 95°C. This was followed by 35 cycles of 5 sec at 96°C and 20 sec at 60°C. CNA assays were completed in quadruplicates, and SNV or fusion assays were completed in duplicates.
Single-cell Q-PCR analysis
The BioMark HD generates a CT value for each reaction. A heterozygous mutation was considered to be present if the signals from the mutant and wild-type sequence probes (FAM and VIC, respectively) had a CT value <28 in a single cell. A homozygous mutation was considered to be present if there was no wild-type sequence signal.
To ensure robust DNA CNA data from a system that can be influenced by assay efficiency and experimental variation, we used the ΔΔCT method (Applied Biosystems) to determine a copy number for each locus with modifications to incorporate data from three distinct assays targeting the control region (B2M) and the region of interest. The ΔΔCT value was calculated for every target gene assay using each of the three reference gene CT values generating nine estimated DNA copy number results for a region of interest. A confidence metric was assigned to the estimated copy number inferring the confidence with which an estimated copy number could be deemed true (according to Applied Biosystems CopyCaller Software v2) (details of this approach can be found in the Supplemental Material). The weighted mean of the nine estimated DNA copy numbers (for a region of interest) was used as the final DNA copy number taking into consideration the confidence metric attributed to each. This reduced the contribution of less-reliable estimated DNA copy numbers to the final DNA copy number. Estimated copy number results were not considered if the confidence value was <50% or the estimated copy number was greater than four (with only quadruplicates per assay, the results are not robust enough to accurately detect DNA copy numbers greater than four) (Weaver et al. 2010). At least two of the nine estimated copy numbers must have a confidence value above 50% to calculate the final copy number for a region of interest.
Defining subclonal populations at low frequencies
Each assay type (gene fusion, CNA, or SNV) varied in error rate demanding careful consideration when defining subclonal populations at low frequencies. Gene fusion assays and SNV assays did not yield any false-positive results in the control experiments, except EZH2 and BCHE for which we saw one cell (Supplemental Table 2). However, CNA assays had an average error rate of 5.4% (Supplemental Table 3). Consequently, the following criterion was used to define a bona fide subclonal population: An observed signal pattern (character state) must be attributed to four or more cells (in this experiment, the equivalent of ∼1%). A population present at less than the error rate for a given CNA assay cannot be defined by that single CNA alone, but if two CNA alterations define a subclonal population, it was deemed to be true. A single fusion event or SNV can distinguish a minor subclonal population. For example, in Case B, subclonal B6 is defined from the ancestral subclone B2 by a KRAS mutation.
Interphase fluorescence in situ hybridization (FISH)
Methanol–acetic acid fixed cells, prepared by standard cytogenetic techniques, were used for all FISH studies. Interphase FISH for the ETV6–RUNX1 fusion gene in combination with probes for regions of copy number alteration was performed using a commercial LSI ETV6-RUNX1 extra signal (ES) probe (Vysis, Abbott Laboratories) as previously described (Horsley et al. 2008; Bateman et al. 2010). The P2RY8-CRLF2 gene fusion was identified using a dual color break-apart probe consisting of two bacterial artificial chromosome (BAC) probes flanking the CRLF2 locus (Supplemental Table 5). BAC and fosmid probes for this and other regions of interest were obtained from the BACPAC Resource Center (Children's Hospital, Oakland Research Institute) (http://bacpac.chori.org). Probes were labeled by nick translation with either biotin-16-dUTP (Roche Ltd.), SpectrumRed, or SpectrumOrange (Vysis) and hybridized in combination. FISH was performed by standard protocols (Horsley et al. 2008; Bateman et al. 2010) with a single detection layer of streptavidin-Cy5 for biotinylated probes. Fluorescent signals were viewed using an Olympus AX2 fluorescence microscope equipped with narrow bandpass filters for FITC, SpectrumOrange, TexasRed, and Cy5. Images were captured and analyzed using a charge-coupled device (Photometrics) and SmartCapture 3 software version 3.0.4 (Digital Scientific UK). In each case, 100 nuclei were scored for the presence of the fusion gene and other targets of interest. Nuclei from karyotypically normal cells (peripheral blood samples from normal individuals) were used to assess probe hybridization efficiency. The percentage of cells with the expected normal signal pattern was 97%–100% (mean = 98%) for each probe (Supplemental Table 5).
Phylogenetic analysis and clonal evolution
Clonal groups
Copy numbers and genotypes for each interrogated cell were concatenated in a linear array of nine characters and then aligned to identify the same motif in the array. Cells sharing identical alterations were grouped together in the same clonal group. Clones were then aligned and used to infer the evolutionary history of each patient's leukemia.
Maximum parsimony
Maximum parsimony searches were conducted using heuristic searches. Heuristic searches were performed with a series of 1 million random additional clones and tree branch-swapping using a bisection-reconnection (TBR) algorithm. All characters except the initiating genetic lesion (ETV6-RUNX1 fusion gene) were weighted equally, and state graphs and step matrices were used to assign equal costs to each character state transition in different genes. The cost assigned for each transition is linear and results from the equation yi = 2xi. We assigned a transition cost equal to 1 (y0 = x0) only to the transition 0 (no fusion) to 1 (one fusion) for the ETV6-RUNX1 fusion. The character state graphs and corresponding matrices used are shown in Supplemental Table 6. The normal clone (according to the alterations interrogated) found in each case was assumed to be the ancestral clone and included in the analysis as the root of the tree/trees. All parsimony analyses were performed using the computer software PAUP* version 4.0b10 for Linux (Swofford 2005). Trees were visualized using Dendroscope Software version 3 (Huson and Scornavacca 2012).
Node support: jackknife
Support for the internal branches was assessed in PAUP* by jackknife with 1000 pseudo-replicates. Heuristic searches with randomly added taxa followed by applying tree bisection and reconnection algorithms were used for each jackknifing iteration deleting 12.5% of the characters in each pseudo-replica. A jackknife 50% majority-rule consensus tree (Margush and McMorris 1981) was used to support the node of phylogenetic trees inferred.
Mimicking the bootstrap procedure
We wrote an R in-house script to mimic the bootstrap resampling method. The script samples without replacement from the aligned clones and generates replicas with columns shuffled in different orders.
The script was run on 50 separate occasions for Cases A and B, using R software for statistical computing version 2.15 (R Core Team 2013). Each resulting replica was used as input for a maximum parsimony search employing the same settings as described in the Supplemental Material.
Data access
The whole-exome sequencing data generated in this study have been submitted to The European Genome-phenome Archive (EGA; http://www.ebi.ac.uk/ega/) under accession number EGAD00001000636. Single nucleotide polymorphism and copy number analysis by SNP array data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE49215.
Acknowledgments
This work was supported by Leukaemia & Lymphoma Research United Kingdom and the Kay Kendall Leukaemia Fund United Kingdom.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.159913.113.
References
- Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. 2011. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469: 356–361 [DOI] [PubMed] [Google Scholar]
- Baslan T, Kendall J, Rodgers L, Cox H, Riggs M, Stepansky A, Troge J, Ravi K, Esposito D, Lakshmi B, et al. 2012. Genome-wide copy number analysis of single cells. Nat Protoc 7: 1024–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman CM, Colman SM, Chaplin T, Young BD, Eden TO, Bhakta M, Gratias EJ, van Wering ER, Cazzaniga G, Harrison CJ, et al. 2010. Acquisition of genome-wide copy number alterations in monozygotic twins with acute lymphoblastic leukemia. Blood 115: 3553–3558 [DOI] [PubMed] [Google Scholar]
- Chin L, Andersen JN, Futreal PA 2011. Cancer genomics: From discovery science to personalized medicine. Nat Med 17: 297–303 [DOI] [PubMed] [Google Scholar]
- Citri A, Pang ZP, Sudhof TC, Wernig M, Malenka RC 2012. Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat Protoc 7: 118–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J, Attard G, Jhavar S, Flohr P, Reid A, De-Bono J, Eeles R, Scardino P, Cuzick J, Fisher G, et al. 2008. Complex patterns of ETS gene alteration arise during cancer development in the human prostate. Oncogene 27: 1993–2003 [DOI] [PubMed] [Google Scholar]
- Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486: 537–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpet P, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366: 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M 2013. Cancer stem cells as ‘units of selection.’ Evol Appl 6: 102–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves M, Maley CC 2012. Clonal evolution in cancer. Nature 481: 306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsley SW, Colman S, McKinley M, Bateman CM, Jenney M, Chaplin T, Young BD, Greaves M, Kearney L 2008. Genetic lesions in a preleukemic aplasia phase in a child with acute lymphoblastic leukemia. Genes Chromosomes Cancer 47: 333–340 [DOI] [PubMed] [Google Scholar]
- Huson DH, Scornavacca C 2012. Dendroscope 3: An interactive tool for rooted phylogenetic trees and networks. Syst Biol 61: 1061–1067 [DOI] [PubMed] [Google Scholar]
- Kitagawa Y, Inoue K, Sasaki S, Hayashi Y, Matsuo Y, Lieber MR, Mizoguchi H, Yokota J, Kohno T 2002. Prevalent involvement of illegitimate V(D)J recombination in chromosome 9p21 deletions in lymphoid leukemia. J Biol Chem 277: 46289–46297 [DOI] [PubMed] [Google Scholar]
- Klein CA, Stoecklein NH 2009. Lessons from an aggressive cancer: Evolutionary dynamics in esophageal carcinoma. Cancer Res 69: 5285–5288 [DOI] [PubMed] [Google Scholar]
- Klemm L, Duy C, Iacobucci I, Kuchen S, von Levetzow G, Feldhahn N, Henke N, Li Z, Hoffmann TK, Kim YM, et al. 2009. The B cell mutator AID promotes B lymphoid blast crises and drug resistance in chronic myeloid leukemia. Cancer Cell 16: 232–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, Chen L, Ewing CM, Eisenberger MA, Carducci MA, et al. 2009. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med 15: 559–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, Basu U, Alt F, Storb U 2006. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol 18: 164–174 [DOI] [PubMed] [Google Scholar]
- Maley CC, Galipeau PC, Finley JC, Wongsurawat VJ, Li X, Sanchez CA, Paulson TG, Blount PL, Risques RA, Rabinovitch PS, et al. 2006. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet 38: 468–473 [DOI] [PubMed] [Google Scholar]
- Margush T, McMorris FR 1981. Consensus n-trees. Bull Math Biol 43: 239–244 [Google Scholar]
- Marusyk A, Almendro V, Polyak K 2012. Intra-tumour heterogeneity: A looking glass for cancer? Nat Rev Cancer 12: 323–334 [DOI] [PubMed] [Google Scholar]
- Merlo LMF, Pepper JW, Reid BJ, Maley CC 2006. Cancer as an evolutionary and ecological process. Nat Rev Cancer 6: 924–935 [DOI] [PubMed] [Google Scholar]
- Merlo LM, Shah NA, Li X, Blount PL, Vaughan TL, Reid BJ, Maley CC 2010. A comprehensive survey of clonal diversity measures in Barrett's esophagus as biomarkers of progression to esophageal adenocarcinoma. Cancer Prev Res (Phila) 3: 1388–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroz EA, Tward AD, Pickering CR, Myers JN, Ferris RL, Rocco JW 2013. High intratumor genetic heterogeneity is related to worse outcome in patients with head and neck squamous cell carcinoma. Cancer 119: 3034–3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, et al. 2007. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446: 758–764 [DOI] [PubMed] [Google Scholar]
- Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR 2008. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322: 1377–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, et al. 2009. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 41: 1243–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Krasnitz A, Rodgers L, Cook K, Meth J, Kendall J, Riggs M, Eberling Y, Troge J, Grubor V, et al. 2010. Inferring tumor progression from genomic heterogeneity. Genome Res 20: 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. 2012. The life history of 21 breast cancers. Cell 149: 994–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE 2011. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469: 362–367 [DOI] [PubMed] [Google Scholar]
- Nowell PC 1976. The clonal evolution of tumor cell populations. Science 194: 23–28 [DOI] [PubMed] [Google Scholar]
- Page RMD, Holmes EC 1998. Molecular evolution: A phylogenetic approach. Wiley-Blackwell, New York [Google Scholar]
- Park SY, Gönen M, Kim HJ, Michor F, Polyak K 2010. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest 120: 636–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina C, Fugazza C, Tipping AJ, Brown J, Soneji S, Teles J, Peterson C, Enver T 2012. Inferring rules of lineage commitment in haematopoiesis. Nat Cell Biol 14: 287–294 [DOI] [PubMed] [Google Scholar]
- R Core Team. 2013. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
- Stephens PJ, McBride DJ, Lin M-L, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, et al. 2009. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462: 1005–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton C 2012. Intratumor heterogeneity: Evolution through space and time. Cancer Res 72: 4875–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D. 2005. PAUP*: Phylogenetic analysis using parsimony (and other methods), Version 4.0, Beta 10. Sinauer, Sunderland, MA. [Google Scholar]
- Van Loo P, Nordgard SH, Lingjaerde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, et al. 2010. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci 107: 16910–16915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, Davies H, Jones D, Lin ML, Teague J, et al. 2011. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 469: 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu V, Zhou S, Diaz L, Kinzler K 2013. Cancer genome landscapes. Science 339: 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waanders E, Scheijen B, van der Meer LT, van Reijmersdal SV, van Emst L, Kroeze Y, Sonneveld E, Hoogerbrugge PM, van Kessel AG, Van Leeuwen FN, et al. 2012. The origin and nature of tightly clustered BTG1 deletions in precursor B-cell acute lymphoblastic leukemia support a model of multiclonal evolution. PLoS Genet 8: e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Lin M, Crenshaw A, Hutchinson A, Hicks B, Yeager M, Berndt S, Huang W-Y, Hayes R, Chanock S, et al. 2009. High-throughput single nucleotide polymorphism genotyping using nanofluidic dynamic arrays. BMC Genomics 10: 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver S, Dube S, Mir A, Qin J, Sun G, Ramakrishnan R, Jones RC, Livak KJ 2010. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods 50: 271–276 [DOI] [PubMed] [Google Scholar]
- Wiemels JL, Greaves M 1999. Structure and possible mechanisms of TEL-AML1 gene fusions in childhood acute lymphoblastic leukemia. Cancer Res 59: 4075–4082 [PubMed] [Google Scholar]
- Wolman SR 1986. Cytogenetic heterogeneity: Its role in tumor evolution. Cancer Genet Cytogenet 19: 129–140 [DOI] [PubMed] [Google Scholar]
- Yates LR, Campbell PJ 2012. Evolution of the cancer genome. Nat Rev Genet 13: 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z 2009. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, Xie XS 2012. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338: 1622–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]