

Abstract
We present a tightly controlled process for strand-specific amplification of circularized DNA molecules. Tandem repeated complements of DNA circles are generated by rolling-circle replication, and converted to monomer circles of opposite polarity to that of the starting material. These circles are then subjected to one more round of rolling-circle replication and circularization, and the process can be further repeated. The method can be directed to produce single-stranded circular or linear monomers, or linear concatemers of the desired polarity. The reaction is not product inhibited, and can yield ≈100-fold higher concentrations of monomer products than PCR. Each generation of the amplification process proceeds in a linear fashion, ensuring precise quantification. The procedure is suitable for parallel amplification of large numbers of DNA circles, because the few cycles and the robust reaction mechanism preserves the proportion of amplified molecules. We demonstrate the utility of the method for multiplexed genotyping of polymorphic loci and for quantitative DNA analysis.
Circularized DNA strands can be formed in highly specific detection reactions using so-called padlock probes (1). These unimolecular, dual-recognition ligation probes allow analysis of single-nucleotide variation directly in unamplified genomic DNA samples (2, 3), and the probe design permits multiplexed genotyping of thousands of loci in individual reactions (4, 5). Reacted probes can be amplified by PCR, or the DNA circles can be replicated by rolling-circle replication (RCR) (2, 6–8). RCR proceeds in a linear fashion, and the highly processive Φ29 DNA polymerase (9) can copy a 100-nt circular probe into a DNA strand containing ≈1,000 complements of the circularized molecule in 1 h (8). The mechanism is promising for detection of circularized DNA probes, but the limited amplification rate has proven insufficient for many applications. By including a second primer, complementary to the RCR product, these products are in turn replicated by a strand-displacement mechanism, and the process can yield a billion-fold amplification in an isothermal reaction of <1 h (2). The amplification kinetics of this so-called hyperbranched rolling-circle amplification technique is difficult to control and often produces circle-independent amplification artifacts (10–12). The reaction has not proven useful for sensitive quantitative analysis or for multiplex amplification.
We demonstrate here that a billionfold or greater amplification of reacted probes is possible in a highly controlled RCR-based process that we call circle-to-circle amplification (C2CA). We characterize the mechanism and investigate its suitability for applications like parallel genotyping of sets of loci and for quantification of specific DNA sequences.
Materials and Methods
Oligonucleotides. Oligonucleotide sequences are shown in Table 1. The polarity of the padlock probes is referred to as (+), and that of their complements as (–), and we identify the polarities of molecules by the appropriate superscripts or colors in Figs. 1 and 2. The padlock probe ppMT+ was designed to detect the mitochondrial DNA A3243G sequence variation involved in the MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes, OMIM 540000) and MIDD (maternally inherited diabetes and deafness, OMIM 520000) syndromes (13). DNA circles were prepared for characterization of the amplification mechanism from padlock probes ppFD+, ppREF+, and ppMT+ by using synthetic ligation templates tFD–, tREF–, and tMT–, respectively. Accumulation of RCR products of ppMT+ and ppREF+ was monitored in real-time by using molecular beacons mbMT+ and mbREF+, respectively, whereas amplification products of ppFD+ of the two polarities were monitored with molecular beacons mbFD+ or mbFD–. Monomerization of RCR products of either polarity was directed by replication oligonucleotides (RO) RSA– and RSA+, allowing restriction digestion by RsaI (New England Biolabs). We used a set of 26 padlock probes in a multiplexed genotyping experiment, targeting 13 single-nucleotide polymorphisms and disease related mutations in the human ATP7B gene. The probes 1216(G/T), 1366(C/G), 1877(C/G), 2128(A/G), 2495(G/A), 2855(G/A), 2930(C/T), 2973(G/A), 3207(C/A), 3317(A/T), 3419(T/C), 3457(C/T), 3955(T/C) are described in Banér et al. (5). ROs for this set of probes were FNU– and FNU+, directing restriction digestion by Fnu4H I (New England Biolabs). Oligonucleotides deposited on microarrays were complementary to locus tag sequences in the padlock probes with an additional (TC)7T-NH2 3′ tail. The tag sequences were selected from the GeneFlex Tag Array sequence collection (Affymetrix, Santa Clara, CA). Two allele-specific oligonucleotides, WDP-F+ and WDP-T+, labeled with fluorescein and Tamra, respectively, were used to detect monomerized RCR products on the microarrays in a sandwich hybridization procedure. Preparation of microarrays was described in Banér et al. (5). The molecular beacons were from Eurogentec (Seraing, Belgium). All other oligonucleotides were from Interactiva (Ulm, Germany), DNA Technology A/S (Aarhus, Denmark), or Eurogentec.
Table 1. Oligonucleotides used in C2CA.
| Sequence 5′-3′ | |
|---|---|
| Padlock probes | |
| ppFD+ | P-CCTCCCATCATATTAAAGGCTTTCTCTATGTTAAGTGACCTACGACCTCAATGCTGCTGCTGTACTACTCTTCCTAAGGCATTCTGCAAACAT |
| ppMT+ | P-CTGCCATCTTAACAAACCCTCATGATTAGAGCGCATCTGCAATAGCTGCTGTACTACTCTCTTCTTCTATGCGATTACCGGGCC |
| ppREF+ | P-CTGCCATCTTAACAAACCCTTACATCTCGGAATCATGCTGGCTGCTGTACTACTCTCTTCTTCTATGCGATTACCGGGCT |
| Targets | |
| tFD- | GCCTTTAATATGATGGGAGGATGTTTGCAGAATGCCTTAG |
| tMT- | GTTTGTTAAGATGGCAGGGCCCGGTAATCG |
| tREF- | TTGTTAAGATGGCAGAGCCCGGTAATCGCA |
| Molecular beacons | |
| mbFD+ | HEX-ccucAAUGCUGCUGCUGUACUACgagg-DABCYL |
| mbFD- | HEX-ccucCGUAGUACAGCAGCAGCAUUgagg-DABCYL |
| mbREF+ | HEX-cagctACATCTCGGAATCAagcug-DABCYL |
| mbMT+ | FAM-gcagCATGATTAGAGCGCATcugc-DABCYL |
| Replication oligonucleotides | |
| RSA- | GCTGCTGTACTACTCTCTT |
| RSA+ | AAGAGAGTAGTACAGCAGC |
| FNU- | GACCGTTAGCAGCATGATTCCG |
| FNU+ | GAATCATGCTGCTAACGGT |
| Sandwich primers | |
| WDP-F+ | FITC-CCGAGATGTACCGCTATCGT |
| WDP-T+ | TAMRA-AGAGCGCATGAATCCGTAGT |
P, 5′ phosphate; the stem parts of 2′-O-Me-RNA molecular beacons are shown in lowercase.
Fig. 1.

The C2CA mechanism. (A) The procedure starts with circularized probes of (+) polarity (red). An initial RCR is primed by a replication oligonucleotide (RO–, blue) resulting in a product composed of repeated copies of the circle (blue). The products are restriction digested to monomers and then circularized. These circles then serve as templates for the next RCR, primed by an RO+ (red), complementary to the RO–. After a second round of monomerization and circularization, identical copies of the starting DNA circles are formed that can be further amplified in additional cycles of C2CA. (B) A detailed description of one cycle of the C2CA procedure. In step I, a circular DNA molecule is replicated by RCR primed by an RO–. In step II, the polymerase is heat-inactivated, and an RO+ is added, which hybridizes once per monomer of the RCR product, forming double-stranded substrates for a restriction enzyme (RE). In step III, the restriction enzyme is heat-inactivated, causing digested fragments of the RO to dissociate from the monomerized RCR product. As the temperature is lowered, remaining intact RO hybridize to monomer RCR products. In step IV, upon addition of a DNA ligase, the RO guide the ligation of the monomers to DNA circles, ready to be replicated in RCR primed by the RO. (C) Gel electrophoretic analysis demonstrating efficient circle-to-circle conversion in the C2CA. DNA circles of (+) and (–) polarity were replicated in the presence of radiolabeled dCTP. Concatemer RCR products were monomerized and circularized, and the products were separated on a denaturing 6% polyacrylamide gel, and then analyzed by using a PhosphorImager.
Fig. 2.

Real-time analyses of RCR reactions demonstrating strand-specific amplification during three generations of C2CA. One, two, and three generations (C2CA1, C2CA2, and C2CA3, respectively) of C2CA were monitored in real-time in separate reaction tubes by using the molecular beacons mbFD+ (red), specific for the (–) strand, and mbFD– (blue), specific for the (+) strand, respectively. The amount of start circles is indicated in the upper left corner in each graph. The hair-pin shaped molecular beacon probes report the presence of RCR products by emitting fluorescence upon hybridization. The fluorescence reaches a plateau when all molecular beacons have hybridized to the RCR product. The maximum slope of the accumulating fluorescence in the graph reflects the amount of circles templating the RCR.
Preparation of DNA Circles. In experiments where DNA circles were not formed in a target-specific padlock probe ligation reaction, DNA circles were prepared by ligation of 20 nM padlock probes in φ29 polymerase buffer [50 mM Tris·HCl,pH7.5/10 mM MgCl2/20 mM (NH4)2SO4/0.2 μg/μl BSA], with 1 mM ATP and 0.02 units/μl T4 DNA ligase (Amersham Pharmacia) templated by 60 nM synthetic target oligonucleotides at 37°C for 10 min.
C2CA. Circularized DNA strands were replicated in φ29 polymerase buffer with 1 mM dNTP and 3 units of φ29 DNA polymerase (New England Biolabs) at 37°C for 1 h, if not otherwise stated. In this and all other reaction steps, additions were made in volumes of 5 μl to an initial volume of 5 μl of probe ligation reaction. Replication of probes ligated on genomic or mitochondrial DNA was primed by 25 nM replication oligonucleotides FNU– and RSA–, respectively. In all other experiments, RCR was primed by the synthetic ligation templates. The replication reactions were terminated by 5-min incubation at 65°C. To monomerize RCR products, ROs complementary to the replication sequence in the RCR products were added together with 10 units of restriction enzyme (Fnu4HI or RsaI) in φ29 polymerase buffer, followed by incubation at 37°Cfor 5 min. After monomerization, the enzyme was inactivated at 65°C for 10 min. Next, the monomerized RCR products were circularized by adding 1 unit of T4 DNA ligase in φ29 polymerase buffer with 5 mM ATP and incubating for 5 min at 37°C. Polymerization, monomerization, and circularization were further repeated, as desired. The concentration of RO was 0.1 μM in the first-generation monomerization reactions, and it was increased 3-fold for each subsequent monomerization reaction.
Radiolabeling of RCR Products. Circularized ppFD+ (100 amol) or its complement were C2C amplified as described above, but including 20 μCi [α-32P]dCTP (3,000 mCi/mmol, Amersham Pharmacia; 1 Ci = 37 GBq) in the RCR reactions. The products were separated on a 6% polyacrylamide gel with 7 M urea in TBE buffer (45 mM Tris-borate, pH 8.3/1 mM EDTA), and the gel was analyzed on a PhosphorImager (Fuji Bas-1800II).
Real-Time Monitoring of RCR. RCR was performed in the presence of 100 nM molecular beacons in 50 μl for 60 min at 37°C. Reactions were followed in an ABI 7700 real-time PCR instrument and analyzed by using the sds v1.7 software package. Data analysis was performed according to Nilsson et al. (14).
Multiplex Genotyping. Padlock probe ligation for multiplexed genotyping of genomic DNA samples was performed as described in Banér et al. (5). Fifty-nanogram aliquots of restriction-digested genomic DNA were mixed with a pool of 400 pM of each of the 26 ATP7B probes and 400 milliunits of Ampligase (Epicentre) in a volume of 10 μl of ligase buffer (20 mM Tris·HCl,pH9.0/100 mM KCl/10 mM MgCl2/1 mM EDTA/1 mM DTT/0.1% Triton X-100). The ligation reactions were placed in a thermal cycler at 95°C for 5 min, and cycled 20 times between 95°C for 2 min and 55°C for 20 min, followed by 95°C for 2 min. After ligation, 0.35 unit/μl exonuclease I (New England Biolabs) and exonuclease III (Amersham Pharmacia) in 10 μl of 110 mM Tris·HCl pH 9.0, 3 mM MgCl2, and 0.1 μg/μl BSA were added and the reactions were incubated for 2 h at 37°C, followed by 95°C for 10 min. Five microliters of the reactions were amplified in three generations of C2CA, performed with 3-h replication reactions, and analyzed on tag microarrays.
RCR Product Hybridization on Oligonucleotide Microarrays. Microarray hybridization and analysis of PCR products are described in detail in Banér et al. (5). The same conditions were used for C2CA products with the following modifications: 30 μl of monomerized RCR products were combined with 20 μl of 5× SSC, 0.1% Triton X-100, and 200 nM of the fluorescent hybridization probes WDP-F+ and WDP-T+. A 45-μl aliquot was added to a microarray with locus tag sequences complementary to the probes and incubated at 55°C for 3 h.
Analysis of Variation in Amplification Efficiency. For the comparison of C2CA with PCR, the ATP7B set of 26 padlock probes was circularized by using 40-mer oligonucleotide targets. The circularized probes were diluted to either 4 nM or 4 fM and subjected to one or three generations of C2CA, respectively, or to 7 or 27 cycles of PCR according to Banér et al. (5). All amplification products were analyzed on tag microarrays.
Quantitative Analysis. DNA was prepared from the M50 cell line (15) by using a genomic DNA extraction kit (Wizard, Promega). The DNA was reduced in size by digestion with RsaI (New England Biolabs) for 1 h in the recommended buffer. Padlock probes were mixed with a dilution series of sample DNA in triplicate 10-μl ligation reactions containing 100 pM padlock probe ppMT+ and 400 milliunits of Ampligase in ligase buffer. The ligation reactions were subjected to five temperature cycles of 95°C for 5 min and 55°C for 20 min in a thermal cycler. After ligation, 10 μl of 10 units exonuclease I (New England Biolabs) in 67 mM Tris·HCl, pH 9.0/10 mM MgCl2/2 μg of BSA was added to the reactions. The samples were incubated at 37°C for 2 h, followed by 95°C for 10 min. Five-microliter aliquots of each reaction were amplified in three generations of C2CA, including 300 zeptomole (zmol) ppREF+ reference DNA circles added in the first RCR. Accumulation of products in the third-generation replication reaction was monitored in real-time as described above. A standard curve was prepared by analyzing a dilution series of ppMT+ circles with a fixed amount of 300 zmol ppREF+ reference circles. The dilution series was amplified and monitored in real-time.
Results
C2CA provides a means to specifically amplify circular DNA strands to very large numbers. First, DNA circles are copied by RCR to generate single-stranded products composed of repeats of probe complements. In a series of reaction steps, the products are then monomerized, converted to new DNA circles, and used to template subsequent rounds of RCRs. The reactions can be repeated in a cyclical procedure where the polarity of the amplified DNA alternates between cycles (Fig. 1A).
In greater detail, the C2CA is initiated by using a replication oligonucleotide that hybridizes to a replication sequence in the circular DNA strand. After heat inactivation of the polymerase, a restriction enzyme is added along with another RO of the opposite polarity to form double-stranded segments along the RCR product that are cleaved by the restriction enzyme. This RO is added in concentrations that assure saturation of complementary replication sequences in the concatemer product as well as any previously added RO. After a brief incubation, the restriction enzyme is heat-inactivated, causing the fragmented ROs to dissociate from the monomerized product. As temperature decreases, intact ROs anneal to the ends of the monomerized product. The RO preferentially hybridizes to both ends of the same monomer, forming circular monomer products rather than linear concatemers upon addition of a DNA ligase (1). These circles are then ready to serve as templates for a new round of RCR, primed by the same RO that templated the circularization (Fig. 1B). The C2CA copies circular DNA with a factor of amplification that can be modeled as a multiplicative function 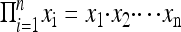 for n generations of polymerization reactions, each one producing xi monomers, where xi is directly proportional to the replication rate of the DNA polymerase and the time allowed for each RCR reaction, and inversely proportional to the length of the circular DNA.
for n generations of polymerization reactions, each one producing xi monomers, where xi is directly proportional to the replication rate of the DNA polymerase and the time allowed for each RCR reaction, and inversely proportional to the length of the circular DNA.
Efficient circle-to-circle conversion is crucial to obtain reproducible and quantitative results in this multistep procedure. To investigate this efficiency we monomerized and circularized first- and second-generation radiolabeled C2CA products, and analyzed the products by gel electrophoresis (Fig. 1C). For both strands, the conversion of RCR products to monomer circles was apparently complete. In particular, no dimeric ligation products were detected, demonstrating that, at this concentration of monomers, ligation only generates intramolecular circularization products. This ensures that different circles amplified in multiplex will be independently replicated.
To characterize the amplification procedure, we followed the accumulation of RCR products of either polarity in real-time by using two strand-specific molecular beacon hybridization probes (14, 16). Dilutions of circularized ppFD+ were used to initiate one, two, or three generations of C2CA. For each C2CA, the last replication phase was monitored in two separate reactions with either of the two molecular beacons (Fig. 2). Clearly, the first- and third-generation amplification reaction predominantly generated padlock probe-complementary strands. Conversely, the padlock probe strand predominated in the second-generation amplification reaction, consistent with the mechanism proposed in Fig. 1. The last replication phase of the one- and three-generation C2CA produced similar amounts of DNA, despite the millionfold lower concentration of starting DNA circles in the latter reaction, indicating that the RCR yielded a thousandfold amplification in each replication reaction. We calculated that the 93-nt circle was replicated at a rate of 17 revolutions per minute (rpm), establishing a polymerization rate for the φ29 polymerase of 1.6 × 103 nt/min under these conditions.
Only one polarity of DNA strands is produced in each generation, avoiding product inhibition by strand reassociation as seen toward the end of PCR. We compared the amount of product obtained from three generations of C2CA and a corresponding 27 cycles of PCR, both templated by 500 zmol ppFD+ circles. The amount of C2CA product was estimated by molecular beacon hybridization analysis and by comparison with known amounts of single-stranded DNA separated in an agarose gel. The amount of PCR product was estimated in a similar gel analysis using known amounts of double-stranded DNA. The PCR was also monitored in real-time to verify that it had reached plateau phase. At least 15 μM monomer products were formed in the C2CA, ≈100-fold higher yield than in the corresponding PCR (data not shown).
Padlock probes are suitable for highly parallel genetic analyses because multiple sequence-tagged padlock probes can be added to individual DNA samples and then amplified with a common set of PCR primers (4, 5), without the problems of cross reactivity seen when multiple pairs of PCR primers are combined in one reaction. To investigate whether the C2CA procedure could provide an alternative mechanism to amplify large sets of reacted padlock probes, we genotyped nine human genomic DNA samples by using the same set of 26 padlock probes specific for 13 single nucleotide variants of the ATP7B gene, as described in Banér et al. (5). The probe set was reacted with 10 ng of genomic DNA, followed by exonuclease digestion of remaining unreacted probes. Circularized probes were then amplified in three generations of C2CA, monomerized, and then analyzed by hybridization to tag microarrays (Fig. 3A). A total of 111 of 117 genotypes were concordant with the results of Banér et al., for an overall success rate of 95%. The results from four loci are shown in Fig. 3B. No C2CA products were detected in control reactions without ligase, in contrast to PCR-amplified padlock probes, where some ligation-independent amplification artifacts were observed (data not shown).
Fig. 3.

PCR-independent multiplexed genotyping. (A) Allele-specific padlock probes were constructed with 20 target-complementary nucleotides at both the 5′ and 3′ ends (gray) and with the allele-specific nucleotide positioned at the 3′ end. Target-specific ends were connected by the following three segments: a replication sequence common for all probes (black), either of two allele tag sequences (red or green), and finally a locus tag sequence (blue). Probe arms correctly hybridized to a target sequence (black line) are efficiently joined by a DNA ligase. A subsequent exonuclease treatment degrades unreacted probes while preserving circularized ones. Circularized probes serve as templates in a replication reaction primed by the general replication oligonucleotide (RO). The monomerized third-generation RCR products were applied to a tag microarray along with TAMRA- or FITC-labeled (red or green star) allele-tag specific oligonucleotides in a sandwich hybridization. (B) Results from a multiplexed genotyping experiment of nine different genomic DNA samples using a pool of 26 padlock probes. Signals from the different allele-specific amplification products were plotted in log–log diagrams for 4 of the 13 loci. The genotypes of the samples were known from previous studies. Green squares represent individuals homozygous for allele 1, red squares represent individuals homozygous for allele 2, and yellow squares represent heterozygous individuals. Locus identities are identified in the lower left corners.
To investigate whether different DNA circles are amplified at similar rates in the C2CA procedure, two dilutions of 26 circularized ATP7B probes with a millionfold difference in concentration were amplified in one or three generations of C2CA. The same sets of DNA circles were also amplified in 7 or 27 cycles of PCR. All reactions were analyzed on microarrays, and fluorescence intensities from individual products from the two amplification methods were plotted in two graphs. Data from the first dilutions were plotted on the x axis, and the more strongly amplified millionfold dilutions were plotted on the y axis (Fig. 4). Correlation coefficients (R2) were calculated for the signals from the two dilutions. The correlation was significantly higher with C2CA (R2 = 0.84) than with PCR (R2 = 0.36). Because both amplification methods were templated by the same dilutions of circles and had similar variation in microarray read-out between triplicate reactions, the difference in correlation reflects a considerably greater consistency in amplification efficiency of different DNA circles during C2CA compared to PCR.
Fig. 4.

Preservation of the proportion of amplification products after million-fold amplification by C2CA or PCR. (A) A pool of 26 different of DNA circles was subjected to one generation of C2CA or they were diluted one-million-fold and amplified in three generations of C2CA. The products were then analyzed on tag microarrays. (B) The same sets of circles were amplified in 7 or 27 cycles of PCR and then analyzed in the same manner. Fluorescence values of the amplification products were plotted in two graphs with results from the undiluted pool of circles on the x axis and the million-fold dilution on the y axis. The correlation coefficients for the two dilutions are indicated for each amplification procedure.
We also investigated the suitability of C2CA for quantitative analyses by determining the precision of the method. Mitochondrial DNA (mtDNA) was detected by using the padlock probe ppMT+ in a dilution series of total cellular DNA. Circularized ppMT+ probes were subjected to three generations of C2CA together with ppREF+ reference circles. The ppREF+ circle is added as an internal reference to normalize for differences in reaction and detection conditions. The last replication reaction was monitored in real-time by using two product-specific molecular beacons. The maximal rate of fluorescence increase reflects the number of replicating DNA circles and was determined for each of the two molecular beacons according to Nilsson et al. (14). The ratios of these two values were converted to numbers of circularized probes by using a standard curve, and finally plotted against the amount of cellular DNA (Fig. 5). There was a direct linear correlation between number of circles formed in the ligation reactions and the amount of cellular DNA with a precision (CV) of 10%.
Fig. 5.

Quantitative precision of C2CA. (A) A dilution series of ppMT+ test circles was amplified together with a constant amount of ppREF+ reference circles in three generations of C2CA. The third RCR reaction was monitored in real-time by using two differently labeled molecular beacons, one specific for the product from the test circle (mbMT+), and one specific for the reference product (mbREF+). Normalized values (ΔmaxmbMT+/ΔmaxmbREF+) were plotted against the number of test circles in the dilution series. Error bars indicate standard deviations of triplicate reactions, and an average CV of 6.5% was calculated. (B) A dilution series of total cellular DNA was analyzed in triplicate ligation reactions by using the mtDNA-specific padlock probe ppMT+. Circularized padlock probes were then amplified in three generations of C2CA along with a constant amount of ppREF+ reference circle, added in the first RCR. The resulting ΔmaxmbMT+/ΔmaxmbREF+ values were converted to numbers of ligated probes by using the standard curve, and then plotted against pg total cellular DNA. Error bars indicate standard deviations.
Discussion
We present a procedure for amplification of circular DNA strands that mimics the RCR of bacteriophages, iteratively copying their circular genomes to multiple new circular genomes. C2CA provides a robust and precise method to detect and quantify DNA circles. C2CA amplification products can be obtained in three different forms as required: concatemers, linear monomers, and circles. For example, individual RCR products can be recorded and enumerated by microscopy (17); monomerized products can be sorted by hybridization to DNA microarrays as shown herein; and replication of DNA circles can also be precisely quantified in real-time (14). The degree of amplification can be tuned by adjusting the number of generations of amplification or the time allowed for polymerization in each generation. The fact that C2CA generates single-stranded products of a defined polarity facilitates hybridization-based analyses. The procedure is suited for automation because it only involves additions of general amplification reagents that can be performed with an automated liquid dispenser with temperature control.
We demonstrate that the C2CA method is sufficiently powerful and specific to amplify and analyze sets of padlock probes reacted with 10-ng aliquots of genomic DNA. Regardless of whether the DNA circles are amplified by PCR or C2CA, it is important to remove excess unreacted linear probe molecules, for example, by exonuclease treatment, but the reasons are different. In contrast to PCR (4, 5), the C2CA reaction does not produce any detectable ligase-independent, probe-dependent amplification artifacts. However, remaining linear padlock probes can hybridize to the replication products and be extended by the polymerase, leading to the accumulation of single- and double-stranded products in a poorly controlled hyperbranched rolling-circle amplification reaction (2). Even though reagents are added several times to the reaction tubes, we have not detected contamination artifacts in control reactions where ligation templates for probes have been omitted.
Analogous to the parallel genotyping approach (Fig. 3A), cDNA samples could be reacted with transcript-specific padlock probes, C2C amplified and quantified on tag-sequence microarrays for highly specific expression profiling. C2CA preserves the proportion among different amplicons better than PCR, and because C2CA also produces higher concentration of products than PCR, the signals in highly multiplexed array-based analyses can be enhanced, extending the dynamic range of the analysis. The padlock probes used for parallel expression analyses using tag microarrays can also be used as needed for precise quantitative analysis of individual transcripts by real-time detection, using the same standard amplification and detection reagents as used for expression profiling. Padlock probes designed for C2CA can also be made shorter than corresponding PCR amplifiable probes because C2CA only requires a single 20-nt replication sequence, whereas two 20-nt sequences typically are required for amplification of padlock probes by PCR.
The ability to amplify circular DNA with great precision could prove of value also in applications other than detection of reacted padlock probes. We have recently described a so-called proximity ligation mechanism where the detection of specific target proteins resulted in the formation of signature DNA sequences that can be amplified by PCR for specific and sensitive detection (18). Also, circular DNA molecules, suitable for amplification by C2CA, can be created in proximity ligation reactions (M.G., unpublished data). It is furthermore possible to circularize specific sets of genomic DNA fragments, allowing these to be subjected to parallel amplification by C2CA for DNA analyses (F.D., M.G., U.L., and M.N., unpublished data). Finally, preparative applications of the C2CA method can be envisioned, further extending the utility of this new DNA amplification mechanism.
Acknowledgments
Dr. George Janssen kindly provided the M50 cell line. The work was supported by the Beijer and Wallenberg Foundations, the Foundation for Medical Research in Uppsala, the Research Councils of Sweden for Natural Science and for Medicine, the Swedish Defense Nanotechnology Program, Amersham Pharmacia, and Polysaccharide Research AB (Uppsala).
Abbreviations: RCR, rolling-circle replication; C2CA, circle-to-circle amplification; RO, replication oligonucleotide.
References
- 1.Nilsson, M., Malmgren, H., Samiotaki, M., Kwiatkowski, M., Chowdhary, B. P. & Landegren, U. (1994) Science 265, 2085–2088. [DOI] [PubMed] [Google Scholar]
- 2.Lizardi, P. M., Huang, X., Zhu, Z., Bray-Ward, P., Thomas, D. C. & Ward, D. C. (1998) Nat. Genet. 19, 225–232. [DOI] [PubMed] [Google Scholar]
- 3.Antson, D.-O., Isaksson, A., Landegren, U. & Nilsson, M. (2000) Nucleic Acids Res. 28, e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardenbol, P., Baner, J., Jain, M., Nilsson, M., Namsaraev, E. A., Karlin-Neumann, G. A., Fakhrai-Rad, H., Ronaghi, M., Willis, T. D., Landegren, U. & Davis, R. W. (2003) Nat. Biotechnol. 21, 673–678. [DOI] [PubMed] [Google Scholar]
- 5.Baner, J., Isaksson, A., Waldenström, E., Jarvius, J., Landegren, U. & Nilsson, M. (2003) Nucleic Acids Res. 31, e103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fire, A. & Xu, S.-Q. (1995) Proc. Natl. Acad. Sci. USA 92, 4641–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu, D., Daubendiek, S. L., Zillman, M. A., Ryan, K. & Kool, E. T. (1996) J. Am. Chem. Soc. 118, 1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banér, J., Nilsson, M., Mendel-Hartvig, M. & Landegren, U. (1998) Nucleic Acids Res. 26, 5073–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanco, L., Bernad, A., Lazaro, J. M., Martin, G., Garmendia, C. & Salas, M. (1989) J. Biol. Chem. 264, 8935–8940. [PubMed] [Google Scholar]
- 10.Faruqi, A. F., Hosono, S., Driscoll, M. D., Dean, F. B., Alsmadi, O., Bandaru, R., Kumar, G., Grimwade, B., Zong, Q., Sun, Z., et al. (2001) BMC Genomics 2, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickering, J., Bamford, A., Godbole, V., Briggs, J., Scozzafava, G., Roe, P., Wheeler, C., Ghouze, F. & Cuss, S. (2002) Nucleic Acids Res. 30, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alsmadi, O. A., Bornarth, C. J., Song, W., Wisniewski, M., Du, J., Brockman, J. P., Faruqi, A. F., Hosono, S., Sun, Z., Du, Y., et al. (2003) BMC Genomics 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wallace, D. C. (1999) Science 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 14.Nilsson, M., Gullberg, M., Dahl, F., Szuhai, K. & Raap, A. K. (2002) Nucleic Acids Res. 30, E66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Ouweland, J. M. W., Maechler, P., Wollheim, C. B., Attardi, G. & Maassen, J. A. (1999) Diabetologia 42, 485–492. [DOI] [PubMed] [Google Scholar]
- 16.Tyagi, S. & Kramer, F. R. (1996) Nat. Biotechnol. 14, 303–308. [DOI] [PubMed] [Google Scholar]
- 17.Blab, G. A., Schmidt, T. & Nilsson, M. (2003) Anal. Chem. 76, 495–498. [DOI] [PubMed] [Google Scholar]
- 18.Fredriksson, S., Gullberg, M., Jarvius, J., Olsson, C., Pietras, K., Gustafsdottir, S. M., Ostman, A. & Landegren, U. (2002) Nat. Biotechnol. 20, 473–477. [DOI] [PubMed] [Google Scholar]