

 This work is licensed under a
This work is licensed under a Abstract
3-M syndrome is an autosomal recessive disorder characterised by pre- and post-natal growth restriction, facial dysmorphism, normal intelligence and radiological features (slender long bones and tall vertebral bodies). It is known to be caused by mutations in the genes encoding cullin 7, obscurin-like 1 and coiled-coil domain containing 8. The mechanisms through which mutations in these genes impair growth are unclear. The aim of this study was to identify novel pathways involved in the growth impairment in 3-M syndrome. RNA was extracted from fibroblast cell lines derived from four 3-M syndrome patients and three control subjects, hybridised to Affymetrix HU 133 plus 2.0 arrays with quantitative real-time PCR used to confirm changes found on microarray. IGF-II protein levels in conditioned cell culture media were measured by ELISA. Of the top 10 downregulated probesets, three represented IGF2 while H19 was identified as the 23rd most upregulated probeset. QRT-PCR confirmed upregulation of H19 (P<0.001) and downregulation of IGF2 (P<0.001). Levels of IGF-II secreted into conditioned cell culture medium were higher for control fibroblasts than those for 3-M fibroblasts (10.2±2.9 vs 0.6±0.9 ng/ml, P<0.01). 3-M syndrome is associated with a gene expression profile of reduced IGF2 expression and increased H19 expression similar to that found in Silver–Russell syndrome. Loss of autocrine IGF-II in the growth plate may be associated with the short stature seen in children with 3-M syndrome.
Keywords: SGA, IGF2, 3-M syndrome, CUL7, OBSL1, CCDC8
Introduction
3-M syndrome (named after the first three authors to describe the condition) is an autosomal recessive disorder characterised by impaired pre- and post-natal growth, facial dysmorphism (triangular shaped face, anteverted nares, full fleshy lips), prominent heels, normal intelligence and, in some, radiological features (slender long bones and tall vertebral bodies). It is caused by loss of function mutations in the genes encoding cullin 7 (CUL7) (1), obscurin-like 1 (OBSL1) (2) and coiled-coil domain containing 8 (CCDC8) (3). CUL7 is a scaffold protein forming part of an E3 ubiquitin ligase enzyme responsible for cytoplasmic protein degradation (4), while OBSL1 is a cytoskeletal adaptor protein which localises to the perinuclear region (5). The function of CCDC8 is unknown, but it binds to OBSL1 (3) and is required for p53-mediated apoptosis (6).
The mechanisms leading to the growth impairment seen in 3-M syndrome remain unclear, but are likely to relate to abnormalities in basic cellular growth as well as alterations in cellular responses to growth factor stimulation. The Cul7−/− mouse displays impaired pre-natal growth and abnormalities in placental vasculature, but dies from respiratory distress after birth (7). Suggested targets for the CUL7 containing E3 ubiquitin ligase enzyme include cyclin D1 (8) and IRS1 (9). Altered IGF-I signalling with increased activation of the downstream signalling molecule AKT was identified in Cul7−/− mouse embryonic fibroblasts (MEFs) (9), associated with poor cell growth and senescence. Overexpression of CUL7 in an immortalised cancer cell line leads to decreased p53-mediated apoptosis (10, 11, 12). In contrast to the data in MEFs, AKT signalling was reduced in human skin fibroblast cell lines derived from 3-M syndrome patients (13) (including one patient with a CUL7 nonsense mutation). Alterations in the levels of the insulin-like growth factor-binding proteins (IGFBPs) have been identified in 3-M syndrome patient cell lines, both at the RNA level for IGFBP2 and 5 (14) and at the protein level for IGFBP2, 5 and 7 (13). Alterations in IGFBP levels and IGF-I signal transduction are seen in cell lines with OBSL1 and CCDC8 mutations (13) as well as CUL7 mutations; there is, however, a paucity of other data on the link between OBSL1, CCDC8 and CUL7 and the mechanism of growth impairment.
Although 3-M syndrome is considered to be a relatively uncommon disorder, it is probably an under recognised condition (6); its core characteristics of pre- and post-natal growth impairment are shared with all small for gestational age (SGA) children with failure of catch up growth. This includes many children in whom there is as yet no clear mechanism of growth impairment. The aim of this study was to identify novel potential mechanisms of growth impairment in 3-M syndrome, as an exemplar condition for SGA, by examining the transcriptome of skin fibroblast cell lines derived from 3-M patients. Skin fibroblast cell lines have previously been useful in the study of other growth disorders (15, 16). An understanding of the mechanisms of growth impairment in 3-M syndrome could lead to insights into the causation of poor growth in other SGA children and potential targets for molecular diagnostics.
Subjects and methods
Patients
Skin fibroblast cell lines were derived from four 3-M syndrome patients and three control subjects. Biopsies were obtained from the forearm after application of EMLA cream (AstraZeneca). The patients included one male with a homozygous CUL7 mutation (c.4191delC p.H1379HfsX11), one male with a homozygous OBSL1 mutation (c.1273insA, p.T425NfsX40, referred to as OBSL1M here), one female with a homozygous OBSL1 mutation (c.1273insA, p.T425NfsX40, referred to as OBSL1F) and one female with a homozygous CCDC8 mutation (c.84dup, p.L29X). The three control fibroblast cell lines (two males and one female) were derived from skin obtained during removal of skin tags. All patients and control subjects were prepubertal at the time the skin samples were obtained. All patients with 3-M syndrome had clinical features of the condition including growth impairment.
Cell culture
Fibroblast cells were cultured in 75 cm2 cell culture flasks (Corning, Tewkesbury, MA, USA) in DMEM (Invitrogen Paisley, Renfrewshire, UK) supplemented to a final concentration with 10% foetal bovine serum (Invitrogen), 50 units/ml penicillin, 50 μg/ml streptomycin, 2 mM glutamine and 2.5 μg/ml amphoteracin B (Invitrogen).
WST-8 cell growth assay
Cells were seeded at a density of 1000 cells/cm2 in 96-well cell culture plates (Corning) in 100 μl cell culture media: 24 and 72 h after seeding, 10 μl WST-8 was added to each well, the plate was incubated for 2 h at 37 °C before measuring absorbance at 450 nm on a u.v. spectrophotometer (Bio-Rad Benchmark microplate reader, Bio-Rad UK). For each cell line at each time measurement, a minimum of eight independent wells were examined on three separate occasions.
5-Ethynyl-2′-deoxyuridine incorporation
Cells were seeded at a density of 1000 cells/cm2 into 8-well chamberslides (Scientific Laboratory Supplies, Hessle, Yorkshire, UK) and incubated for 24 h in 600 μl cell culture media at 37 °C in 5% CO2. After 24 h, the culture medium was removed and replaced with media containing 40 μM 5-ethynyl-2′-deoxyuridine (EdU) for 3 h with the cells incubated in standard conditions. The media was then removed and the cells washed, fixed and permeabilised. EdU incorporation was assessed using the Click-iT EdU Alexa Fluor 488 Imaging Kit *for 50 coverslips (Invitrogen) as per the manufacturer's instructions. DAPI was used to identify the total number of cells present. Three independent fields containing at least 50 cells per field were examined for each cell line and the experiment was repeated on three occasions. A Leica CTR 5000 microscope was used to visualise the cells incorporating EdU.
Cleaved caspase-3 ELISA
Cleaved caspase-3 was measured with the PathScan Cleaved Caspase-3 ELISA (New England Biolabs, Hitchin, Hertfordshire, UK). Cells were seeded in 6-well plates at 1000 cells/cm2 for each cell line: 48 h after seeding, the media were removed and cell lysate was generated as per the manufacturer's instructions. Absorbance at 450 nm was measured on a u.v. spectrophotometer (Bio-Rad Benchmark microplate reader, Bio-Rad UK).
RNA extraction and transcriptome analysis
RNA was extracted from the fibroblasts using the RNAEasy kit (Qiagen, Manchester, UK) as per the manufacturer's instructions and supplied to the University of Manchester Microarray Facility (Faculty of Life Sciences, University of Manchester, UK). RNA quality was assessed using an Agilent 2100 Bioanalyser: 500 ng of total RNA per cell line was reverse transcribed using a T7 Oligo dT primer. An in vitro transcription reaction was used to generate biotinylated cRNA, which was purified, fragmented and hybridised to an Affymetrix HU-133 Plus 2.0 chip (Affymetrix, Santa Clara, CA, USA).
Microarray data were analysed using Propagating Uncertainty Microarray Analysis (PUMA – http://www.bioinf.manchester.ac.uk/resources/puma/). This process obtains a value for expression for each probeset on the microarray chip and involves normalising gene expression both within and between chips. Probesets were defined as being up- or downregulated if there was a ±1.5-fold difference in the expression between the control and 3-M samples with an expression level >50 (arbitrary units) in at least one cell line.
PUMA was also used to undertake principle component analysis (PCA) with probability of positive log-ratio (PPLR) to examine any differences in gene expression between control and 3-M fibroblasts. PPLR values closer to +1 indicate those probesets that are most likely to be upregulated and values closer to −1 indicate those most likely to be downregulated. In addition to PCA, quality control of the arrays was assessed with dCHIP (http://biosun1.harvard.edu/complab/dchip/).
Gene ontology and pathway analysis were performed with the use of the National Institute's Health Database for Annotation, Visualisation, Integrated Discovery (NIH DAVID) (http://david.abcc.ncifcrf.gov/).
Quantitative PCR
One microgram RNA (derived independently from the samples used for microarray) was reverse transcribed using the high capacity RNA to cDNA kit (Applied Biosystems). IGF2 and H19 mRNA levels were assessed using TaqMan assays (Hs01005963_m1 and Hs00262142_g1) with a cyclophyllin A probe (4333763T) for control gene expression. The other genes were assayed using SYBR green with GAPDH as the control gene (primer sequences available on request). Relative fold gene expression for the target gene in the 3-M cell lines was calculated as 2−ΔΔCT. For each experiment, three independent RNA extractions were assayed with three technical replicates.
Methylation analysis of H19 and IGF2
One microgram peripheral leukocyte or fibroblast-derived genomic DNA was treated with bisulphite using the EZ-DNA Methylation kit, according to manufacturer's instructions (Zymo Research, Orange, CA, USA), except that DNA was eluted in 50 μl. Methylation-specific PCRs were performed in duplicate within the H19 promoter and IGF2 DMR0 (as described in Poole et al. (17)) and the products were visualised by capillary electrophoresis on an ABI 3130 Genetic Analyzer (Applied Biosystems). Peak height ratiometry was performed and normalised to control samples.
Pyrosequencing was performed in duplicate, interrogating both the H19 ICR (as described in Poole et al. (17)) and IGF2P0 DMR0 (as described in Murrell et al. (18)). Primer sequences for all assays are provided in Tables 1 and 2.
Table 1.
Primer sequences for methylation-specific PCR.
| DMR | Chr | Chromosomal location GRCh37 | Methylated allele size | Unmethylated allele size | Methylated primer | Unmethylated primer | Universal FAM-labelled primer |
|---|---|---|---|---|---|---|---|
| H19 promotor | 11p15 | chr11: 2 019 455–2 019 764 | pat 295 | mat 305 | CGTTTGTTAGTAGAGTGCGTTCGCGAGTCG | GGTTGTTTATTGTTTGTTAGTAGAGTGTGTTTGTG | ATAACAGAAAAAACCCCTTCCTACCACCATCAC |
| IGF2P0 | 11p15 | chr11: 2 169 485–2 169 651 | pat 155 | mat 163 | GTTTGACGAGGTTAGTGAGGGACGGCG | ATAGTTTTGTTTGAtGAGGTTAGTGAGGGATGGTG | CCAAAACAATTTCCCTAAAAATACTCATTCATAC |
Table 2.
Primer sequences for pyrosequencing.
| DMR | Chr | Chromosomal location GRCh37 | Methylated allele size | Unmethylated allele size | Primer 1 | Primer 2 (biotinylated) | Sequencing primer(s) |
|---|---|---|---|---|---|---|---|
| IGF2 DMR0 | 11p15 | chr11: 2 169 328–2 169 582 | NA | NA | TGAGGATGGGTTTTTGTTTGGTAT | TCCTCAATCCACCCAAAATAATAT | AAAAGTTATTGGATATATAGT or GGGGTGGAGGGTGTA |
| H19 ICR1 | 11p15 | chr11: 1 977 650–1 977 877 | NA | NA | GTATAGTATATGGGTATTTTTGGAGG | CCATAAATATCCTATTCCCAAATAACC | GTTTYGGGTTATTTAAGTT |
Measurement of IGF-II in cell culture medium
Conditioned cell culture medium was obtained by incubating serum-free media with the relevant skin fibroblast cell line for 7 days. IGF-II levels were measured using an Active Non-Extraction IGF-II ELISA (Beckman Coulter, High Wycombe, Buckinghamshire, UK). IGF-II concentrations are much lower in cell culture medium than serum (for which the kit was designed) and this required some amendment to the standard kit protocol. Up to 400 μl conditioned cell culture medium was added to 550 μl sample buffer 1 and incubated at room temperature for 30 min: 950 μl sample buffer 2 was then added and the tube was vortexted. Fifty microlitre of the treated samples were added to each well of the ELISA plate and the remainder of the process was performed as per the manufacturer's instructions. Total protein concentration was measured in the conditioned media using Bio-Rad protein assay dye reagent (Bio-Rad) as per the manufacturer's instructions. Protein concentration of the control media was normalised to 1 and the IGF-II concentration adjusted for the total protein concentration in each cell line's conditioned media.
Results
Whole transcriptome analysis
There were 644 probesets identified as being upregulated and 658 identified as being downregulated in all three groups of 3-M patients compared with controls (Fig. 1). The top 10 up- and downregulated probesets are listed in Tables 3 and 4 respectively. Table 5 lists the top 20 up- and downregulated probesets for each cell line (CUL7, OBSL1 or CCDC8 mutation) and indicates which of these probesets are shared between more than one group. The majority of probesets is shared between more than one group and this suggests that there is a common set of genes dysregulated in 3-M syndrome.
Figure 1.
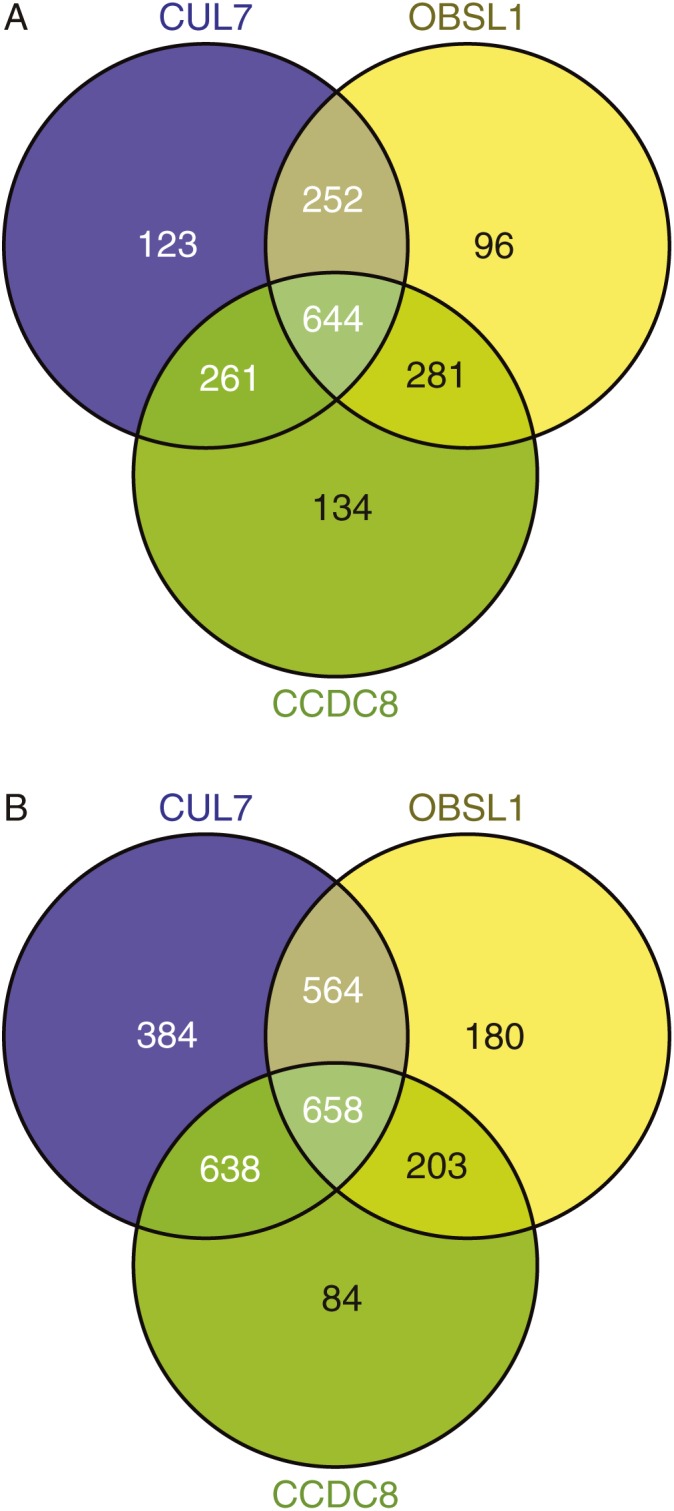
Venn diagrams of (A) up- and (B) downregulated probesets with expression level >50 and fold change (FC) >±1.5 compared to control.
Table 3.
Top 10 upregulated probesets comparing all 3-M cell lines (n=4) with control (n=3).
| Gene title | Gene symbol | Mean expression level control | Mean expression level 3-M | Fold difference 3-M/control | PPLR |
|---|---|---|---|---|---|
| Zic family member 1 | ZIC1 | 0.16 | 171.85 | 1087.41 | 1.00 |
| Purkinje cell protein 4 | PCP4 | 0.36 | 185.70 | 513.42 | 1.00 |
| Homeobox C6 | HOXC6 | 2.40 | 615.78 | 256.07 | 0.99 |
| Homeobox A10 | HOXA10 | 1.40 | 311.76 | 223.27 | 0.55 |
| Homeobox A9 | HOXA9 | 1.83 | 346.95 | 189.98 | 1.00 |
| Interleukin 13 receptor, alpha 2 | IL13RA2 | 6.27 | 779.20 | 124.27 | 1.00 |
| Collagen, type XIV, alpha 1 | COL14A1 | 1.28 | 145.45 | 113.88 | 1.00 |
| Glypican 6 | GPC6 | 5.51 | 591.28 | 107.25 | 1.00 |
| Clusterin | CLU | 7.84 | 795.86 | 101.45 | 1.00 |
| Solute carrier member 15 | SLC6A15 | 1.42 | 117.15 | 82.29 | 1.00 |
PPLR, probability of positive log-ratio.
Table 4.
Top 10 downregulated probesets comparing all 3-M cell lines (n=4) with control (n=3).
| Gene title | Gene symbol | Mean expression level control | Mean expression level 3-M | Fold difference 3-M/control | PPLR |
|---|---|---|---|---|---|
| Insulin-like growth factor 2 | IGF2 | 2118.84 | 0.06 | −38 253.37 | 0.00 |
| Leptin | LEP | 64.69 | 0.10 | −642.51 | 0.00 |
| Insulin-like growth factor 2 | IGF2 | 94.15 | 0.17 | −549.78 | 0.00 |
| Brain expressed, X-linked 1 | BEX1 | 369.40 | 1.30 | −283.28 | 0.00 |
| Prostaglandin D2 synthase 21 kDa (brain) | PTGDS | 136.49 | 0.62 | −219.06 | 0.00 |
| Collagen, type IV, alpha 1 | COL4A1 | 199.37 | 1.09 | −183.08 | 0.03 |
| Leucine-rich repeat-containing G protein-coupled receptor 5 | LGR5 | 270.20 | 1.51 | −179.24 | 0.01 |
| Insulin-like growth factor 2 | IGF2 | 44.95 | 0.29 | −157.09 | 0.00 |
| Glutamate receptor, ionotropic, kainate 2 | GRIK2 | 44.54 | 0.35 | −126.79 | 0.00 |
| WAP four-disulfide core domain 1 | WFDC1 | 37.84 | 0.37 | −101.50 | 0.00 |
PPLR, probability of positive log-ratio.
Table 5.
Top 20 up- and downregulated probesets in the 3-M group as a whole and in each cell line by mutation.
| Up-regulated probesets | Down-regulated probesets | ||||||
|---|---|---|---|---|---|---|---|
| 3-M | CUL7 | OBSL1 | CCDC8 | 3-M | CUL7 | OBSL1 | CCDC8 |
| ZIC1a | PCP4a | ZIC1a | XIST | IGF2a | IGF2a | IGF2a | IGF2a |
| PCP4a | ZIC1a | PCP4a | ZIC1a | LEPa | LGR5b | LEPa | CADM1 |
| HOXC6a | HOXA10a | HOXC6a | HOXC6a | IGF2a | COL4A1b | EDIL3c | PSG2 |
| HOXA10a | COL14A1b | COL14A1b | HOXA10a | – | SFRP2 | IGF2a | IGF2a |
| HOXA9a | HOXA9a | SNCAc | HOXA9a | BEX1a | LEPa | – | DDX3Y |
| IL13RA2a | IL13RA2a | HOXA10a | PCP4a | PTGDSb | DIO2 | COL4A1b | LGR5 |
| COL14A1b | THBS4 | HOXA9a | PAX6 | COL4A1b | APOEc | BEX1a | PTGDS |
| GPC6a | – | CLUb | EMCN | LGR5b | RARRES2b | PTGDSb | PSG3 |
| CLUb | HOXA11b | – | XIST | IGF2a | NID2 | IGF2a | BEX1a |
| SLC6A15b | HOXC6a | CYP3A5 | PAX6 | GRIK2c | TFAP2Ab | HAPLN1 | RPS4Y1 |
| HOXA10a | TNXB | SLC6A15b | GPC6a | WFDC1c | – | IGFBP5 | MAOA |
| HOXA11b | KCNB1 | IL13RA2a | CLUb | TFAP2Ab | LXN | RARRES2b | GRIK2 |
| – | WIF1 | GPC6a | IL13RA2a | RARRES2b | IGF2a | WFDC1c | TFAP2Ab |
| CLUb | GPC6a | CLUb | HOXA10a | APOEc | TLR4a | HAPLN1 | LEPa |
| – | HOXA10a | SPON1c | XIST | WNT5Ac | APOEc | SYNPO2c | IGF2a |
| ABCA6b | ST8SIA1 | SCARA3c | WISP1 | EDIL3c | ITIH5c | WNT5Ac | TRPC6 |
| SCARA3c | ACE | HOXA10a | THBD | SIM2 | PMEPA1 | IGFBP7 | FAM19A5 |
| HOXA9a | SLC6A15b | ABCA6b | THBD | – | PSG7 | TPD52L1 | ITIH5c |
| SPON1c | COL14A1b | SPON1c | H19 | SYNPO2c | DIO2 | MRVI1 | ITIH5c |
| TBX5 | ABCA6b | SNX10 | SNCAc | PPP1R14A | BEX1a | STXBP6 | USP9Y |
aProbesets present in all groups; bprobesets present in the 3-M group and two mutation groups; cprobesets present in the 3-M group and one mutation group. –, probeset is designed to a gene which was not named at the time of the study.
Among the upregulated probesets were two homeobox genes, HOXC6, a transcription factor expressed in the developing skeleton (19), and HOXA9, a transcription factor involved in myeloid differentiation linked with increased cell proliferation in leukaemia (20). Other upregulated genes included GPC6 (loss of function mutations result in the short stature condition omodysplasia (21)) as well as zinc finger protein of cerebellum 1 (ZIC1) and PCP4 both of which are known to be differentially expressed in tumours (22, 23). Three out of the top 10 downregulated probesets represented IGF2 which encodes a 7.5 kDa secreted hormone known to be a regulator of intra-uterine growth (24).
Q-PCR validation of a six up-and downregulated genes was undertaken (Table 6). In all cases, the Q-PCR result confirmed the findings on the microarray.
Table 6.
Additional validation of gene expression data. Expression of genes identified as being up- or downregulated in the microarray were assessed with Q-PCR. Relative fold expression for each of the seven genes analysed is given for each of the four 3-M cell lines. Expression was normalised to GAPDH and mean control cell line expression.
| Gene | CUL7 | OBSL1 F | OBSL1 M | CCDC8 | ||||
|---|---|---|---|---|---|---|---|---|
| Relative expression | P | Relative expression | P | Relative expression | P | Relative expression | P | |
| BEX1 | 0.06±0.01 | <0.001 | 0.15±0.20 | <0.001 | 0.03±0.02 | <0.001 | 0.20±0.24 | <0.001 |
| LEP | 0.07±0.04 | <0.001 | 0.06±0.03 | <0.001 | 0.01±0.00 | <0.001 | 0.04±0.01 | <0.001 |
| ZIC1 | 946±462 | <0.001 | 496±186 | 0.004 | 1200±891 | <0.001 | 759±498 | 0.002 |
| HOXC6 | 31±24 | 0.006 | 88±55 | 0.001 | 51±38 | 0.005 | 100±87 | 0.009 |
| HOXA9 | 690±586 | 0.008 | 501±466 | 0.012 | 349±296 | 0.008 | 399±342 | 0.008 |
| GPC6 | 35±6 | <0.001 | 44±10 | <0.001 | 7±2 | <0.001 | 5±1 | <0.001 |
Gene ontology analysis of the top 500 up- and downregulated probesets comparing all four 3-M cell lines with controls identified terms including skeletal system morphogenesis, cell adhesion and cell–cell signalling as being over represented (Benjamini–Hochberg adjusted P value <0.05). Cellular compartment terms significantly over-represented all related to the extra-cellular region.
Of the differentially regulated genes identified, the gene most closely linked with impaired growth was IGF2. Hypomethylation of the H19 differentially methylated region leads to IGF2 silencing in the Silver–Russell syndrome (SRS) (25). SRS shares key features with 3-M syndrome, namely the pre- and post-natal growth restriction with normal head size and triangular facies. It was therefore decided to focus further studies on IGF2.
IGF2 expression and protein concentrations in conditioned cell culture media
Q-PCR using three independently extracted RNA samples (each sample run in triplicate) confirmed the decrease in IGF2 expression with relative fold expression of 0.0019±0.0009 for CUL7 (P<0.001), 0.0155±0.0021 for OBSL1M (P<0.001), 0.0497±0.0170 for OBSL1F (P<0.001) and 0.1355±0.0146 for CCDC8 (P<0.001) compared with controls (Fig. 2).
Figure 2.

Relative fold expression of (A) IGF2 and (B) H19 measured using quantitative PCR in all 3-M cell lines. Expression of IGF2 is reduced while H19 is increased. *P<0.05.
Although not present in the top 10 upregulated probesets, the H19 non-coding RNA was represented by the 23rd most upregulated probeset (FC 38, PPLR 1). Q-PCR of H19 confirmed that it was upregulated in all four 3-M cell lines (Fig. 2). Relative fold expression was 2.5±0.8 for CUL7 (P<0.001), 140±53 for OBSL1M (P<0.001), 72±12 for OBSL1F (P<0.001) and 1106±435 for CCDC8 (P<0.001).
Concentrations of IGF-II were reduced in conditioned cell culture media from all four 3-M cell lines compared with control cell lines (Fig. 3). The mean IGF-II concentration for the three control cell lines after adjustment for total protein concentration in the media was 10.2±2.9 ng/ml, compared with 0.1±0.2 ng/ml for the CUL7 cell line (P<0.001), 0.3±0.4 ng/ml for the OBSL1M cell line (P<0.001), 0.4±0.5 ng/ml for the OBSL1F cell line (P<0.001) and 1.6±1.3 ng/ml for the CCDC8 cell line (P<0.001).
Figure 3.

IGF-II concentrations in conditioned cell culture media are significantly reduced for all 3-M syndrome fibroblast cell lines. *P<0.05 compared to control. IGF-II concentrations are adjusted for total protein concentration in the cell culture media.
Overexpression of H19 and silencing of IGF2 in SRS are caused by changes in methylation in the H19 differentially methylated region. Methylation-specific PCR and pyrosequencing of the H19 ICR, H19 promotor and IGF2 DMR0 identified no differences in methylation between control and 3-M syndrome subjects, for both peripheral leucocyte and fibroblast-derived DNA.
Cell proliferation and apoptosis
Cell proliferation was assessed via incorporation of EdU and by a WST-8 assay. Incorporation of EdU 48 h after its addition to cell culture media was reduced for all 3-M fibroblast cell lines compared with control (Fig. 4A, P<0.05), while cell proliferation as measured by colorimetric change induced by WST-8 was reduced at 48 and 72 h after seeding for 3-M fibroblast cell lines compared with control (Fig. 4B, P<0.05). Cleaved caspase-3, a biomarker of apoptosis was not significantly different between control and 3-M fibroblasts (Fig. 4C).
Figure 4.

Reduced cell proliferation with no increase in apoptosis is seen in fibroblasts from 3-M syndrome patients. (A) Decreased incorporation of EdU measured at 48 h. (B) Decreased cell proliferation at 24 and 72 h after seeding measured by WST-8. (C) No difference in apoptosis as measured by Cleaved Caspase-3 ELISA. *P<0.05.
Discussion
The aim of this study was to identify novel pathogenic mechanisms underlying the growth failure of patients with 3-M syndrome, which could potentially be relevant to other patients born SGA with the failure of post-natal growth but no defined aetiology. Previous work has examined the role of CUL7 and OBSL1 either in mouse studies or using gene overexpression or knockdown strategies in immortalised cancer cell lines. The limitations of mouse studies are clear from the death of the mice in the neonatal period (a feature not commonly seen in humans with 3-M syndrome). The mouse thus gives no opportunity to study the effects on post-natal growth and also indicates significant differences in the result of loss of CUL7 between species. Studies using temporary over/under expression strategies in immortalised cells yield useful data, but the extrapolation from these findings to normal human growth is not clear. This study therefore used patient-derived fibroblast cell lines.
It is clear that there is a common set of genes dysregulated in 3-M syndrome. The top upregulated gene was Zic1, a transcription factor which, in mouse, is predominantly expressed within the nervous system with the highest levels of expression in the cerebellum (26). ZIC1 expression is downregulated in gastric carcinomas (23) and increased in desmoid tumour fibroblasts (27) and brain tumours (medulloblastomas and meningiomas) (28, 29). Several other genes in the top 10 upregulated probesets are also overexpressed in tumours including PCP4 in leiomyomas (22), HOXC6 in oesophageal (30), breast (31) and lung carcinomas (32) and IL16RA2 in glioblastomas (33), prostate cancer and adrenocortical tumours (34). This indicates that their overexpression in 3-M fibroblasts could be an attempt to increase cell proliferation. Data on siRNA-mediated knockdown and overexpression of HOXC6 in a gastric carcinoid cell line are consistent with this hypothesis as overexpression leads to improved growth while loss of HOXC6 leads to impaired cell growth (35).
Glypican 6 (GPC6), in the top 10 downregulated probesets, is a heparan sulphate proteoglycan, which is linked to the extracellular surface of the cell membrane. Glypicans are expressed during development and are thought to control availability of local growth factors (36). Loss of function mutations in GPC6 lead to impaired endochondral ossification and cause the short stature condition omodysplasia (21). Loss of function mutations in glypican 3 (GPC3) causes the overgrowth disorder Simpson–Golabi–Behmel syndrome (SGBS). GPC3 interacts with IGF-II and it was initially hypothesised that GPC3 binds to and sequesters IGF-II; thus the overgrowth in SGBS is caused by increased availability of IGF-II (37). More recent data on mouse indicate that the overgrowth of Gpc3 null mice is independent of IGF-II (38), while data on the role of GPC3 in the growth of cancer cell lines are inconsistent with some studies suggesting that GPC3 suppressed growth in an IGF-dependant manner (39) while others identified GPC3 as a growth-promoting protein (40, 41). Of note, of the two probesets in our microarray designed to detect the expression of GPC3, one did not detect expression of GPC3 (defined in this study as an expression level >50) in any cell line while the other probeset identified a modest downregulation (FC −1.76 PPLR 0.44).
Three of the top 10 downregulated probesets represented IGF2 with the smallest fold change being −157. The second most downregulated probeset represented leptin, a 16 kDa adipocyte-derived hormone which plays a central role in the regulation of body weight, both by inhibiting food intake and increasing energy expenditure (42, 43). Downregulation of leptin in 3-M syndrome may represent a response to the patients slim body habitus or be a signal to drive energy intake in order to promote growth. BEX1, PTGDS, GRIK2 and WFDC1, also in the top 10 downregulated probesets, have all been identified as downregulated in tumours (44, 45) or as inhibitors of cell proliferation in immortalised cell lines (46, 47, 48). Thus in common with several of the genes identified as being upregulated these changes are likely to represent a response to increase cell proliferation.
While many of the identified changes in gene expression, such as the overexpression of GPC6, are likely to be a compensatory response to the growth impairment, we hypothesised that there would be a smaller number of genes with altered expression which are key to the pathogenic process underlying 3-M syndrome. The most obvious of such candidate was IGF2, which was represented by three of the top 10 downregulated probesets. The encoded protein IGF-II is a 7.5 kDa secreted hormone that acts to increase cell proliferation via stimulation of the IGF1R. It is widely expressed during the development and is a major regulator of intra-uterine growth. IGF2 expression is regulated by methylation of the H19 region (H19 DMR); hypomethylation of the H19 DMR with subsequent IGF2 gene silencing leads to the short stature condition SRS (25). SRS shares several key features with 3-M syndrome: intra-uterine growth retardation, post-natal growth impairment, relatively normal head size, normal intelligence and a triangular shaped face. Other downregulated genes identified which could potentially be implicated in the pathogenesis of 3-M syndrome included LGR5 (a member of the G-protein-coupled receptor superfamily) and COL4A1 (the main component of type IV collagen which forms basement membrane); silencing of expression of these genes is associated with decreased cell proliferation (49) while in tumours (50, 51), they have been found to be upregulated. Of the top 10 upregulated probesets, the only gene identified as potentially being involved in the pathogenesis of 3-M syndrome was COL14A1, a large glycoprotein of the extracellular matrix which has an anti-proliferative effect on fibroblasts (52) and knockdown in renal cancer cells, which results in increased growth (53).
Given the findings of reduced IGF-II production from 3-M syndrome fibroblasts, it is likely that local production of IGF-II is reduced with loss of its autocrine/paracrine effects. Loss of local IGF-II in the growth plates and other tissues leads to growth impairment both pre- and post-natally. The reduced cell proliferation with no change found in a biomarker of apoptosis would be consistent with a reduction in the presence of a growth factor. While there is significant phenotypic overlap between 3-M syndrome and SRS, there are also phenotypic differences. These are likely to be due to additional functions of the proteins affected in 3-M syndrome.
The mechanisms through which IGF2 expression is reduced in 3-M syndrome remain unclear. It does not appear to be via the same mechanism as is found in SRS, i.e. hypomethylation at H19 DMR. It is possible that there may be an epigenetic change which has not been recognised but this appears unlikely. There may be another mechanism, such as alteration in CCCTC-binding factor concentrations or activity that could lead to the same gene expression pattern.
Height at presentation in 3-M syndrome is lowest in patients with CUL7 mutations and highest in those with CCDC8 mutations (13). Of interest, the pattern of IGF2 expression and IGF-II production mirrored the growth phenotype of the patients with the lowest IGF-II production in the CUL7 cell line and the highest IGF-II production in the CCDC8 cell line.
In conclusion, this study demonstrates that there is reduced expression of IGF2 in 3-M syndrome linking the pathogenesis to that of SRS. The mechanisms underlying the silencing of IGF2 in 3-M syndrome are unclear, but do not appear to involve hypomethylation at the H19 DMR.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
P G Murray was a MRC Clinical Training Fellow. The support of the Manchester NIHR Biomedical Research Centre is acknowledged.
References
- 1.Huber C, Dias-Santagata D, Glaser A, O'Sullivan J, Brauner R, Wu K, Xu X, Pearce K, Wang R, Uzielli ML, et al. Identification of mutations in CUL7 in 3-M syndrome. Nature Genetics. 2005;37:1119–1124. doi: 10.1038/ng1628. [DOI] [PubMed] [Google Scholar]
- 2.Hanson D, Murray PG, Sud A, Temtamy SA, Aglan M, Superti-Furga A, Holder SE, Urquhart J, Hilton E, Manson FD, et al. The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. American Journal of Human Genetics. 2009;84:801–806. doi: 10.1016/j.ajhg.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanson D, Murray PG, O'Sullivan J, Urquhart J, Daly S, Bhaskar SS, Biesecker LG, Skae M, Smith C, Cole T, et al. Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. American Journal of Human Genetics. 2011;89:148–153. doi: 10.1016/j.ajhg.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dias DC, Dolios G, Wang R, Pan ZQ. CUL7: a DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. PNAS. 2002;99:16601–16606. doi: 10.1073/pnas.252646399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geisler SB, Robinson D, Hauringa M, Raeker MO, Borisov AB, Westfall MV, Russell MW. Obscurin-like 1, OBSL1, is a novel cytoskeletal protein related to obscurin. Genomics. 2007;89:521–531. doi: 10.1016/j.ygeno.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clayton PE, Hanson D, Magee L, Murray PG, Saunders E, Abu-Amero SN, Moore GE, Black GC. Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clinical Endocrinology. 2012;77:335–342. doi: 10.1111/j.1365-2265.2012.04428.x. [DOI] [PubMed] [Google Scholar]
- 7.Arai T, Kasper JS, Skaar JR, Ali SH, Takahashi C, DeCaprio JA. Targeted disruption of p185/Cul7 gene results in abnormal vascular morphogenesis. PNAS. 2003;100:9855–9860. doi: 10.1073/pnas.1733908100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okabe H, Lee SH, Phuchareon J, Albertson DG, McCormick F, Tetsu O. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE. 2006;1:e128. doi: 10.1371/journal.pone.0000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, Zhu W, Nakajima H, Nakajima HO, Field LJ, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Molecular Cell. 2008;30:403–414. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrews P, He YJ, Xiong Y. Cytoplasmic localized ubiquitin ligase cullin 7 binds to p53 and promotes cell growth by antagonizing p53 function. Oncogene. 2006;25:4534–4548. doi: 10.1038/sj.onc.1209490. [DOI] [PubMed] [Google Scholar]
- 11.Kim SS, Shago M, Kaustov L, Boutros PC, Clendening JW, Sheng Y, Trentin GA, Barsyte-Lovejoy D, Mao DY, Kay R, et al. CUL7 is a novel antiapoptotic oncogene. Cancer Research. 2007;67:9616–9622. doi: 10.1158/0008-5472.CAN-07-0644. [DOI] [PubMed] [Google Scholar]
- 12.Jung P, Verdoodt B, Bailey A, Yates JR, III, Menssen A, Hermeking H. Induction of cullin 7 by DNA damage attenuates p53 function. PNAS. 2007;104:11388–11393. doi: 10.1073/pnas.0609467104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanson D, Murray PG, Coulson T, Sud A, Omokanye A, Stratta E, Sakhinia F, Bonshek C, Wilson LC, Wakeling E, et al. Mutations in CUL7, OBSL1 and CCDC8 in 3-M syndrome lead to disordered growth factor signalling. Journal of Molecular Endocrinology. 2012;49:267–275. doi: 10.1530/JME-12-0034. [DOI] [PubMed] [Google Scholar]
- 14.Huber C, Fradin M, Edouard T, Le Merrer M, Alanay Y, Da Silva DB, David A, Hamamy H, van Hest L, Lund AM, et al. OBSL1 mutations in 3-M syndrome are associated with a modulation of IGFBP2 and IGFBP5 expression levels. Human Mutation. 2009;31:20–26. doi: 10.1002/humu.21150. [DOI] [PubMed] [Google Scholar]
- 15.Freeth JS, Ayling RM, Whatmore AJ, Towner P, Price DA, Norman MR, Clayton PE. Human skin fibroblasts as a model of growth hormone (GH) action in GH receptor-positive Laron's syndrome. Endocrinology. 1997;138:55–61. doi: 10.1210/en.138.1.55. [DOI] [PubMed] [Google Scholar]
- 16.Westwood M, Tajbakhsh SH, Siddals KW, Whatmore AJ, Clayton PE. Reduced pericellular sensitivity to IGF-I in fibroblasts from girls with Turner syndrome: a mechanism to impair clinical responses to GH. Pediatric Research. 2011;70:25–30. doi: 10.1203/PDR.0b013e31821b570b. [DOI] [PubMed] [Google Scholar]
- 17.Poole RL, Baple E, Crolla JA, Temple IK, Mackay DJ. Investigation of 90 patients referred for molecular cytogenetic analysis using aCGH uncovers previously unsuspected anomalies of imprinting. American Journal of Medical Genetics. Part A. 2010;152A:1990–1993. doi: 10.1002/ajmg.a.33530. [DOI] [PubMed] [Google Scholar]
- 18.Murrell A, Ito Y, Verde G, Huddleston J, Woodfine K, Silengo MC, Spreafico F, Perotti D, De Crescenzo A, Sparago A. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer. PLoS ONE. 2008;3:e1849. doi: 10.1371/journal.pone.0001849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharpe PT, Miller JR, Evans EP, Burtenshaw MD, Gaunt SJ. Isolation and expression of a new mouse homeobox gene. Development. 1988;102:397–407. doi: 10.1242/dev.102.2.397. [DOI] [PubMed] [Google Scholar]
- 20.Whelan JT, Ludwig DL, Bertrand FE. HoxA9 induces insulin-like growth factor-1 receptor expression in B-lineage acute lymphoblastic leukemia. Leukemia. 2008;22:1161–1169. doi: 10.1038/leu.2008.57. [DOI] [PubMed] [Google Scholar]
- 21.Campos-Xavier AB, Martinet D, Bateman J, Belluoccio D, Rowley L, Tan TY, Baxová A, Gustavson KH, Borochowitz ZU, Innes AM, et al. Mutations in the heparan-sulfate proteoglycan glypican 6 (GPC6) impair endochondral ossification and cause recessive omodysplasia. American Journal of Human Genetics. 2009;84:760–770. doi: 10.1016/j.ajhg.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanamori T, Takakura K, Mandai M, Kariya M, Fukuhara K, Kusakari T, Momma C, Shime H, Yagi H, Konishi M, et al. PEP-19 overexpression in human uterine leiomyoma. Molecular Human Reproduction. 2003;9:709–717. doi: 10.1093/molehr/gag088. [DOI] [PubMed] [Google Scholar]
- 23.Wang LJ, Jin HC, Wang X, Lam EKY, Zhang JB, Liu X, Chan FKL, Si JM, Sung JJY. ZIC1 is downregulated through promoter hypermethylation in gastric cancer. Biochemical and Biophysical Research Communications. 2009;379:959–963. doi: 10.1016/j.bbrc.2008.12.180. [DOI] [PubMed] [Google Scholar]
- 24.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 25.Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, Danton F, Thibaud N, Le Merrer M, Burglen L, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver–Russell syndrome. Nature Genetics. 2005;37:1003–1007. doi: 10.1038/ng1629. [DOI] [PubMed] [Google Scholar]
- 26.Aruga J, Yokota N, Hashimoto M, Furuichi T, Fukuda M, Mikoshiba K. A novel zinc finger protein, zic, is involved in neurogenesis, especially in the cell lineage of cerebellar granule cells. Journal of Neurochemistry. 1994;63:1880–1890. doi: 10.1046/j.1471-4159.1994.63051880.x. [DOI] [PubMed] [Google Scholar]
- 27.Pourebrahim R, Van Dam K, Bauters M, De Wever I, Sciot R, Cassiman JJ, Tejpar S. ZIC1 gene expression is controlled by DNA and histone methylation in mesenchymal proliferations. FEBS Letters. 2007;581:5122–5126. doi: 10.1016/j.febslet.2007.09.061. [DOI] [PubMed] [Google Scholar]
- 28.Aruga J, Nozaki Y, Hatayama M, Odaka YS, Yokota N. Expression of ZIC family genes in meningiomas and other brain tumors. BMC Cancer. 2010;10:79. doi: 10.1186/1471-2407-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokota N, Aruga J, Takai S, Yamada K, Hamazaki M, Iwase T, Sugimura H, Mikoshiba K. Predominant expression of human zic in cerebellar granule cell lineage and medulloblastoma. Cancer Research. 1996;56:377–383. [PubMed] [Google Scholar]
- 30.Chen KN, Gu ZD, Ke Y, Li JY, Shi XT, Xu GW. Expression of 11 HOX genes is deregulated in esophageal squamous cell carcinoma. Clinical Cancer Research. 2005;11:1044–1049. [PubMed] [Google Scholar]
- 31.Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. Immunocytochemical detection of the homeobox B3, B4, and C6 gene products in breast carcinomas. Anticancer Research. 2000;20:3281–3286. [PubMed] [Google Scholar]
- 32.Bodey B, Bodey B, Jr, Groger AM, Siegel SE, Kaiser HE. Immunocytochemical detection of homeobox B3, B4, and C6 gene product expression in lung carcinomas. Anticancer Research. 2000;20:2711–2716. [PubMed] [Google Scholar]
- 33.Marie SK, Okamoto OK, Uno M, Hasegawa AP, Oba-Shinjo SM, Cohen T, Camargo AA, Kosoy A, Carlotti CG, Jr, Toledo S, et al. Maternal embryonic leucine zipper kinase transcript abundance correlates with malignancy grade in human astrocytomas. International Journal of Cancer. 2008;122:807–815. doi: 10.1002/ijc.23189. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Ranvier GG, Weng J, Yeh RF, Khanafshar E, Suh I, Barker C, Duh QY, Clark OH, Kebebew E. Identification of biomarkers of adrenocortical carcinoma using genomewide gene expression profiling. Archives of Surgery. 2008;143:841–846. doi: 10.1001/archsurg.143.9.841. discussion 6. [DOI] [PubMed] [Google Scholar]
- 35.Fujiki K, Duerr EM, Kikuchi H, Ng A, Xavier RJ, Mizukami Y, Imamura T, Kulke MH, Chung DC. Hoxc6 is overexpressed in gastrointestinal carcinoids and interacts with JunD to regulate tumor growth. Gastroenterology. 2008;135:907–16, 916.e1–2. doi: 10.1053/j.gastro.2008.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Filmus J. Glypicans in growth control and cancer. Glycobiology. 2001;11:19R–23R. doi: 10.1093/glycob/11.3.19R. [DOI] [PubMed] [Google Scholar]
- 37.Pilia G, Hughes-Benzie RM, MacKenzie A, Baybayan P, Chen EY, Huber R, Neri G, Cao A, Forabosco A, Schlessinger D. Mutations in GPC3, a glypican gene, cause the Simpson–Golabi–Behmel overgrowth syndrome. Nature Genetics. 1996;12:241–247. doi: 10.1038/ng0396-241. [DOI] [PubMed] [Google Scholar]
- 38.Chiao E, Fisher P, Crisponi L, Deiana M, Dragatsis I, Schlessinger D, Pilia G, Efstratiadis A. Overgrowth of a mouse model of the Simpson–Golabi–Behmel syndrome is independent of IGF signaling. Developmental Biology. 2002;243:185–206. doi: 10.1006/dbio.2001.0554. [DOI] [PubMed] [Google Scholar]
- 39.Sakurai M, Shibata K, Umezu T, Kajiyama H, Yamamoto E, Ino K, Nawa A, Kikkawa F. Growth-suppressing function of glypican-3 (GPC3) via insulin like growth factor II (IGF-II) signaling pathway in ovarian clear cell carcinoma cells. Gynecologic Oncology. 2010;119:332–336. doi: 10.1016/j.ygyno.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 40.Cheng W, Tseng CJ, Lin TT, Cheng I, Pan HW, Hsu HC, Lee YM. Glypican-3-mediated oncogenesis involves the insulin-like growth factor-signaling pathway. Carcinogenesis. 2008;29:1319–1326. doi: 10.1093/carcin/bgn091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun CK, Chua MS, He J, So SK. Suppression of glypican 3 inhibits growth of hepatocellular carcinoma cells through up-regulation of TGF-β2. Neoplasia. 2011;13:735–747. doi: 10.1593/neo.11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 44.Arslan AA, Gold LI, Mittal K, Suen TC, Belitskaya-Levy I, Tang MS, Toniolo P. Gene expression studies provide clues to the pathogenesis of uterine leiomyoma: new evidence and a systematic review. Human Reproduction. 2005;20:852–863. doi: 10.1093/humrep/deh698. [DOI] [PubMed] [Google Scholar]
- 45.Wu CS, Lu YJ, Li HP, Hsueh C, Lu CY, Leu YW, Liu HP, Lin KH, Hui-Ming Huang T, Chang YS. Glutamate receptor, ionotropic, kainate 2 silencing by DNA hypermethylation possesses tumor suppressor function in gastric cancer. International Journal of Cancer. 2010;126:2542–2552. doi: 10.1002/ijc.24958. [DOI] [PubMed] [Google Scholar]
- 46.Vilar M, Murillo-Carretero M, Mira H, Magnusson K, Besset V, Ibanez CF. Bex1, a novel interactor of the p75 neurotrophin receptor, links neurotrophin signaling to the cell cycle. EMBO Journal. 2006;25:1219–1230. doi: 10.1038/sj.emboj.7601017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larsen M, Ressler SJ, Gerdes MJ, Lu B, Byron M, Lawrence JB, Rowley DR. The WFDC1 gene encoding ps20 localizes to 16q24, a region of LOH in multiple cancers. Mammalian Genome. 2000;11:767–773. doi: 10.1007/s003350010135. [DOI] [PubMed] [Google Scholar]
- 48.Madar S, Brosh R, Buganim Y, Ezra O, Goldstein I, Solomon H, Kogan I, Goldfinger N, Klocker H, Rotter V. Modulated expression of WFDC1 during carcinogenesis and cellular senescence. Carcinogenesis. 2009;30:20–27. doi: 10.1093/carcin/bgn232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanese K, Fukuma M, Yamada T, Mori T, Yoshikawa T, Watanabe W, Ishiko A, Amagai M, Nishikawa T, Sakamoto M. G-protein-coupled receptor GPR49 is upregulated in basal cell carcinoma and promotes cell proliferation and tumor formation. American Journal of Pathology. 2008;173:835–843. doi: 10.2353/ajpath.2008.071091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen C, Méndez E, Houck J, Fan W, Lohavanichbutr P, Doody D, Yueh B, Futran ND, Upton M, Farwell DG, et al. Gene expression profiling identifies genes predictive of oral squamous cell carcinoma. Cancer Epidemiology, Biomarkers & Prevention. 2008;17:2152–2162. doi: 10.1158/1055-9965.EPI-07-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Carson-Walter EB, Cooper A, Winans BN, Johnson MD, Walter KA. Vascular gene expression patterns are conserved in primary and metastatic brain tumors. Journal of Neuro-Oncology. 2010;99:13–24. doi: 10.1007/s11060-009-0105-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruehl M, Erben U, Schuppan D, Wagner C, Zeller A, Freise C, Al-Hasani H, Loesekann M, Notter M, Wittig BM, et al. The elongated first fibronectin type III domain of collagen XIV is an inducer of quiescence and differentiation in fibroblasts and preadipocytes. Journal of Biological Chemistry. 2005;280:38537–38543. doi: 10.1074/jbc.M502210200. [DOI] [PubMed] [Google Scholar]
- 53.Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F, Maher ER. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. 2010;29:2104–2117. doi: 10.1038/onc.2009.493. [DOI] [PMC free article] [PubMed] [Google Scholar]