

Abstract
The recent human infections caused by H5N1, H9N2, and H7N7 avian influenza viruses highlighted the continuous threat of new pathogenic influenza viruses emerging from a natural reservoir in birds. It is generally believed that replication of avian influenza viruses in humans is restricted by a poor fit of these viruses to cellular receptors and extracellular inhibitors in the human respiratory tract. However, detailed mechanisms of this restriction remain obscure. Here, using cultures of differentiated human airway epithelial cells, we demonstrated that influenza viruses enter the airway epithelium through specific target cells and that there were striking differences in this respect between human and avian viruses. During the course of a single-cycle infection, human viruses preferentially infected nonciliated cells, whereas avian viruses as well as the egg-adapted human virus variant with an avian virus-like receptor specificity mainly infected ciliated cells. This pattern correlated with the predominant localization of receptors for human viruses (2-6-linked sialic acids) on nonciliated cells and of receptors for avian viruses (2-3-linked sialic acids) on ciliated cells. These findings suggest that although avian influenza viruses can infect human airway epithelium, their replication may be limited by a nonoptimal cellular tropism. Our data throw light on the mechanisms of generation of pandemic viruses from their avian progenitors and open avenues for cell level-oriented studies on the replication and pathogenicity of influenza virus in humans.
Influenza pandemics originate from birds which provide a reservoir for a large variety of influenza A viruses containing at least 15 hemagglutinin (HA) and nine neuraminidase (NA) subtypes (1, 2). Fortunately, both initial human infections by avian viruses and the emergence of pandemic viruses are restricted by a limited fitness of avian viruses in humans. In the case of virus genes other than the HA gene, this restriction can be overcome by gene reassortment between avian and human viruses similar to that which occurred in the 1957 and 1968 pandemic viruses (1, 3-5). However, any pandemic virus must carry the HA from a nonhuman virus. Therefore, one of the key themes in the study of pandemic influenza is to understand the mechanisms of HA-mediated restriction and how avian viruses manage to breach it.
Avian influenza viruses bind to cell-surface glycoproteins or glycolipids containing terminal sialyl-galactosyl residues linked by 2-3-linkage [Neu5Ac(α2-3)Gal], whereas human viruses, including the earliest available isolates from the 1957 and 1968 pandemics, bind to receptors that contain terminal 2-6-linked sialyl-galactosyl moieties [Neu5Ac(α2-6)Gal] (6-8). The 1918 influenza pandemic viruses presumably also had a human-virus-like receptor specificity (8, 9). By contrast, H5N1 chicken viruses that caused the influenza outbreak in humans in Hong Kong in 1997 (10, 11) had an avian virus-like receptor specificity (12) and were unable to transmit efficiently from human to human (13). It is believed, therefore, that an alteration of the receptor specificity is essential for the emergence of pandemic viruses from avian progenitors (8, 14), but the mechanism of this phenomenon is not clear.
In humans, influenza viruses replicate in the ciliated epithelium of conducting airways (15), which consists of several distinct cell types with different functions (16). Observations made late in the infectious process in humans and monkeys show that influenza viruses infect many different types of airway epithelial cells (17-19). However, neither initial targets of the virus attack nor specific cell types that are essential for virus replication have been defined. Our knowledge on the receptor equipment of human airway epithelium is also limited. Predominant expression of 2-6-linked sialic acids on the apical surface of human tracheal epithelial cells has been reported (20, 21), but little is known about the presence of 2-3-linked sialic acids on such cells or about variations in sialic acid expression on different cell types.
To address these questions and to understand how avian-virus-like receptor specificity limits efficiency of virus replication in humans, we studied cellular tropism of human and avian influenza viruses and distribution of virus receptors in differentiated cultures of human tracheobronchial epithelium (HTBE) (22). These cultures are pseudostratified and polarized, contain ciliated, secretory, and basal cells, and they resemble human airway epithelium in vivo both morphologically and functionally (22-24). Here we show that different receptor-binding specificity of human and avian viruses determines their preferential tropism to nonciliated cells and ciliated cells, respectively. This finding suggests that infection of nonciliated epithelial cells in human airways is essential for the efficient replication and/or transmission of influenza viruses and that fitness of avian viruses in humans may be compromised by their nonoptimal cellular tropism.
Materials and Methods
Airway Epithelial Cultures. Primary human epithelial cells from tracheal/bronchial and nasal tissues were purchased from Clonetics and Promo Cell, Heidelberg, respectively. Cells of passage 2 were grown on membrane supports (12-mm Transwell-Clear, Corning) at the air-liquid interface in serum-free and hormone- and growth factor-supplemented medium as described (22). Fully differentiated 4- to 8-wk-old cultures were used for all experiments.
Viruses and Abs. Human influenza A/Bayern/7/95-like virus (H1N1), A/Sydney/5/97-like virus (H3N2), and B/Sichuan/379/99-like virus were isolated from clinical material and passaged in Madin-Darby canine kidney (MDCK) cells. Sheep antisera against human viruses were provided by Alexander Klimov at the Centers for Disease Control and Prevention, Atlanta. Human clinical isolates A/Memphis/14/96-M (H1N1), A/Memphis/5/98 (H3N2), and B/Memphis/25/99 in MDCK cells, avian influenza viruses, and rabbit antisera against avian viruses were kindly provided by Robert Webster of St. Jude Children's Research Hospital, Memphis, TN.
Virus Infection. The apical surfaces of the cultures were washed 10 times to remove accumulated mucins before inoculation with 0.2 ml of virus dilutions in the complete growth medium. The inoculum was removed after 1 h of incubation at 35°C; the cultures were incubated either for 6 h or for 23 h at the air-liquid interface and fixed with 4% paraformaldehyde for 1 h at 20°C. No trypsin was added to the cultures because previous studies in similar cultures demonstrated efficient proteolytic activation of influenza viruses by endogenous proteases (25, 26).
Double Immunostaining and Light Microscopy. Fixed cultures were permeabilized with 0.2% Triton X-100 and immunostained by sequential incubations with reagents from the apical side. Cilia were stained by using the β-tubulin-specific mAb (Sigma), horseradish peroxidase (HRP)-labeled secondary Abs, and Vector SG substrate (Vector Laboratories). Virus-infected cells were stained by using antiviral Abs, followed by corresponding HRP-labeled secondary Abs (Dianova, Hamburg, Germany) and aminoethylcarbazole substrate (Sigma). Some cultures were additionally stained by Alcian blue (pH 3) and periodic acid-Schiff reagent (Sigma) to identify secretory cells. Immunostained cultures were mounted by using Crystal Mount (Biomeda, Foster City, CA) and photographed en face by using a Nikon Optiphot-2 microscope equipped with a charge-coupled device camera. For cell counting, the cultures were observed en face with oil-immersion at ×1,000 magnification. In microscopic fields containing between 5% and 25% ciliated cells with respect to the total amount of superficial cells, each infected cell was classified as ciliated (based on the presence of cilia), nonciliated, or undefined. Percentages of ciliated and nonciliated infected cells with respect to the total amount of infected cells were calculated. For each sample, 25-35 fields were analyzed and the results were averaged.
Fluorescent Confocal Microscopy. Infection and immunostaining were performed as described above, using FITC-labeled and Texas red-labeled secondary Abs (Dianova) to detect cilia and the virus antigen, respectively. The cultures were mounted and photographed by using an LSM 510 laser scanning microscope (Zeiss).
Receptor-Binding Activity. Monospecific HRP-labeled fetuin containing either 2-6-linked or 2-3-linked sialic acid was prepared, and its binding to the viruses was determined by using a solid-phase binding assay as described (27).
Detection of 2-6- and 2-3-Linked Sialic Acids. HTBE cultures were washed 10 times with growth medium to remove overlying mucus and incubated for 1 h on ice with digoxigenin-labeled lectins Sambucus nigra agglutinin (SNA; 4 μg/ml) or Maackia amurensis agglutinin (MAA; 20 μg/ml) (Roche Molecular Biochemicals) in Tris-buffered saline (pH 7.2) containing 1% BSA and 1 mM Mg2+, Ca2+, and Mn2+. After washing and fixation with 4% paraformaldehyde, the cultures were incubated for 1 h at 20°C with HRP-labeled anti-digoxigenin Abs (Roche Molecular Biochemicals) in 1% BSA followed by incubation with aminoethylcarbazole substrate. The cells were permeabilized, and cilia were immunostained as described above. Control cultures were treated with lectin solutions that had been preincubated for 1 h with 10 mg/ml bovine fetuin. They showed no significant staining.
Results
Differential Cellular Tropism of Human and Avian Influenza Viruses. To identify cell types in human airway epithelium most susceptible to influenza viruses, we infected HTBE cultures from the apical side with either the human virus A/Memphis/14/96-M (H1N1) (Mem96-M) or avian virus A/Mallard/Alberta/119/98 (H1N1) (DkH1) and fixed the cells 6-8 h after infection after a single cycle of virus replication. The cultures were then double-immunostained for ciliated cells and for the virus antigen. The patterns of infection with these two viruses were strikingly different. The human virus infected mainly nonciliated cells (typically <5% of infected cells were ciliated cells), whereas the avian virus infected predominantly ciliated cells (Fig. 1; for cell counts, see Fig. 6). These patterns were reproducible in multiple replicate experiments in which the multiplicity of infection (moi) varied from 0.02 to 1. The nonciliated cells infected by human influenza virus were typically periodic acid-Schiff-positive (Fig. 2), suggesting that they are of a secretory type.
Fig. 1.

Differential cellular tropism of human and avian influenza viruses in HTBE cultures. The cultures were infected with Mem96-M (a and c) or DkH1 (b and d) viruses at the moi of ≈1, fixed 7 h after infection, and double-immunostained for virus antigen and for cilia of ciliated cells as described in Materials and Methods.(a and b) Light microscopy [virus (red), cilia (gray)]. (c and d) Fluorescent confocal microscopy [virus (red), cilia (green)]. Virus antigen was not observed in the negative controls in which infected cultures were either immunostained with omission of the antiviral Abs or fixed 2 h after infection. (Bars = 10 μm.)
Fig. 6.

Receptor specificity and cell tropism of influenza viruses. (a) Association constants (Kass) of virus complexes with monospecific HRP-labeled fetuin, which carried either 2-3-linked or 2-6-linked sialic acids (filled and hatched bars, respectively). Higher Kass values indicate stronger binding. (b) HTBE cultures were infected, fixed 7 h after infection, and double-immunostained for light microscopy. Percentages of infected ciliated cells (filled bars) and infected nonciliated cells (hatched bars) with respect to the total amount of infected cells were determined as described in Materials and Methods. For Mem96-M and DkH1, the experiments were repeated three times with different lots of HTBE cultures. Mem96-E1 was tested in parallel with two other viruses in the first two experiments.
Fig. 2.

Human influenza virus targets secretory cells. The HTBE cultures were infected with human virus Mem96-M, fixed 7 h after infection, and immunostained for cilia (gray) and virus antigen (red). Secretory cells were identified by using Alcian blue (pH 3)-periodic acid-Schiff staining (purple). The cultures were counterstained with hematoxylin, embedded (Crystal Mount, Biomeda), and sectioned (5 μm). (Bar = 10 μm.)
To corroborate the observed differences between the avian and human viruses, we tested several other avian and human virus strains. A/Mallard/Alberta/279/98 (H3N8), A/Duck/Minnessota/1525/81 (H5N1), and A/Chicken/FPV/Rostock/34 (H7N1) preferentially infected ciliated cells, whereas an H1N1, two H3N2, and two type B isolates obtained from human clinical specimens infected mainly nonciliated cells (data not shown). These results confirmed that the tropism for a specific type of airway epithelial cells depends on the virus host species, rather than on virus type, subtype, or strain.
To test whether patterns of virus infections observed in HTBE cultures will be reproduced in analogous cultures derived from both a different human donor and a different region of airway epithelium, we used differentiated cultures of human nasal epithelial cells. The virus tropism in human nasal epithelial cell cultures resembled that in HTBE cultures, namely, Mem96-M and DkH1 viruses showed preferential infection of nonciliated and ciliated cells, respectively (Fig. 3).
Fig. 3.
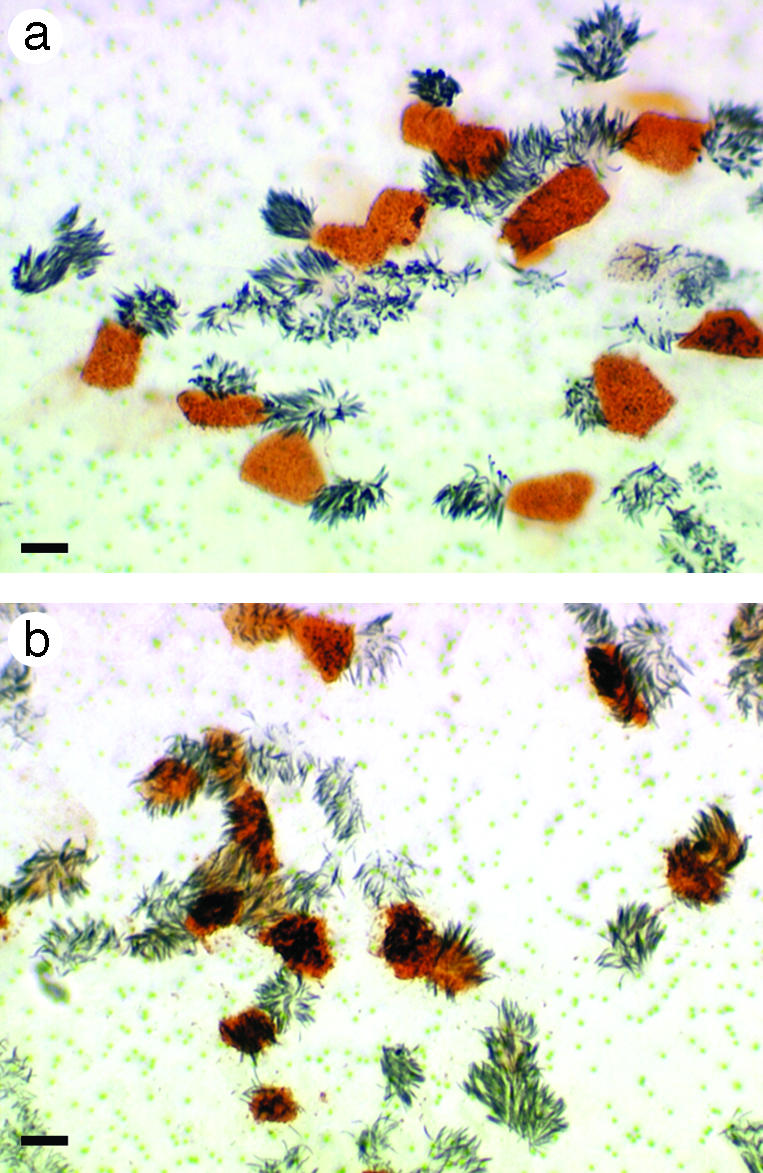
Cellular tropism of human and avian influenza viruses in differentiated cultures of nasal epithelial cells. The cultures were infected with either Mem96-M (a) or DkH1 (b) viruses, fixed 7 h after infection, and immunostained for cilia (gray) and virus antigen (red). (Bars = 10 μm.)
Virus Spread in HTBE Cultures. Infected ciliated cells are abundant in samples obtained from the airway mucosa of influenza patients (17, 18). To understand how ciliated cells become infected despite their apparent low sensitivity to human influenza viruses during the initial stages of infection, we studied the virus spread in HTBE cultures (Fig. 4). After infection (24 h), human virus produced continuous foci of infected cells, which included both nonciliated and ciliated cells, suggesting that the virus spreads from cell to cell and that it infects any encountered cell irrespective of its type. Thus, although ciliated cells are relatively refractory to infection with the human virus, this can be overcome by high local concentrations of the virus released from neighboring cells. Under the same conditions, the avian virus did not form continuous foci, with the viral antigen being found mainly in ciliated cells.
Fig. 4.

Spread of human and avian viruses in HTBE cultures in the course of multicycle infection. Cultures were infected with Mem96-M (a and c) or DkH1 (b and d) at a moi of ≈0.02, fixed 24 h after infection, and immunostained [virus (red), cilia (gray)]. [Objectives, ×10 (a and b) and ×40 (c and d). Bars = 25 μm.]
Distribution of 6- and 3-Linked Sialic Acids. To test for the presence of sialic acid-containing receptors for influenza viruses on the apical surface of airway epithelium, we used the lectins SNA and MAA, which bind 2-6- and 2-3-linked sialic acids, respectively (Fig. 5). Although both types of receptors were present, SNA stained cultures more efficiently than MAA (Fig. 5 a and b), suggesting that airway epithelial cells express more receptor moieties for human viruses than for avian viruses. Interestingly, SNA strongly stained nonciliated cells, with much weaker if any staining of neighboring ciliated cells (Fig. 5c). By contrast, MAA stained the surface of some (but not all) ciliated cells (Fig. 5d), so that the number of positively stained cells was much lower than in the case of SNA. Although we extensively washed the apical surfaces of cultures to remove mucous secretions before incubation with lectins, some of the staining could be due to lectin binding to cell surface-associated mucins rather than to integral glycoproteins or glycolipids. Still, the pattern of localization of 2-6- and 2-3-linked sialic acids in washed HTBE cultures (Fig. 5 c and d) clearly correlated with the sensitivity of cells to human and avian influenza viruses, respectively (Fig. 1), suggesting that differential cell tropism of these viruses is receptor-dependent.
Fig. 5.

Localization of 2-6- and 2-3-linked sialic acids on the surface of HTBE cultures. Cultures were incubated with digoxigenin-labeled lectins SNA (a and c) or MAA (b and d), followed by fixation and double-immunostaining for digoxigenin (red) and cilia (gray). (a and b) Scanned images of stained cultures (membrane diameter 12 mm). (c and d) Micrographs of mounted cultures. (Bars = 10 μm.)
Cellular Tropism Depends on the Virus Receptor-Binding Specificity. To examine whether differential cellular tropism of influenza viruses is mediated by the receptor specificity of the viral HA, we used a receptor binding variant of the original clinical isolate Mem96-M which was obtained by passaging the virus twice in embryonated chicken eggs followed by plaque purification in MDCK cells (27). The egg-adapted variant Mem96-E1 differed from Mem96-M by a single amino acid substitution in the receptor-binding site of the HA, Gln226Arg (H3 numbering). This mutation enabled the Mem96-E1 variant to bind to 2-3-linked sialic acid receptors (an effect that is typical of egg-adapted viruses) and abrogated its binding to 2-6-linked receptors (Fig. 6a). Mem96-E1 was reproducibly less infectious for HTBE cultures than either Mem96-M or DkH1. Namely, 20- to 100-fold higher mois were required in the case of Mem96-E1 than in the case of the other two viruses to infect comparable numbers of cells in HTBE cultures (data not shown). This effect was consistent with the weaker binding of Mem96-E1 to both 2-3- and 2-6-linked sialic acids as compared with the avian and human viruses, respectively. Importantly, the pattern of relative infectivity of Mem96-E1 for ciliated and nonciliated cells more closely resembled that of the duck virus than the relative infectivity of the human virus (Fig. 6b). This finding was consistent with the avian virus-like binding preference of Mem96-E1 for 2-3-linked sialic acids (Fig. 6a), thus confirming that viral cellular tropism in airway epithelium depends on the virus receptor specificity.
Discussion
In recent years, techniques have been developed for the preparation of differentiated airway epithelial cells in a configuration resembling airway epithelium in vivo (e.g., see refs. 22-26 and references therein). Here we used this experimental system to specify cellular targets for human and avian influenza viruses in human airway epithelium.
Previous studies on sections of formalin-fixed, paraffin-embedded samples of human trachea revealed abundant expression of 2-6-linked sialic acids on the apical surface of the tracheal epithelium and predominant expression of 2-3-linked sialic acids on the intracellular mucin granules (20, 21). In accordance with this pattern, a human influenza virus bound efficiently to the apical epithelial surface, whereas its variant with the avian-virus-like receptor specificity similar to that of our virus Mem96-E1 showed no detectable binding (21). These results raised the hypothesis that avian influenza viruses fail to replicate efficiently in humans because of a general deficiency of cell surface receptors for such viruses in human airway epithelium (14, 21).
Our data agree with the previous notion on the overall predominance of 2-6-linked sialic acids on the surface of human tracheal epithelium. However, we found that 2-6-linked receptors are mainly expressed on nonciliated cells and that ciliated cells, a substantial cellular subset of the respiratory epithelium, express 2-3-linked sialic acid receptors in sufficient density to allow entry and replication of avian viruses. Ciliated cells therefore most likely serve as primary target cells in those rare cases where avian viruses cause human disease, with sometimes even fatal outcome (10, 11, 28). Nevertheless, our finding that epidemic human influenza A and B viruses specifically target nonciliated cells clearly indicates that ciliated cells are suboptimal for influenza virus replication and/or transmission in humans. This could explain why avian influenza viruses usually fail to cause disease in humans and why human-to-human transmission of avian H5N1 and H7N7 viruses was inefficient (13, 28). Furthermore, our data suggest that a shift in the receptor specificity of the avian virus HA, an apparent prerequisite for the generation of human pandemic viruses (8), may be required primarily for the virus to adapt to nonciliated cells and, thus, to optimize its cellular tropism in humans. The question remains why targeting of nonciliated cells as a primary entry site is an advantage for the human influenza virus. In HTBE cultures, the human virus appeared to target a secretory subset of nonciliated cells. Such cells would likely be characterized by a higher rate of protein synthesis and, potentially, by a higher virus yield than ciliated cells.
Our data also add a dimension to the long-standing question concerning the necessity of an intermediate animal host for the generation of pandemic viruses (1-5, 14). Because late in infection ciliated cells from human airways are permissive to both avian and human influenza viruses, these cells themselves can provide a milieu for gene reassortment between the viruses, leading to a gene constellation optimal for replication and spread in humans. In this case, the receptor specificity of the viral HA may be the only barrier separating humans from a new pandemic virus. Alternatively, the avian virus can first change its receptor specificity during limited replication in human airways followed by reassortment with human virus in nonciliated cells.
The interplay between influenza viruses and their target cells involves a variety of mechanisms which, on the one hand, trigger defensive cellular responses and, on the other hand, are necessary to support virus replication and to counteract cellular defense (29-31). Most studies in this field have been performed by using undifferentiated cell lines. Whereas some mechanisms may be universal, others can vary in a species-, tissue-, and cell-specific manner. Analysis of these interactions in distinct subsets of differentiated airway epithelial cells will be critically important to better understand the virus replication, transmission, and pathogenicity in humans.
Acknowledgments
We thank Robert Webster and Alexander Klimov for providing viruses and immunoreagents, and Larissa Kolesnikova and Andrea Meissner for advice on microscopic analysis. This work was supported by Roche Products, Ltd.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HA, hemagglutinin; HRP, horseradish peroxidase; SNA, Sambucus nigra agglutinin; MAA, Maackia amurensis agglutinin; HTBE, differentiated human tracheobronchial epithelial cells; Mem96-M, A/Memphis/14/96-M (H1N1); Mem96-E1, A/Memphis/14/96-E1 (H1N1); DkH1, A/Mallard/Alberta/119/98 (H1N1); moi, multiplicity of infection.
References
- 1.Webster, R. G., Bean, W. J., Gorman, O. T., Chambers, T. M. & Kawaoka, Y. (1992) Microbiol. Rev. 56, 152-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander, D. J. & Brown, I. H. (2000) Rev. Sci. Tech. 19, 197-225. [DOI] [PubMed] [Google Scholar]
- 3.Cox, N. J. & Subbarao, K. (2000) Annu. Rev. Med. 51, 407-421. [DOI] [PubMed] [Google Scholar]
- 4.De Jong, J. C., Rimmelzwaan, G. F., Fouchier, R. A. & Osterhaus, A. D. (2000) J. Infect. 40, 218-228. [DOI] [PubMed] [Google Scholar]
- 5.Horimoto, T. & Kawaoka, Y. (2001) Clin. Microbiol. Rev. 14, 129-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paulson, J. C. (1985) in The Receptors, ed. Conn, M. (Academic, Orlando, FL), Vol. 2, pp. 131-219. [Google Scholar]
- 7.Connor, R. J., Kawaoka, Y., Webster, R. G. & Paulson, J. C. (1994) Virology 205, 17-23. [DOI] [PubMed] [Google Scholar]
- 8.Matrosovich, M., Tuzikov, A., Bovin, N., Gambaryan, A., Klimov, A., Castrucci, M. R., Donatelli, I. & Kawaoka, Y. (2000) J. Virol. 74, 8502-8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid, A. H., Fanning, T. G., Hultin, J. V. & Taubenberger, J. K. (1999) Proc. Natl. Acad. Sci. USA 96, 1651-1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Jong, J. C., Claas, E. C., Osterhaus, A. D., Webster, R. G. & Lim, W. L. (1997) Nature 389, 554 (lett.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webster, R. G. & Hay, A. J. (1998) in Textbook of Influenza, eds. Nicholson, K. G., Webster, R. G. & Hay, A. J. (Blackwell, London), pp. 561-565.
- 12.Matrosovich, M., Zhou, N., Kawaoka, Y. & Webster, R. (1999) J. Virol. 73, 1146-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mounts, A. W., Kwong, H., Izurieta, H. S., Ho, Y., Au, T., Lee, M., Buxton, B. C., Williams, S. W., Mak, K. H., Katz, J. M., et al. (1999) J. Infect. Dis. 180, 505-508. [DOI] [PubMed] [Google Scholar]
- 14.Ito, T., Couceiro, J. N. S. S., Kelm, S., Baum, L. G., Krauss, S., Castrucci, M. R., Donatelli, I., Kida, H., Paulson, J. C., Webster, R. G. & Kawaoka, Y. (1998) J. Virol. 72, 7367-7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hers, J. F. (1966) Am. Rev. Respir. Dis. 93, Suppl., 162-177. [DOI] [PubMed] [Google Scholar]
- 16.Jeffery, P. K. & Li, D. (1997) Eur. Respir. J. 10, 1655-1662. [DOI] [PubMed] [Google Scholar]
- 17.Tateno, I., Kitamoto, O. & Kawamura, A., Jr. (1966) N. Engl. J. Med. 274, 237-242. [DOI] [PubMed] [Google Scholar]
- 18.Ebisawa, I. T., Kitamoto, O., Takeuchi, Y. & Makino, M. (1969) Am. Rev. Respir. Dis. 99, 507-515. [DOI] [PubMed] [Google Scholar]
- 19.Rimmelzwaan, G. F., Kuiken, T., van Amerongen, G., Bestebroer, T. M., Fouchier, R. A. & Osterhaus, A. D. (2001) J. Virol. 75, 6687-6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baum, L. G. & Paulson, J. C. (1990) Acta Histochem. Suppl. 40, 35-38. [PubMed] [Google Scholar]
- 21.Couceiro, J. N., Paulson, J. C. & Baum, L. G. (1993) Virus Res. 29, 155-165. [DOI] [PubMed] [Google Scholar]
- 22.Gray, T. E., Guzman, K., Davis, C. W., Abdullah, L. H. & Nettesheim, P. (1996) Am. J. Respir. Cell Mol. Biol. 14, 104-112. [DOI] [PubMed] [Google Scholar]
- 23.Nettesheim, P., Koo, J. S. & Gray, T. (2000) J. Aerosol Med. 13, 207-218. [DOI] [PubMed] [Google Scholar]
- 24.Thornton, D. J., Gray, T., Nettesheim, P., Howard, M., Koo, J. S. & Sheehan, J. K. (2000) Am. J. Physiol. 278, L1118-L1128. [DOI] [PubMed] [Google Scholar]
- 25.Endo, Y., Carroll, K. N., Ikizler, M. R. & Wright, P. F. (1996) J. Virol. 70, 2055-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slepushkin, V. A., Staber, P. D., Wang, G., McCray, P. B. & Davidson, B. L. (2001) Mol. Ther. 3, 395-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matrosovich, M., Matrosovich, T., Carr, J., Roberts, N. A. & Klenk, H. D. (2003) J. Virol. 77, 8418-8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Centers for Disease Control and Prevention (2003) Morbid. Mortal. Wkly. Rep. 52, 516-521. [Google Scholar]
- 29.Krug, R. M., Yuan, W., Noah, D. L. & Latham, A. G. (2003) Virology 309, 181-189. [DOI] [PubMed] [Google Scholar]
- 30.Geiss, G. K., An, M. C., Bumgarner, R. E., Hammersmark, E., Cunningham, D. & Katze, M. G. (2001) J. Virol. 75, 4321-4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ludwig, S., Pleschka, S. & Wolff, T. (1999) Viral Immunol. 12, 175-196. [DOI] [PubMed] [Google Scholar]