

Abstract
Cardiac myosin binding protein C (cMyBP-C) phosphorylation is differentially regulated in the normal heart and during disease development. Our objective was to examine in detail three phosphorylatable sites (Ser-273, Ser-282, and Ser-302) present in the protein’s cardiac-specific sequences, as these residues are differentially and reversibly phosphorylated during normal and abnormal cardiac function. Three transgenic lines were generated: DAA, which expressed cMyBP-C containing Asp-273, Ala-282, Ala302, in which a charged amino acid was placed at residue 273 and the remaining two sites rendered nonphosphorylatable by substituting alanines for the two serines; AAD containing Ala-273, Ala-282, Asp-302), in which aspartate was placed at residue 302 and the remaining two sites rendered nonphosphorylatable; and SDS containing Ser-273, Asp-282, Ser-302. These mice were compared to mice constructed previously along similar lines: wild type, in which normal cMyBP-C is transgenically expressed, AllP−, in which alanines were substituted and ADA mice as well. DAA and AAD mice showed pathology that was more severe than cMyBP-C nulls. DAA and AAD animals exhibited left ventricular chamber dilation, interstitial fibrosis, irregular cardiac rhythm and sudden cardiac death. Our results define the effects of the sites’ post-translational modifications on cMyBP-C functionality and together, give a comprehensive picture of the potential consequences of site-specific phosphorylation. Ser-282 is a key residue in controlling S2 interaction with the thick and thin filaments. The new DAA and AAD constructs show that phosphorylation at one site in the absence of the ability to phosphorylate the other sites, depending upon the particular residues involved, can lead to severe cardiac remodeling and dysfunction.
Keywords: cardiac function, sarcomere, phosphorylation, cardiac myosin binding protein C
1. Introduction
Myosin binding protein C (MyBP-C) is a thick filament protein that helps regulate the structural integrity and actomyosin movement of sarcomeres [1]. Mutations in the cardiac isoform are a frequent cause of familial hypertrophic cardiomyopathy, with a recent review reporting that approximately 200 mutations in MYBPC3 have been associated with human disease [2].
Three isoforms of MyBP-C are expressed in mammals: slow skeletal, fast skeletal and cardiac, with each encoded by a separate gene (MYBPC1, MYBPC2, MYBPC3, respectively). Structurally, the isoforms are closely related, with all containing seven immunoglobulin-like (IgG) domains and three fibronectin-like motifs (Fig. 1A). cMyBP-C constitutes 1–2% of the total myofibrillar protein [3, 4] and is located in the A-band of the cardiac sarcomere, but is uniquely arrayed and sequestered, appearing as 7–9 bands adjacent on each side of the central M line [5]. The N-terminus of the protein interacts with both actin and myosin, while the mid-regions (C5–C8) of the protein may interact with one another to form a trimeric “collar”-like structure [6], although the actual architecture of the protein remains unresolved and a “strut” or “tether” model has also been proposed [7]. C9–C10 binds to titin and the last domain, C10, binds to the light meromyosin (LMM) region of myosin heavy chain [8]. High resolution atomic force microscopy suggests that cMyBP-C is a mixture of ordered and disordered structures, transiting from one form to other form depending upon environmental factors [9]. Recently, an actin-binding site has also been reported in the COOH half of the molecule but this has not been independently confirmed [10].
Figure 1.

Cardiomyocyte-specific replacement with altered cMyBP-C. A) Schematic diagram showing the domain structure of cMyBP-C, with the 8 IgG domains (blue), 3 fibronectin domains (green) and cardiac specific M domain (pink). B) The phosphorylatable serine residues that are mutated in the different constructs are indicated in color. C) Expression of intact, TG protein was confirmed by western blot analysis with anti-cMyBP-C and anti-myc antibodies. D) SDS-PAGE analyses of the myofilament proteins. E) Expression and sarcomere incorporation of the myc-tagged, TG cMyBP-C was confirmed by immunofluorescent staining with anti-cMyBP-C and anti-myc antibody. F) The spatially correct incorporation of the mutant protein in the sarcomere was confirmed by immunofluorescent staining of cardiac muscle with anti-cMyBP-C and anti α-actinin antibody. All TG samples were derived from 8 week old hearts.
Long term, cMyBP-C is a critical component for maintaining normal cardiomyocyte function, as demonstrated by a number of loss-of-function genetic models [5, 11, 12] as well as expression of modified cMyBP-C proteins [13, 14]. MRI studies of cMyBP-C null hearts suggest that the protein’s ablation leads to depressed LV function and reduced LV torsion and circumferential strain, underscoring the requirement of this sarcomeric protein for normal mechanical function [15]. Numerous reductionist-based approaches have shown significant alterations in crossbridge kinetics in the absence of normal or altered cMyBP-C [15–17].
The cardiac isoform differs from the other two isoforms in that it contains an extra N-terminal domain (C0) and a unique structural motif termed the ‘M’ insertion, which is located between the C1 and C2 domains (Fig. 1A). It is well established that this N-terminal portion interacts with the head region of myosin heavy chain, which is contained within the “subfragment 2” (S2) [1, 18]. The M domain has at least three potential phosphorylation sites; Ser-273, Ser-282 and Ser-302. Recent in vitro studies confirmed that several kinases, including PKA, PKC, RSK, CK2 and CaMKII, can differentially phosphorylate these residues [19–23]. Biochemical analyses showed that, in response to β-adrenergic stimulation, PKA phosphorylates Ser-273, Ser-282, and Ser-302 whereas PKC phosphorylates only Ser-273 and Ser-302 [19, 20]. In vitro studies suggested that CaMKII can phosphorylate Ser-273, Ser-282 and Ser-302 in a calcium concentration dependent manner [14, 23]. In vitro, cMyBP-C phosphorylation by PKA alters the interaction of myosin’s S2 domain with cMyBP-C, changes the myofibrillar Ca2+ sensitivity and enhances actin interaction [24, 25]. Additionally, protein kinase D (PKD) can specifically phosphorylate cMyBP-C at Ser-302 and this may play an important role in accelerating cross bridge kinetics, although the precise mechanism is not known [21]. Recently, it was reported that CK2 can also phosphorylate Ser-282 but the function of this phosphorylation was not explored [23].
Studies from both our laboratory and others have shown that M domain phosphorylation can affect cMyBP-C functionality and cardiac function, presumably by modulating the protein’s interactions with the thick and thin filament proteins [24, 26]. Phosphorylation of the cardiac myofilament proteins represents an important post-translational mechanism that both regulates cardiac function and the heart’s response to internal and external stresses. Phosphorylation levels of cMyBP-C are decreased significantly during the development of heart failure [27–29]. Using a series of mouse models in which the phosphorylatable sites were replaced either by residues that could not be phosphorylated, or by amino acids whose charges served as effective phosphomimetics, we showed the importance of the 3 sites for normal cardiac function [14, 24, 30]. Using site-specific antibodies, we found that phosphorylation at serine residues 273, 282 and 302 are differentially regulated during cardiac stress [14], and recently it was reported that Ser-282 phosphorylation was significantly reduced in heart failure samples when compared to Ser-273 and Ser-302 phosphorylation [31]. Our laboratory has also shown that N-terminal cMyBP-C phosphorylation can protect the heart from I/R injury [24]. However, a comprehensive analysis of each of these sites mutated singly or in combination was not done.
Transgenic (TG) mice expressing non-phosphorylatable cMyBP-C (AllP−), in which Ser-273, 282, 302 were all mutated to alanines were unable to rescue the cMyBP-C knockout phenotype, whereas mice expressing only cMyBP-C in which the 3 serines were mutated to aspartates (AllP+) restored normal cardiac function. cMyBP-C phosphorylation clearly plays an important role in regulating thick and thin filament interaction but the processes are undefined and the physiological consequences of the individual phosphorylation sites are unknown [14, 30, 32]. All these data underscore the importance of a detailed functional study of these three cMyBP-C phosphorylation sites. In this study, TG mice were generated to explore the functionality of these sites at high resolution. We mutated the sites singly and in combination to establish their roles in normal cardiac function. The effects of phosphorylation on thin and thick filament interactions were also determined.
2. Materials and Methods
2.1. Transgenic constructs
The cDNA for mouse cMyBP-C was subjected to site-directed mutagenesis to generate three constructs (DAA, AAD, SDS). When serines 273 or 282 were substituted, the two adjacent threonines, 272 and 281, were also replaced with aspartic acid or alanine in order to rule out any confounding effects of unknown modification at those sites as threonine residues are also subject to post-translational modification [23]. The modified cDNA was cloned downstream of the α-myosin heavy chain (MyHC) promoter, which drives high levels of cardiomyocyte-specific expression in the adult ventricles and atria [33]. The myc epitope, EQKLISEEDL, was incorporated at the N-terminus to distinguish the TG cMyBP-C from endogenous protein. These constructs were used to generate multiple TG founders with cardiomyocyte specific expression and bred with the cMyBP-C null mice (t/t) [12]. Animals were handled in accordance with the principles and procedures of the Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee at Cincinnati Children’s Hospital approved all experimental procedures.
2.2. Microscopy
For immunohistochemistry, frozen cardiac sections were prepared from hearts as described previously [30]. Tissue sections were stained with anti-myc antibody (1:1000 dilution, Cell Signaling Tech), anti-a-actinin antibody (1:500 dilution, Sigma) and anti-cMyBP-C antibody, which was custom synthesized (1:1000 dilution, ProSci Incorporated). Secondary antibodies were used at a 1:100 dilution (Molecular Probes). Paraffin sections of formalin-fixed hearts were stained with H&E and Gomori’s Trichrome. Sarcomere ultrastructure was analyzed via transmission electron microscopy (TEM) as described earlier [13, 34]. For quantification of the tissue area and line scans of sarcomeres, Image J (NIH, Public Domain) or MetaMorph (Molecular Devices) were used to quantitate signal as described previously [24].
2.3. Myofibrillar protein isolation
Myofibrillar proteins were isolated from the ventricular tissue as described [30]. Proteins were separated in SDS-PAGE and stained with Pro-Q Diamond (Molecular Probe) stain according to the manufacturer’s protocol and scanned in a Typhoon 9400 Scanner (GE Healthcare). The same gel was further stained with Bio-Safe Coomassie stain (Bio-Rad).
2.4. Immunoblotting
Frozen ventricular tissue was homogenized in a Bead Beater (Bertin Technologies) in CelLytic tissue homogenizer buffer (Sigma), freshly supplemented with protease and phosphatase inhibitor cocktails (Roche Applied Sciences). Proteins were separated by SDS-PAGE and transferred onto PVDF membranes (Bio-Rad). For western blotting, the following antibodies were used: anti-myc antibody (1:1000 dilution, Cell Signaling Tech), anti-cMyBP-C (1:10000, ProSci Incorporated), anti phospho specific antibody Ser-273 (1:2500), anti phospho specific antibody Ser-282 (1:2500) and anti phospho specific antibody Ser-302 (1:5000).
2.5. Cardiac hemodynamics and electrophysiology
Cardiac function was analyzed as described [30]. Age-matched mice were anaesthetized using 1.5% isoflurane and a continuous supply of 1.5% isoflurane was maintained during assessment using a nose cone. A Vevo2100 (VisualSonics) echocardiography system with a 40 MHz transducer was used to assess left ventricular dimensions and function using M-mode imaging. Early diastolic maximal velocity (E′) and late diastolic maximal velocities (A′) were measured using tissue Doppler and used as noninvasive indicators of left ventricular (LV) diastolic function [24]. ECG was monitored on isoflurane-anesthetized mice using Biopac ECG telemetry. Electrodes were inserted subcutaneously in a lead II configuration and data recorded for 1 minute to determine the baseline cardiac rhythm. In vivo cardiac function and β-adrenergic responsiveness were analyzed as described [30].
2.6. Statistical analyses
All functional studies used 8 week old mice of mixed gender. Results are shown as means±SEM or ±SD. Paired data were evaluated by Student’s t test. For multiple comparisons, ANOVA with post hoc Tukey’s test, or Dunnet’s test was used. P<0.05 was considered significant.
3. Results
3.1. Generation of TG Mice in a cMyBP-C null background
While phosphorylation of sites within cMyBP-C’s M motif (Fig. 1A, B) plays an important role in cardiac function [30], the role of the individual phosphorylatable sites, Ser-273, Ser-282 and Ser-302 remain obscure. To investigate whether a single site phosphorylation could rescue the cMyBP-C null phenotype, three separate constructs were made and subsequently used to generate TG mice (Fig. 1B). In one line, termed DAA, we mutated Ser-273 to aspartate to create a phosphomimetic, and then mutated Ser-282 and Ser-302 to alanine in order to prevent those sites from being phosphorylated. In the AAD line, Ser-273 and Ser-282 were mutated to alanine, while Ser-302 was mutated to aspartate. DAA was designed to test the functionality of Ser-273 phosphorylation in isolation, while AAD was designed to do the same, but for Ser-302. Finally, in the SDS line, Ser-282 was mutated to aspartate in order to determine the effect, if any, that this site’s chronic phosphorylation might have on the basal and stimulated phosphorylation states of the adjacent residues.
For each TG mouse model, we generated at least three lines with varying levels of expression to rule out any insertion site effects on the phenotype. Transgenically-encoded cMyBP-C was tagged with a myc epitope as in our previous studies so that it could be easily differentiated from endogenous protein when necessary [13, 24]. The lines selected for these experiments, which had between 30–45% replacement, showed no insertion site specific phenotype and had TG protein levels approximately equal to those detected for TG mice prepared earlier that expressed intact, wild type cMyBP-C (WT). Previously, we showed those mice had normal levels of cMyBP-C and that 50% of the endogenous protein was replaced with the TG protein [30]. We also included TG mice in which endogenous cMyBP-C was completely replaced by cMyBP-C in which the 3 serines were replaced by non-phosphorylatable alanines, termed AllP− [30]. All TG lines were then bred into the (t/t) (cMyBP-C null) background [12] to generate complete replacement of the endogenous protein with transgenically encoded cMyBP-C [30]. The subsequent analysis of myofilament proteins from total heart lysates confirmed that expression of the TG cMyBP-C’s achieved levels characteristic of normal, endogenous cMyBP-C expression, with no obvious perturbations in the steady state protein levels for the other myofilament proteins (Fig. 1C, D).
The ability of the altered cMyBP-C to incorporate correctly into the sarcomere was determined using immunohistochemistry and the staining compared to sarcomeres containing transgenically expressed normal cMyBP-C (WT) and AllP− (Fig. 1D, E). The data showed that the different cMyBP-C proteins all incorporated correctly, with well-defined intercalation when co-stained with the Z-disc protein, α-actinin, although minor misalignments could be discerned in some of the filament structures associated with incorporation of the ADA and AllP− proteins (Fig. 1E).
3.2. Phosphorylation status is communicative between the sites and dependent upon Ser-282
Myofibrillar proteins were isolated and their steady state levels analyzed using gradient SDS-PAGE (Fig. 2A). Contractile protein expression appeared unaffected by replacement of the endogenous cMyBP-C with the transgenically encoded species and myofilament protein stoichiometry was maintained in all TG mice. Phosphorylation levels were first analyzed using Pro-Q Diamond staining and showed that the total phosphorylation of mutant cMyBP-C in DAA (t/t), AAD (t/t) and AllP− (t/t) was significantly reduced compared to the NTG and WT mice (Fig. 2B and C). It should be noted that total phosphorylation levels in the mutated cMyBP-C’s may reflect increased phosphorylation of other sites in the protein as additional sites capable of being phosphorylated are present in both the M domain and in the N terminus [23]. We also observed that the phosphorylation of the other myofilament proteins appeared unaffected by transgenic replacement (Fig. 2B, D, E). We did note that in our hands, we observed some limited staining of proteins that are not routinely stained by Pro-Q Diamond, such as actin, raising the possibility that some non-specificity of the staining might be occurring (Figure 2). However, this would not impact on the central conclusion of the data, as stated above. Previously we showed that Ser-282’s inability to be phosphorylated resulted in the chronic dephosphorylation of Ser-302 as well [14]. To understand the consequences of the charged residue in terms of Ser-273 and Ser-302 phosphorylation, we determined the relative phosphorylation states of these residues in the SDS mice using our phosphorylation residue-specific antibodies [30]. We found that phosphomimetic substitution at Ser-282 led to high levels of phosphorylation at Ser-273 with Ser-302 phosphorylation unaffected relative to normal levels. The hyper-phosphorylated (relative to wild type) Ser-273 versus normal phosphorylated Ser-302 underscores the uniqueness of each site, both in its autonomous behavior and when functionally linked to the other phosphorylation sites in the molecule. This observation supports the hypothesis that Ser-282 phosphorylation can effectively and differentially modulate spatially distinct phosphorylation sites and points to the unique control exerted at each phosphorylatable residue in this region (Fig. 2F and G).
Figure 2.

Phosphorylation of myofilament proteins in the TG lines. A) SDS-PAGE stained with coomassie blue. B) Pro-Q diamond stain detects phosphorylated protein in the myofilaments. C) Total phosphorylation of cMyBP-C was significantly reduced in DAA/(t/t), AAD/(t/t) and in tt mice. D, E. Phosphorylation levels of other myofilament proteins were also quantitated. D) Desmin phosphorylation levels. E) TnI phosphorylation levels. F, G) Phosphorylation of Ser-273 and Ser-302 in SDS/(t/t) mice was assayed using phosphorylation-residue specific antibodies. Data are expressed as mean ± SD. *P<0.005 versus WT/(t/t). All samples were derived from 8 week old hearts (n=3).
3.3. Chronic phosphorylation at Ser-273 leads to fibrosis and dilated cardiomyopathy
To understand the effects of single-site chronic phosphorylation/dephosphorylation in the cardiac-specific domain of cMyBP-C, the TG mice were bred, weaned and the early adult heart morphology determined at 8 weeks. Examination of sections of the TG hearts suggested major differences in the phenotypic outcomes of the residue substitutions, even in early adulthood. Chronic phosphorylation of one residue, Ser-273, coupled with the prevention of phosphorylation at residues 282 and 302 (DAA), resulted in significant hypertrophy and chamber dilation (Fig. 3A, B), showing the mutated protein’s inability to function normally. Strikingly, those hearts were more affected than hearts lacking the protein entirely (t/t). In contrast neither AAD, SDS or ADA mice showed a dilated phenotype at this developmental stage although the AAD hearts were affected, with significant fibrosis and modest hypertrophy even at 8 weeks (Fig. 3C, D). The heart to body weight ratios and quantification of cardiomyocyte area using wheat germ agglutinin (WGA) stained cells, also suggested that DAA and AAD mice displayed a hypertrophic phenotype when compared to the NTG or WT mice (Fig. 3C, E). Consistent with the previous studies, cMyBP-C null mice also had hypertrophic hearts [12], but the phenotype could be rescued by the SDS or ADA proteins. We conclude that the phosphorylation status of the three serines is important on an individual basis and that chronic phosphorylation at one site, with the inability to phosphorylate the other sites, is particularly detrimental to the maintenance of normal cardiac structure.
Figure 3.
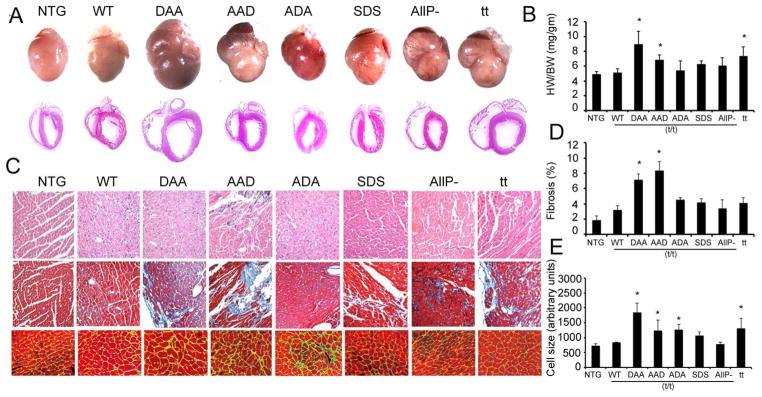
Phenotypic consequences of the expression of altered cMyBP-C protein. A) Shown are 8 week old hearts from NTG and TG animals. B) Heart weight/body weight ratio (HW/BW; mg/gm) (n=6). C) Top to bottom: H&E staining, Masson trichrome staining, and (bottom) delineation of cardiomyocyte membranes using wheat germ agglutinin staining to determine cardiomyocyte hypertrophy in the left ventricle. D) Morphometric analysis of interstitial fibrosis was carried out using Metamorph software (n=6). E) Cell size was determined via wheat germ agglutinin staining (green), with the cardiomyocytes identified using troponin I antibody (red) with quantitation via ImageJ software (n=6). Data are expressed as mean ± SD. * P<0.05 versus WT/(t/t)
3.4. Functional analyses
To address the early functional consequences of mutant protein expression in vivo, echocardiography was performed on 8 week old TG and NTG mice. M-mode echocardiography showed that “mismatched” phosphorylation patterns in the DAA and AAD mice resulted in depressed systolic function and LV dilation (Fig. 4A, Table 1). Fractional shortening (FS) in both the DAA and AAD hearts was decreased to levels equal to, or even less than those present in the cMyBP-C null animals (t/t), emphasizing the need for cooperative and interactive post-translational modification of the 3 sites (Fig. 4B). Systolic LV internal dimensions (LVIDs) were also greater in the DAA and AAD hearts, as compared to the NTG and WT (t/t) mice (Fig. 4C, Table 1). Constitutive phosphorylation of Ser-282 in the SDS and ADA mice resulted in essentially normal values for FS and LVIDs compared to the WT (t/t) mice (Fig. 4B, 4C).
Figure 4.

Ultrasound analyses of chamber function. A) M-mode echocardiography of the TG mice at 8 weeks post-birth. B-D) Cardiac dysfunction as determined by the decreased fractional shortening, increased LV inner diameter in systole and significantly decreased E′ in the DAA/(t/t) and AAD/(t/t), AllP−(t/t) and t/t hearts (n=7–9 as described in Table 1). Data are expressed as mean ± SD. *P<0.05 versus WT/(t/t), **P< 0.005 versus WT/(t/t).
Table 1.
Echocardiographic analyses
| NTG (n=9) | WT (t/t) (n=9) | DAA (t/t) (n=7) | AAD (t/t) (n=7) | ADA (t/t) (n=7) | SDS (t/t) (n=8) | AllP− (t/t) (n=8) | tt (t/t) (n=11) | |
|---|---|---|---|---|---|---|---|---|
| IVSd (mm) | .87±.11 | 0.96±0.05 | 0.91±0.06 | 1.04±0.10 | 0.97±0.21 | 0.98±0.25 | 1.01±0.09 | 1.03±0.26 |
| LVPWd (mm) | .71±.13 | 0.82±0.16 | 0.95±0.24 | 0.89±0.19 | 0.88±0.23 | 0.88±0.15 | 0.84±0.14 | 0.94±0.23 |
| LVIDd (mm) | 4.20±.13 | 4.50±0.31 | 4.83±0.32 | 4.63±0.18 | 4.57±0.33 | 4.48±0.25 | 4.60±0.16 | 4.65±0.32 |
| LVIDs (mm) | 2.66±.24 | 3.20±0.26 | 3.87±0.46* | 3.80±0.21* | 3.30±0.32 | 3.24±0.17 | 3.60±0.26 | 3.76±0.77 |
| LV VOLd (μl) | 79±6 | 93±15 | 110±17 | 99±9 | 96±16 | 92±12 | 98±8 | 103±40 |
| LV VOLs (μl) | 26±5 | 41±8 | 66±20* | 62±8* | 45±10 | 42±5 | 55±9 | 64±38 |
| EF (%) | 66±6 | 56±3 | 41±9* | 37±6*** | 54±4 | 54±2 | 44±7* | 40±8** |
| FS (%) | 36±5 | 29±2 | 20±5** | 18±3*** | 28±2 | 28±1 | 22±4* | 20±4** |
| LV mass (mg) | 100±19 | 133±18 | 157±20 | 155±26 | 142±31 | 140±35 | 144±12 | 162±43 |
| HR (bpm) | 414±70 | 348±120 | 371±122 | 406±63 | 399±125 | 461±100 | 457±46 | 353±56 |
IVSd, diastolic interventricular septum thickness; LVPWd, diastolic left ventricular posterior wall thickness; LVIDd, diastolic left ventricular internal dimension; LVIDs, systolic left ventricular internal dimension; LV VOLd, left ventricular diastolic volume; LV VOLs, left ventricular systolic volume; EF, ejection fraction; FS, fractional shortening, HR heart rate. Values shown are mean±standard deviation.
P<0.05 versus WT,
P<0.01 versus WT,
P<0.001 versus WT.
We assessed LV diastolic function with tissue Doppler imaging at the mitral valve annulus, measuring the velocity of ventricular myocardium during early relaxation (E′) and atrial contraction (A′). We found that, compared to WT(t/t) mice, E′ tissue velocity was significantly depressed in the DAA and AAD hearts as well as in the AllP− and t/t null hearts. Interestingly, while E′ in the SDS hearts showed a decreasing trend, this parameter did not reach statistical significance compared to WT(t/t) hearts (Fig. 4D). The A′ velocity was not significantly different across the groups (Supplemental Fig. 1), but the E′/A′ ratio was significantly depressed in all cMyBP-C mutations studied, again consistent with diastolic dysfunction (Online Supplement Fig. 1) [35].
3.5. Ultrastructural analyses
We previously found that altered cMyBP-C phosphorylation affected sarcomere spacing [24]. This is not surprising, considering the interaction of this protein with all three filament systems in the contractile apparatus. In the absence of cMyBP-C (t/t mice), sarcomeres were relatively disorganized, with filament spacing, as measured by the distances between adjacent myosin filaments in the filament lattice disturbed and irregular [24]. Phosphorylation of cMyBP-C can extend the myosin cross bridge to the surface of the thin filament, loosening the packing of the light meromyosin in the filament lattice and affecting overall sarcomeric structure [24]. We wished to determine if single or double substitutions of the phosphorylatable serines affected these parameters as well, and detailed ultrastructural analyses were carried out using 8 week old mice, before compromised cardiac function could lead to major secondary effects. Consistent with previous data, the t/t mice displayed altered M line architecture (Fig. 5A) and relatively disorganized filament spacing as manifested by irregular distances between adjacent myosin filaments (Fig. 5B, Online Supplement Fig. 2). Incorporation of dephosphorylated cMyBP-C (AllP−) was unable to rescue this defect [24]. Consistent with the gross histological disorganization (Fig. 3), the DAA sarcomeres lacked a distinct M line and showed abnormal Z lines as well (Fig. 5A). While histology of the AAD mice was also abnormal, the sarcomeres appear better conserved (Fig. 5B). Quantitation (Online Supplement Fig. 2) revealed that regular myosin filament spacing was conserved in all of the cMyBP-C-substitutions: only the cMyBP-C null showed statistically significant differences. We conclude that cMyBP-C phosphorylation does not affect thick-thin filament spacing in the absence of other factors, at least at the level of resolution afforded by transmission electron microscopy. This lack of altered filament spacing is consistent with previous data noting that cMyBP-C’s axial distribution was unaffected by site mutations in human cardiac and skeletal muscle [36].
Figure 5.

Transmission electron microscopy of TG sarcomeres. A) Thin sections were cut longitudinally to reveal sarcomere organization, size and architecture in the different TG lines. B) Thick filament line scanning of the corresponding sarcomeres as measured by Metamorph software. All samples were derived from 8 week old animals.
3.6. Surface electrocardiograms are significantly altered in DAA and AAD mice
The above data clearly show that the DAA and AAD hearts are both structurally and anatomically compromised and we noted early deaths in these populations, even in overtly healthy mice (Fig. 6A). In order to determine the cause of sudden cardiac death, we examined the electrical activity of the hearts. Surface ECGs were recorded on NTG and TG mice and the mean RR, PR, QRS, QT and QTc values determined at 8 weeks (n = 6–8 for each group). Both the DAA and AAD mice had irregular cardiac rhythms recorded at baseline, suggesting cardiac arrhythmias (Fig. 6B and Online Supplement, Table 1). The mean RR intervals for the AAD and t/t lines were significantly longer and there was a trend towards increased RR intervals in the DAA hearts as well, although statistical significance was not reached. In addition, the QT interval was prolonged significantly in the DAA, AAD, AllP− and t/t mice (Fig. 6D). Slow ECGs for the most affected lines, DAA and AAD, are shown in the Online Supplement (Fig. 3). QT prolongation reflects impaired cardiac repolarization, leading to the increased risk of arrhythmogenic events and sudden cardiac death. An irregular cardiac rhythm is apparent in the representative tracings from the DAA and AAD animals (Fig. 6B). We hypothesize that these events are at least partially responsible for the sudden cardiac death as manifested in the Kaplan-Meier curves for these two lines (Fig. 6A).
Figure 6.

Residue-specific mutation of the phosphorylation sites leads to decreased survival probability and conduction abnormalities. A) Kaplan-Meier curve of the different TG lines. NTG, WT and ADA were indistinguishable (n=8–11). B) Representative surface ECG recordings of TG mice showed irregular RR and QT intervals in DAA/(t/t) and AAD/(t/t) hearts. C) Quantitation of the RR intervals (n=6–8). D) Quantitation of QT intervals (n=6–8). Data are expressed as mean ± SD. *P<0.05, versus WT/(t/t), †P <0.0001 versus WT/(t/t).
3.7. In vivo hemodynamics
Considering the major anatomical, histologic and electrical conduction effects of forced, uncoordinated cMyBP-C phosphorylation in the AAD and DAA hearts, we looked for changes in the hemodynamics of these hearts at 8 weeks of age as well. cMyBP-C is thought to play a major role in modulating cardiac relaxation [14, 32, 37]. Consistent with this hypothesis, incorporation of altered cMyBP-C led to significant changes in relaxation kinetics, while contraction was relatively unaffected in the non stimulated hearts (Table 2). While all lines were able to respond to β-adrenergic stimulation, the AAD, DAA and t/t mice responses were all significantly blunted, which is not surprising considering the significant fibrosis and disarray in these hearts (Fig. 3).
Table 2.
In Vivo Cardiac Hemodynamics
| NTG | WT | DAA | AAD | SDS | AllP− | tt | ||
|---|---|---|---|---|---|---|---|---|
| Heart rate, Bpm | Basal | 408±8 | 394±12.8 | 407±12.2 | 384±25.21 | 411±28 | 380±10 | 380±9 |
| Dobut. | 579±9 | 565±10.1 | 595±19.2 | 543±11.9 | 573±16.18 | 552±19 | 544±8.18 | |
| MAP, mm Hg/s | Basal | 76±2 | 80±2.06 | 69±3.95 | 64±4.37 | 58±2.82*** ¶ | 80.31±3.3 | 64.65±0.83*¶ |
| Dobut. | 70±2 | 79±3.9 | 59±1.72 | 63±6.07 | 59±2.54 | 74.6±3.3 | 61±4 | |
| Systolic Pressure mm Hg/s | Basal | 94±4 | 94±2.72 | 81±3.90 | 76.6±3.32 | 70±2.32* | 92±4.7 | 76±4.82* |
| Dobut. | 89±4.6 | 94±3.41 | 71±2.24¶ | 76.2±5.86¶¶ | 74±2.05 | 89±5.3 | 75±4.63¶ | |
| LVP, mm Hg/s | Basal | 98±5 | 104±2.72 | 92±3.91 | 83.4±4.13 | 81±2.81 | 106±4.31 | 86±1.30 |
| Dobut. | 101±2 | 106±2.98 | 97±1.6 | 92.96±4.48 | 91±1.54 | 111±3.8 | 93±3.2 | |
| dP/dtmax mm Hg/S | Basal | 9218±508 | 8522±383 | 7085±359* | 7107±507* | 6710±539* | 9051±364 | 7558±319 |
| Dobut. | 20229±717 | 17904±830 | 16473±711* | 14162±1325¶*** | 15556±769**** | 1687±641** | 15751±957**** | |
| dP/dtmin mm Hg/s | Basal | −9203±636 | −6372±514** | −6891±608* | −3436.6±116**** | −5200±221**** | −5638±442**** | −4296.0±156**** |
| Dobut. | −12053±1212 | −8679±436 | −8845±528 | −5167.6±259 | −7078±347 | −7710±584 | −5449±410 | |
| LVEDP, mm Hg/s | Basal | 6.65±0.6 | 5.89±0.74 | 7.78±1.35 | 10.178±3.36 | 4.63±1.34 | 15.4±4.0 | 13.43±3.15 |
| Dobut. | 1.5±0.4 | 2.68±0.78 | 1.69±0.30 | 0.82±0.98 | 1.50±0.36 | 6.0±1.5*** | 3.09±0.43 | |
| dP/dt40, mm Hg/s | Basal | 8310±297 | 7645.5±332 | 6609.3±350 | 6761±563 | 6270±876 | 8391±385 | 7049.2±433 |
| Dobut. | 16499±286 | 14368±451 | 15130.25±718 | 12659±687 | 14297±604* | 13912±418 | 14435±414 |
dP/dtmax, maximum rate of heart contraction and relaxation; dP/dtmin minimum rate of contraction and relaxation; LVEDP left ventricular end-diastolic pressure; LVP, left ventricular pressure; MAP, mean arterial pressure. Ventricular contraction and relaxation rates were measured at baseline and during β-agonist stimulation in the intact closed chest model, including telemetric measurements of heart rate, MAP, systolic pressure, LVP, LVEDP and rate of LVP increase determined at an LVP of 40mm Hg (dP/dt40). Data are presented as mean±SE.
P<0.05 versus NTG,
P<0.01 versus NTG,
P<0.002 versus NTG,
P<0.0001 versus NTG,
P<0.05 versus WT (t/t).
P<0.004 versus WT(t/t).
4. Discussion
Long term, cMyBP-C is a critical component for maintaining normal cardiomyocyte function, as demonstrated by a number of loss-of-function genetic models [5, 11, 12] and expression of modified cMyBP-C proteins [13, 14]. MRI studies of the cMyBP-C null hearts suggest that cMyBP-C ablation leads to depressed LV function, reduced LV torsion and circumferential strain, underscoring the requirement of this sarcomeric protein for normal mechanical function [15]. Numerous reductionist-based approaches showed significant alterations in crossbridge kinetics in the absence of normal or altered cMyBP-C [15–17].
We previously investigated the physiological significance of “global” cMyBP-C phosphorylation/dephosphorylation in TG models in which the 3 phosphorylatable serines were replaced with alanines or a charged amino acid that served as a phosphomimetic [24, 30]. The data confirmed the importance of these sites in modulating normal cMyBP-C function. Independently, other laboratories confirmed much of those data as well [17, 38]. We also showed that phosphorylated cMyBP-C stabilized the intact protein in the presence of cardiac stress, decreasing production of a 40 kD fragment of cMyBP-C that could interfere with normal sarcomere function [24]. Subsequently, we undertook a series of studies in which additional TG lines were generated, SAS, DAD and ADA, in order to study the regulatory role that Ser-282 might play [14]. While those studies confirmed the importance of that residue, they shed little light on understanding the role that the other phosphorylation sites played in the protein’s function, and a comprehensive analysis of understanding the function consequences of discoordinate phosphorylation could not be undertaken without constructing additional lines.
The current manuscript completes and augments our prior studies, providing a comprehensive analysis of single and double site mutations in this critical domain. The creation of a more complete set of TG lines that now include the DAA, AAD and SDS mice, allowed us to test the hypothesis that these phosphorylation sites are non-equivalent and explore in detail the functional consequences of this non-equivalency, focusing on what happens when 1 site is always charged in the absence of the other two sites being phosphorylated. The results are striking and differ from our previous studies: the DAA and AAD hearts are significantly affected. An inability of two residues to be phosphorylated when the other is chronically phosphorylated results in cardiac morbidity, remodeling and fibrosis over a relatively short (15 week) period. Again, comparing the multiple data sets shows that single site phosphorylation of Ser-273 (DAA) and Ser-302 (AAD) did not improve the null phenotype, whereas the single site phosphorylation of Ser-282 (ADA/SDS) could rescue the null phenotype. When these results are compared with our prior data with DAD lines [14] in which two residues were also modified but resulted in charged phosphomimetics, the data taken in total show the point to the importance of residues being phosphorylated in concert.
We approached the general question of phosphorylation non-equivalency by replacing endogenous MyBP-C with intact protein in which the three phosphorylatable serines were individually modified, ablating any coordinated phosphorylation or dephosphorylation of the sites by endogenous kinases and phosphatases. By uncoordinating the three residues’ post-translational modifications, we could then determine the functional effects of de-coupling. Multiple, stable TG lines were constructed and differences in the phenotype, line variability and heart disease, both subtle and striking were noted as being dependent upon the particular phosphorylatable residue or residues modified. Our previous lines in which various residues were substituted, the new, DAA and AAD lines show early mortality, earlier even then mice carrying the null cMyBP-C allele. These data are summarized in Table 2.
In the present study we observed that mimicking constitutive phosphorylation at Ser-282 did not affect Ser-302 phosphorylation, but Ser-273 phosphorylation was upregulated in the SDS mice. Quantification of total phosphorylation by Pro-Q staining suggests that overall phosphorylation is dramatically reduced in the DAA, ADA and AAD mice. Ser-282 phosphorylated mice were overtly healthy with essentially normal cardiac anatomy and cardiac function equivalent to those mice in which we had replaced endogenous cMyBP-C with transgenically encoded wild-type protein. In contrast, mice showed significant pathology when cMyBP-C was completely replaced with either the DAA or AAD cMyBP-C species. Interestingly, both these TG lines showed more fibrosis than mice carrying either cMyBP-C that was unable to be phosphorylated at any of the three residues (AllP−) or mice completely lacking functional cMyBP-C (t/t). We do not believe the observed pathologies are due directly to an artifact of the serine to alanine changes, as replacements in the ADA construct (alanine replacing serines 273 and 302) resulted in mice with no overt fibrosis, pathology or decreased life span.
Despite sarcomere incorporation across the different cMyBP-C species, the functional effects were dramatically different. For example, while normal cardiac architecture and function is largely conserved in the ADA and SDS mutations (eg, Fig. 1, Table 1), DAA and AAD lead to dramatic pathology. This is also not due to simply changing different numbers of sites, as the AAA mutation (AllP−), while leading to morbidity, is less pathogenic. While we do not yet understand the mechanistic consequences of the multiple charged residues interactions with the different filament systems, our data do imply that each residue and combination of residues is unique and the resultant functional differences are not due to the general regional charge in this part of the molecule. It remains for detailed modeling to be carried out with the mutated fragments to resolve the unanswered mechanistic questions.
Reduced phosphorylation of the myofibrillar proteins is often associated with human arrhythmias [39, 40] and phosphorylation of the myofilament proteins can play an important role in myofilament calcium sensitivity [41, 42]. Our ECG data show irregular cardiac rhythms and QT prolongation in the DAA and AAD mice (Fig. 6) as well as RR lengthening in AAD and t/t mice. No significant differences in average heart rate were noted across among the cohorts during the echocardiograms and invasive hemodynamic assessments; however, differences in anesthetic regimen and animal preparation for these procedures may have contributed to the variations noted.
These arrhythmias likely contributed to sudden cardiac death in the animals and played a significant role in the decreased survival probabilities for the DAA and AAD animals (Fig. 6). Decreased E′ tissue Doppler velocities are consistent with invasive hemodynamic data showing altered/affected relaxation as a result of altered cMyBP-C phosphorylation [14, 43]. An alternative explanation may lie in the extensive hypertrophy and atrial enlargement observed in both these lines (Fig. 3). It is well documented in human disease that these types of anatomical alterations, brought about because of altered hemodynamics and fiber kinetics, can cause electrophysiologic abnormalities that can lead to arrhythmogenic events and sudden cardiac death [44].
cMyBP-C can slow the cross-bridge kinetics of the sarcomere and recently, this ability was shown to be restricted specifically to the central region of the sarcomere where cMyBP-C is located [45] In vitro biophysical studies and in vivo imaging suggest that phosphorylation of cMyBP-C regulates its dynamic interactions with myosin and actin [26, 46, 47]. Direct interaction studies with single molecules using the laser trap assay demonstrated that cMyBP-C binds to actin, and phosphorylation of cMyBP-C’s amino terminal residues could inhibit actin interaction [26, 46]. Previously we found that phosphorylation of all three sites (Ser-273, Ser-282 and Ser-302) in cMyBP-C inhibited S2. Taken together, we have now constructed a number of single, double and triple phosphomimetic or phospho-ablated cMyBP-C proteins and can draw a number of conclusions from these data when they are taken as a whole from our multiple studies [14, 16, 21, 24, 26, 30, 32, 45, 46, 48]. First, phosphorylation in the N-terminal “M” domain at 1 or more residues is absolutely essential for cMyBP-C function. A lack of phosphorylation results in altered affinities for both the myosin heavy chain and actin filaments, altering cross bridge kinetics and force production. Second, phosphorylation at Ser-282 is particularly critical: it serves as both a nodal point of control, potentiating phosphorylation at the adjacent residues, and renders a μ-calpain site inaccessible, preventing cleavage of intact cMyBP-C. Third, the data in the present study point to the non-equivalency of each of the phosphorylatable sites, as subtle functional differences present for essentially all of the unique constructs. Fourth, it is clear that phosphorylation of specific residues can impact on the degree of phosphorylation observed, with Ser-282 undoubtedly playing a role in this. Fifth, phosphorylation at one site in the absence of the ability to phosphorylate the other sites, such as occurs in the DAA construct, leads to severe cardiac remodeling and dysfunction: the DAA and AAD constructs prepared for this study result in abnormal cardiac conduction, arrhythmias and premature death. We conclude that the different phosphorylation sites can play different roles, when viewed in isolation and, importantly, in conjunction with the charge of its adjacent, phosphorylatable sites. It now becomes critical to attempt to show, in vivo, which kinases are actually acting on each site. Considering the frequency with which this protein is mutated in human cardiac disease [49], identifying the exact residues that can mediate cMyBP-C function will enable us to understand how the protein functions in the normal and diseased states.
Supplementary Material
Table 3.
Summary of Cardiac and Animal Phenotypes
| Genotype | Heart Morphology | % FS | Cardiac Rhythm | dP/dtmax | dP/dtmin | Total Phosphorylation | Survival (1 year) | ||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Basal. | Dobt. | Basal. | Dobt. | ||||||
| NTG | Heart shape: Normal Fibrosis: Less Cell size: Normal |
↔ | RR interval: Normal QT Interval: Normal |
↔ | ↔ | ↔ | ↔ | No change | No mortality |
| WT (t/t) | Heart shape: Normal Fibrosis: Less Cell size: Normal |
↔ | RR interval: Normal QT Interval: Normal |
↔ | ↔ | ↔ | ↔ | No change | No mortality |
| DAA (t/t) | Heart shape: Dilated Fibrosis: Extensive Cell size: Increased |
↓ | RR interval: Irregular QT Interval: Prolonged |
↓ | ↔ | ↔ | ↔ | Decreased | Early mortality |
| AAD (t/t) | Heart shape: Hypertrophied Fibrosis: Extensive Cell size: Increased |
↓ | RR interval: Irregular QT Interval: Prolonged |
↓ | ↓ | ↔ | ↓ | Decreased | Early mortality |
| ADA (t/t) | Heart shape: Normal Fibrosis: Increased Cell size: Normal |
↔ | RR interval: Normal QT Interval: Normal |
↔ | ↓ | ↓ | ↓ | Slight decrease | No mortality |
| SDS (t/t) | Heart shape: Normal Fibrosis: Increased Cell size: Increased |
↔ | RR interval: Normal QT Interval: Normal |
↔ | ↓ | ↔ | ↔ | Slight decrease | No mortality |
| AllP− (t/t) | Heart shape: Dilated Fibrosis: Increased Cell size: Normal |
↓ | RR interval: Irregular QT Interval: Prolonged |
↔ | ↔ | ↔ | ↔ | Decreased | Late mortality |
| tt | Heart shape: Dilated Fibrosis: Extensive Cell size: Increased |
↓ | RR interval: Irregular QT Interval: Prolonged |
↔ | ↓ | ↓ | ↓ | Not applicable | Early mortality |
Highlights.
Three lines of transgenic mice were made, containing mutated phosphorylation sites
Each of the three sites are uniquely functional
Ser-282 phosphorylation modulates spatially distinct phosphorylation sites
Phosphorylation at one site with an inability to phosphorylate other sites is toxic
Acknowledgments
This work was supported by NIH grants P01HL077101, P01HL049058, R01HL105924 and The Transatlantic Network of Excellence Program grant from Le Fondation Leducq (to J.R.). MKG is a postdoctoral fellow of American Heart Association (Great Rivers Affiliate).
Glossary: Non standard abbreviations and acronyms
- A′
peak atrial velocity
- ADA
cMyBP-C protein with alanines at residues 273, 302 and aspartate at residue 282
- AAD
cMyBP-C protein with alanines at residues 273, 282 and aspartate at residue 302
- AllP−
substitutions of alanine at Ser-273, 282, 302
- AllP+
phosphomimetic aspartates placed at Ser-273, 282, 302
- CaMKII
Ca2/calmodulin-dependent protein kinase II
- CK2
Casein kinase 2
- cMYBP-C
cardiac myosin binding protein C
- DAA
cMyBP-C protein with aspartate at residue 273, and alanines at residues 282 and 302
- dP/dtmax
maximum left ventricle pressure development
- dP/dtmin
minimum left ventricle pressure development
- E′
peak early diastolic wave
- EF
ejection fraction
- FS
fractional shortening
- I/R
ischemia/reperfusion
- IVSd
interventricular septal thickness at diastole
- LVIDd
left ventricular inner diameter in diastole
- LVIDs
left ventricle inner diameter in systole
- MyHC
myosin heavy chain
- PKA
protein kinase A
- PKC
protein kinase C
- PKD
protein kinase D
- PR
time interval between onset of P wave and R wave
- QRS
duration of the interval between Q wave to peak of S wave
- QT
duration of beginning of Q wave the end of the T wave
- QTc
corrected QT interval
- RR
time interval between two consecutive R waves
- RSK
ribosomal S6 kinase
- S2
amino terminal fragment of myosin heavy chain containing the ATPase domain
- SDS
cMyBP-C protein with aspartate at residue 282
- t/t
A cMyBP-C null allele
- TG
transgenic
- WGA
wheat germ agglutinin
Footnotes
Disclosures: none declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oakley CE, Hambly BD, Curmi PM, Brown LJ. Myosin binding protein C: structural abnormalities in familial hypertrophic cardiomyopathy. Cell Res. 2004;14:95–110. doi: 10.1038/sj.cr.7290208. [DOI] [PubMed] [Google Scholar]
- 2.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 2011;108:751–64. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moos C, Offer G, Starr R, Bennett P. Interaction of C-protein with myosin, myosin rod and light meromyosin. Journal of molecular biology. 1975;97:1–9. doi: 10.1016/s0022-2836(75)80017-9. [DOI] [PubMed] [Google Scholar]
- 4.Offer G, Moos C, Starr R. A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification and characterization. Journal of molecular biology. 1973;74:653–76. doi: 10.1016/0022-2836(73)90055-7. [DOI] [PubMed] [Google Scholar]
- 5.Luther PK, Bennett PM, Knupp C, Craig R, Padron R, Harris SP, et al. Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. Journal of molecular biology. 2008;384:60–72. doi: 10.1016/j.jmb.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flashman E, Korkie L, Watkins H, Redwood C, Moolman-Smook JC. Support for a trimeric collar of myosin binding protein C in cardiac and fast skeletal muscle, but not in slow skeletal muscle. FEBS Lett. 2008;582:434–8. doi: 10.1016/j.febslet.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 7.Squire JM, Luther PK, Knupp C. Structural evidence for the interaction of C-protein (MyBP-C) with actin and sequence identification of a possible actin-binding domain. Journal of molecular biology. 2003;331:713–24. doi: 10.1016/s0022-2836(03)00781-2. [DOI] [PubMed] [Google Scholar]
- 8.Gilbert R, Kelly MG, Mikawa T, Fischman DA. The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. J Cell Sci. 1996;109 (Pt 1):101–11. doi: 10.1242/jcs.109.1.101. [DOI] [PubMed] [Google Scholar]
- 9.Karsai A, Kellermayer MS, Harris SP. Mechanical unfolding of cardiac myosin binding protein-C by atomic force microscopy. Biophysical journal. 2011;101:1968–77. doi: 10.1016/j.bpj.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rybakova IN, Greaser ML, Moss RL. Myosin binding protein C interaction with actin: characterization and mapping of the binding site. J Biol Chem. 2011;286:2008–16. doi: 10.1074/jbc.M110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 12.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1771. doi: 10.1172/JCI7377C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. In vivo modeling of myosin binding protein C familial hypertrophic cardiomyopathy. Circ Res. 1999;85:841–7. doi: 10.1161/01.res.85.9.841. [DOI] [PubMed] [Google Scholar]
- 14.Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, et al. A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ Res. 2011;109:141–50. doi: 10.1161/CIRCRESAHA.111.242560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desjardins CL, Chen Y, Coulton AT, Hoit BD, Yu X, Stelzer JE. Cardiac myosin binding protein C insufficiency leads to early onset of mechanical dysfunction. Circ Cardiovasc Imaging. 2012;5:127–36. doi: 10.1161/CIRCIMAGING.111.965772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Palmer BM, Sadayappan S, Wang Y, Weith AE, Previs MJ, Bekyarova T, et al. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophysical journal. 2011;101:1661–9. doi: 10.1016/j.bpj.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974–82. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flashman E, Redwood C, Moolman-Smook J, Watkins H. Cardiac myosin binding protein C: its role in physiology and disease. Circ Res. 2004;94:1279–89. doi: 10.1161/01.RES.0000127175.21818.C2. [DOI] [PubMed] [Google Scholar]
- 19.Mohamed AS, Dignam JD, Schlender KK. Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys. 1998;358:313–9. doi: 10.1006/abbi.1998.0857. [DOI] [PubMed] [Google Scholar]
- 20.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952–60. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bardswell SC, Cuello F, Rowland AJ, Sadayappan S, Robbins J, Gautel M, et al. Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J Biol Chem. 2010;285:5674–82. doi: 10.1074/jbc.M109.066456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cuello F, Bardswell SC, Haworth RS, Ehler E, Sadayappan S, Kentish JC, et al. Novel role for p90 ribosomal S6 kinase in the regulation of cardiac myofilament phosphorylation. J Biol Chem. 2011;286:5300–10. doi: 10.1074/jbc.M110.202713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kooij V, Holewinski RJ, Murphy AM, Van Eyk JE. Characterization of the cardiac myosin binding protein-C phosphoproteome in healthy and failing human hearts. Journal of molecular and cellular cardiology. 2013;60:116–20. doi: 10.1016/j.yjmcc.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, et al. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16918–23. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res. 2000;86:51–8. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- 26.Weith A, Sadayappan S, Gulick J, Previs MJ, Vanburen P, Robbins J, et al. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. Journal of molecular and cellular cardiology. 2012;52:219–27. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh YH, Nattel S, et al. Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure. Journal of molecular and cellular cardiology. 2007;43:223–9. doi: 10.1016/j.yjmcc.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 28.Jacques AM, Copeland O, Messer AE, Gallon CE, King K, McKenna WJ, et al. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. Journal of molecular and cellular cardiology. 2008;45:209–16. doi: 10.1016/j.yjmcc.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 29.Zaremba RMD, Hamdani N, Lamers JM, Paulus WJ, Dos Remedios C, Duncker DJ, Stienen GJ, van der Velden J. Quantitative analysis of myofilament protein phosphorylation in small cardiac biopsies. Proteomics Clin App. 2007;10 doi: 10.1002/prca.200600891. [DOI] [PubMed] [Google Scholar]
- 30.Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, et al. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res. 2005;97:1156–63. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Copeland O, Sadayappan S, Messer AE, Steinen GJ, van der Velden J, Marston SB. Analysis of cardiac myosin binding protein-C phosphorylation in human heart muscle. Journal of molecular and cellular cardiology. 2010;49:1003–11. doi: 10.1016/j.yjmcc.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Sadayappan S, Gulick J, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, et al. Cardiac myosin binding protein-C phosphorylation in a {beta}-myosin heavy chain background. Circulation. 2009;119:1253–62. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.James JF, Hewett TE, Robbins J. Cardiac physiology in transgenic mice. Circ Res. 1998;82:407–15. doi: 10.1161/01.res.82.4.407. [DOI] [PubMed] [Google Scholar]
- 34.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest. 1998;102:1292–300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaefer A, Klein G, Brand B, Lippolt P, Drexler H, Meyer GP. Evaluation of left ventricular diastolic function by pulsed Doppler tissue imaging in mice. Journal of the American Society of Echocardiography: official publication of the American Society of Echocardiography. 2003;16:1144–9. doi: 10.1067/S0894-7317(03)00679-5. [DOI] [PubMed] [Google Scholar]
- 36.Vydyanath A, Gurnett CA, Marston S, Luther PK. Axial distribution of myosin binding protein-C is unaffected by mutations in human cardiac and skeletal muscle. Journal of muscle research and cell motility. 2012;33:61–74. doi: 10.1007/s10974-012-9286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pohlmann L, Kroger I, Vignier N, Schlossarek S, Kramer E, Coirault C, et al. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res. 2007;101:928–38. doi: 10.1161/CIRCRESAHA.107.158774. [DOI] [PubMed] [Google Scholar]
- 38.Tong CWC, Abdalla MI, Wu X, Liu Y, Muthuchamy M, Moss R. Lack of Cardiac Myosin Binding Protein-C Phosphorylation Is a Model of Heart Failure with Preserved Ejection Fraction. J Am Coll Cardiol. 2012;59:E860-E. [Google Scholar]
- 39.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, et al. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation. 2006;114:670–80. doi: 10.1161/CIRCULATIONAHA.106.636845. [DOI] [PubMed] [Google Scholar]
- 40.El-Armouche A, Wittkopper K, Degenhardt F, Weinberger F, Didie M, Melnychenko I, et al. Phosphatase inhibitor-1-deficient mice are protected from catecholamine-induced arrhythmias and myocardial hypertrophy. Cardiovasc Res. 2008;80:396–406. doi: 10.1093/cvr/cvn208. [DOI] [PubMed] [Google Scholar]
- 41.Chen PP, Patel JR, Rybakova IN, Walker JW, Moss RL. Protein kinase A-induced myofilament desensitization to Ca(2+) as a result of phosphorylation of cardiac myosin-binding protein C. J Gen Physiol. 2010;136:615–27. doi: 10.1085/jgp.201010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker JS, Walker LA, Margulies K, Buttrick P, de Tombe P. Protein kinase A changes calcium sensitivity but not crossbridge kinetics in human cardiac myofibrils. Am J Physiol Heart Circ Physiol. 2011;301:H138–46. doi: 10.1152/ajpheart.00838.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J, et al. Control of in vivo left ventricular [correction] contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation. 2007;116:2399–408. doi: 10.1161/CIRCULATIONAHA.107.706523. [DOI] [PubMed] [Google Scholar]
- 44.King JH, Huang CL, Fraser JA. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Frontiers in physiology. 2013;4:154. doi: 10.3389/fphys.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Previs MJ, Previs SB, Gulick J, Robbins J, Warshaw DM. Molecular Mechanics of Cardiac Myosin-Binding Protein C in Native Thick Filaments. Science. 2012 doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weith AE, Previs MJ, Hoeprich GJ, Previs SB, Gulick J, Robbins J, et al. The extent of cardiac myosin binding protein-C phosphorylation modulates actomyosin function in a graded manner. Journal of muscle research and cell motility. 2012 doi: 10.1007/s10974-012-9312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luther PK, Winkler H, Taylor K, Zoghbi ME, Craig R, Padron R, et al. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc Natl Acad Sci U S A. 2011;108:11423–8. doi: 10.1073/pnas.1103216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mun JY, Gulick J, Robbins J, Woodhead J, Lehman W, Craig R. Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C) Journal of molecular biology. 2011;410:214–25. doi: 10.1016/j.jmb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L. How do MYBPC3 mutations cause hypertrophic cardiomyopathy? Journal of muscle research and cell motility. 2012;33:75–80. doi: 10.1007/s10974-011-9268-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.