

Abstract
The development and validation of a LC-MS/MS method is often performed using pooled human plasma, which may fail to account for variations in interindividual matrices. Since calibrator standards and quality control samples are routinely prepared in pooled human plasma, variations in the extraction recovery and/or matrix effect between pooled plasma and individual patient plasma can cause erroneous measurements. Using both pooled human plasma as well as individual healthy donor and cancer patient plasma samples, we evaluated the analytical performance of two classes of internal standards (i.e., non-isotope-labeled and isotope-labeled) in the quantitative LC-MS/MS analysis of lapatinib. After exhaustive extraction with organic solvent, the recovery of lapatinib, a highly plasma protein-bound drug, varied up to 2.4-fold (range, 29 – 70%) in 6 different donors of plasma and varied up to 3.5-fold (range, 16 – 56%) in the pretreatment plasma samples from 6 cancer patients. No apparent matrix effects were observed for lapatinib in both pooled and individual donor or patient plasma samples. The calibration curve range was 5 – 5000 ng/ml of lapatinib in plasma. Both the non-isotope-labeled (zileuton) and isotope-labeled (lapatinib-d3) internal standard methods showed acceptable specificity, accuracy (within 100 ± 10%), and precision (< 11%) in the determination of lapatinib in pooled human plasma. Nevertheless, only the isotope-labeled internal standard could correct for the interindividual variability in the recovery of lapatinib from patient plasma samples. As inter- and intra-patient matrix variability is commonly presented in the clinical setting, this study provides an example underscoring the importance of using a stable isotope-labeled internal standard in quantitative LC-MS/MS analysis for therapeutic drug monitoring or pharmacokinetic evaluation.
Keywords: Lapatinib, High performance liquid chromatography (HPLC), Mass spectrometry, LC-MS/MS, Recovery, Internal standard
1. Introduction
Lapatinib (Tykerb®, GlaxoSmithKline) is a small molecule, orally active dual tyrosine kinase inhibitor that targets both Epidermal Growth Factor Receptor (EGFR or ErbB1) and Human Epidermal Growth Factor Receptor Type 2 (HER2 or ErbB2). Lapatinib inhibits ErbB-driven tumor cell growth in vitro and in various animal models. The combination of lapatinib with capecitabine has been approved by the US Food and Drug Administration (FDA) for the treatment of patients with advanced or metastatic HER2-positive breast cancer who have progressed following therapy with taxanes, anthracyclines, and trastuzumab. The combination of lapatinib with letrozole is FDA-approved for treating postmenopausal women with HER2-positive and estrogen receptor-positive metastatic breast cancer [1].
In addition, lapatinib has been investigated in combination with other cytotoxic or molecularly targeted agents for treating patients with breast cancers. For example, lapatinib is currently being evaluated in combination with MK-2206, a selective allosteric inhibitor of Akt, in patients with HER2-positive advanced breast cancer in a multi-center phase I clinical trial (NCI study #8747 ), with the Karmanos Cancer Institute as the lead institution. Given the fact that multiple metabolizing enzymes and transporters involved in its absorption and disposition, lapatinib exhibits large interindividual pharmacokinetic variability and has the potential for drug interactions with concomitant drugs. Therapeutic drug monitoring may be useful in improving lapatinib therapy. To provide efficient lapatinib therapeutic drug monitoring or support clinical pharmacokinetic studies, a sensitive, specific, reliable, and reproducible bioanalytical method for the quantitation of laptinib in patient plasma samples is required.
Several liquid chromatography with tandem mass spectrometry (LC-MS/MS) methods have been reported for the determination of lapatinib in biological matrices [2–9]. For quantification purpose, internal standardization using either a stable isotope-labeled or non-isotope-labeled internal standard was used in all reported lapatinib assays. Currently, there is no consensus on preferred internal standards in the quantitative LC-MS/MS analysis of lapatinib to support clinical pharmacokinetic studies or therapeutic drug monitoring. Several reported LC-MS/MS methods using non-isotope-labeled internal standards have been demonstrated to have acceptable accuracy and precision for the determination of lapatinib in human plasma [2, 5, 6]. However, these methods were developed and validated in pooled human plasma, which may fail to account for the interindividual variability in the recovery and/or matrix effect of the analyte in patient plasma samples. Since the calibrator standards and quality control samples are routinely prepared in pooled human plasma, variations in recovery and/or matrix effect between pooled plasma and individual patient plasma have the potential to cause erroneous concentration measurements for patient plasma samples.
Matrix effects from patient plasma samples have been well documented to cause differences in the ionization of the analyte of interest in electrospray ionization tandem mass spectrometry analysis. Nevertheless, interindividual variability in the recovery of an analyte from patient plasma samples is often overlooked. In the present study, we demonstrated that the recovery of lapatinib, a highly plasma protein-bound drug, was largely variable from different sources of plasma (i.e., different batches of pooled healthy donor plasma, different individual healthy donor plasma, and different individual cancer patient plasma). By evaluating the analytical performance of two classes of internal standards (i.e., non-isotope-labeled and isotope-labeled) in both pooled human plasma as well as individual healthy donor and cancer patient plasma samples, we demonstrated that although both the non-isotope-labeled and isotope-labeled internal standard methods showed acceptable specificity, accuracy, and precision in the determination of lapatinib in pooled human plasma, only the isotope-labeled internal standard could correct for the interindividual variability in the recovery of lapatinib from patient plasma samples.
2. Experimental
2.1. Chemicals and reagents
The reference standards of lapatinib and deuterated lapatinib (lapatinib-d3) were purchased from Toronto Research Chemicals (Ontario, CA). Zileuton [N-(1-benzobthien-2-ylethyl)-N-hydroxyurea)] was obtained from Rhodia Pharma Solutions Ltd (Northumberland, UK). All other chemicals and reagents were HPLC grade. Water was filtered and deionized with a US Filter PureLab Plus UV/UF system (Siemens, Detroit, MI) and used throughout in all aqueous solutions. Blank pooled human plasma and six different donors of plasma were obtained from Innovative Research Inc. (Novi, MI).
2.2. Stock solutions, calibration standards, and quality control samples
Stock solution of lapatinib, lapatinib-d3, and zileuton were prepared by dissolving an accurately weighed amount of the compound in methanol to obtain a final concentration of 1 mg/ml, and stored in brown glass vials at − 20°C. Working solutions were prepared freshly on each day of analysis as serial dilutions in methanol. The calibration curves were constructed by spiking lapatinib in blank pooled human plasma at concentrations of 5, 10, 50, 200, 500, 1000, 2000 and 5000 ng/ml. The quality control (QC) samples were prepared in blank pooled human plasma at concentrations of 5 (LLOQ), 15, 800, and 4000 ng/ml. All calibration standards and QC samples were prepared freshly. For long-term and freeze-thaw stability, QC samples were prepared as a batch and stored at −80°C.
2.3. Plasma sample preparation
Lapatinib is a strong non-polar hydrophobic compound with a log P > 4.6. Previously studies have shown that acidification of plasma sample followed by organic solvent extraction resulted in a favorable recovery of lapatinib [3]. To optimize the liquid-liquid extraction method in our study, the process efficiency of lapatinib was examined under different extraction conditions. In brief, the plasma samples were acidified with 90% of formic acid or 1 M of ammonium formate (pH 3) followed by extraction with different extraction solvents including t-butyl methyl ether (tBME), ethyl acetate, or the mixture of tBME and acetonitrile (3:1, v/v). It was found that acidification of the sample with 90% of formic acid followed by extraction with ethyl acetate yielded the best process efficiency from pooled human plasma (mean, 63%) (Fig.1). Hence, the optimized extraction procedure was: 250 µl of plasma sample was spiked with 5 µl of internal standard working solution, and acidified with 20 µl of concentrated formic acid (90%) followed by extraction with 1 ml of ethyl acetate. The mixture was vortex-mixed for 1 minute, and centrifuged at 14,000 rpm for 5 minutes at 4 °C. The top layer was transferred to a 1.5-ml polypropylene screw top tube, and evaporated to dryness under a stream of nitrogen in a water bath at 50 ± 2 °C. The residue were reconstituted in 100 µl of mobile phase, vortex-mixed for 30 seconds, and centrifuged at 14,000 rpm for 5 minutes at 4 °C. The supernatant was transferred to an autosampler vial, and 10 µL was injected into the LC-MS/MS system using a temperature-controlled autosampling device (set at 4 °C).
Fig. 1.
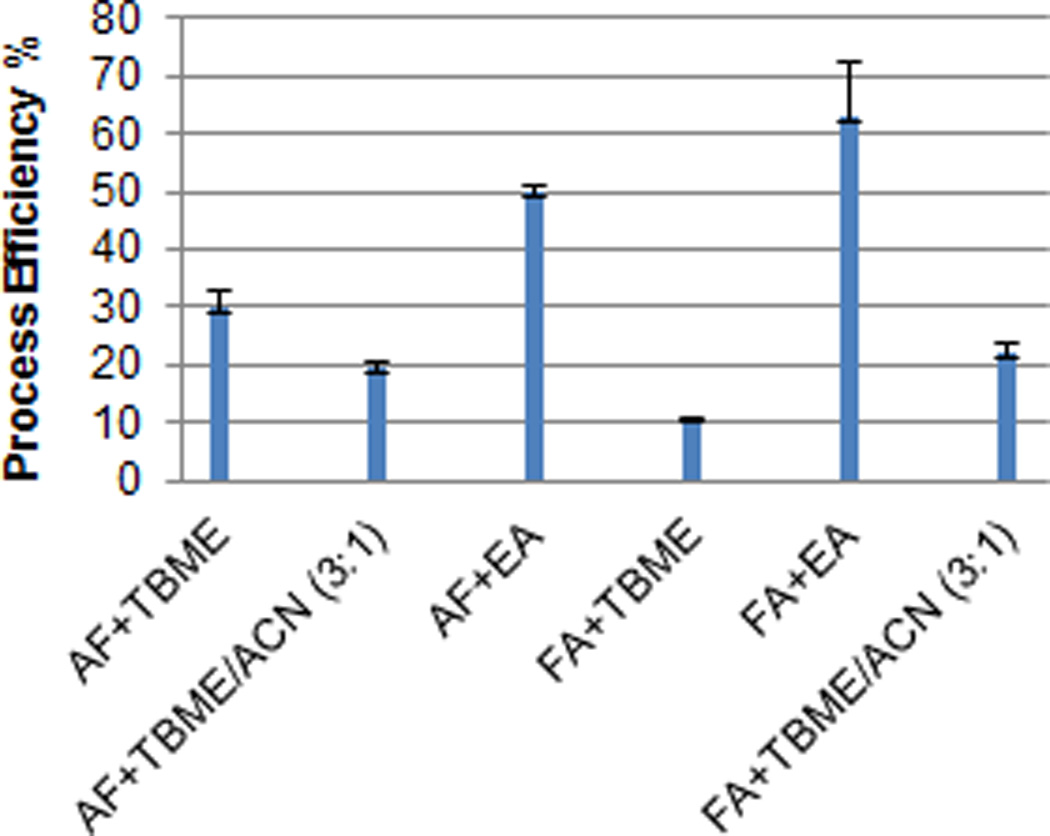
Process efficiency of lapatinib under different extraction conditions. The plasma samples spiked with 800 ng/ml of lapatinib were acidified with 90% of formic acid (FA) or 1 M of ammonium formate (AF) (pH 3) followed by extraction with t-butyl methyl ether (tBME), ethyl acetate, or the mixture of tBME and acetonitrile (3:1, v/v). Process efficiency is estimated as the ratio of the peak area of lapatinib spiked in plasma prior to extraction to the peak area of the same amount of the analyte spiked in the mobile phase. Data are expressed as the mean ± standard deviation of triplicate measurements.
2.4. Chromatographic and mass-spectrometric conditions
Chromatographic analysis was performed on a Waters Alliance 2695 system (Milford, MA, USA). Chromatographic separation was achieved at 30°C on a Waters XTerra C8 columm (3.5 µm, 50 × 2.1 mm i.d.) when zileuton was used as the internal standard or on a Waters XBridge C18 column (3.5 µm, 50 × 2.1 mm i.d.) when lapatinib-d3 was used as the internal standard. The mobile phase consisted of methanol and 0.45% formic acid in water (50:50, v/v), running isocratically at a flow rate of 0.2 ml/min. Mass spectrometric detection was performed on a Waters Quattro Micromass triple quadrupole mass spectrometer equipped with an electrospray ionization source. The instrument was controlled and data were acquired and analyzed by Masslynx 4.1 software. The analyte and internal standard were detected using multiple reaction monitoring (MRM) mode under positive ionization mode. The optimized mass spectrometric conditions and MRM transitions for lapatinib and the internal standard (zileuton or lapatinib-d3) are presented in Table 1.
Table 1.
Optimal mass spectrometric settings and multiple reaction monitoring (MRM) transitions for lapatinib, lapatinib-d3, and zileuton
| Lapatinib | Lapatinib-d3 | Zileuton | |
|---|---|---|---|
| Ionization mode | Positive | Positive | Positive |
| Heated nebulized probe | 350°C | 350°C | 350°C |
| Pressure of collision gas (Argon) (mBar) | 5.92e−003 | 5.92e−003 | 5.92e−003 |
| Dwell time (second) | 0.5 | 0.5 | 0.5 |
| Cone voltage (V) | 60 | 60 | 22 |
| Collision energy (eV) | 34 | 34 | 12 |
| Transition | 581.40 > 364.96 | 584.10 > 366.00 | 237.43 > 161.09 |
2.5. Method validation
The LC-MS/MS method with zileuton or lapatinib-d3 as the internal standard was validated according to the Food & Drug Administration (FDA) Guidance on Bioanalytical Method Validation (http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf).
2.5.1. Specificity and selectivity
The presence of endogenous interfering peaks was inspected by comparing the chromatograms of the extracted blank human plasma samples from 6 different donors and those spiked with lapatinib at the LLOQ (5ng/ml). The interfering peak area should be less than 10% of the peak area for the analyte at the LLOQ.
2.5.2. Calibration curve, accuracy, and precision
Linearity was assessed at lapatinib concentrations ranging from 5 to 5000 ng/ml in pooled human plasma. Calibration curves were built by fitting the analyte concentrations versus the peak area ratios of the analyte to internal standard using least-squares non-linear regression analysis with a weighting scheme of 1/x2. The goodness-of-fit was assessed based on correlation coefficient (R2), deviations of the back-calculated concentrations, and residual plots.
The intra-day and inter-day accuracy and precision were assessed for the calibrator standards (each in duplicate) and QC samples at the concentrations of 5 ng/ml (LLOQ), 15, 800, and 4000 ng/ml in pooled human plasma (each in quintuplicate) on three consecutive days. The accuracy was assessed as the percentage of the mean determined concentration relative to the nominal concentration. The intra- and inter-day precisions were estimated by one-way analysis of variance (ANOVA) using the JMP™ statistical discovery software version 5 (SAS Institute, Cary, NC), as described previously [10].
2.5.3. Stability
The short-term (bench-top) stability of the lapatinib in methanol (working solution) at the concentration of 1 and 100 µg/ml as well as in plasma at the concentration of 15 and 4000 ng/ml were tested at ambient temperature (25 °C) for 6 hour. The autosampler stability of lapatinib in the reconstitution solution (methanol/0. 45% formic acid in water, 50:50, v/v) was examined at 4 °C for 12 hours for the low and high QC plasma samples. The freeze-thaw stability was assessed lapatinib at the concentrations of 15 and 4000 ng/ml in plasma through three freeze-thaw cycles. The long-term stability of lapatinib in methanol stock solution (at 1 mg/ml) and in plasma (at 15 and 4000 ng/ml) was investigated up to 6 months so far. All QCs were run in triplicate.
2.5.4. Matrix effect and recovery
Matrix effect and recovery of lapatinib were assessed in 6 different donors of plasma and in the pre-treatment plasma samples from 6 cancer patients, using the method with zileuton or lapatinib-d3 as the internal standard. Briefly, three sets of QC samples were prepared, triplicate for each sample. Set 1 QC samples were prepared by spiking lapatinib (at 15, 800, and 4000 ng/ml) and zileuton (at 50 ng/ml) or lapatinib-d3 (at 1000 ng/ml) in plasma prior to extraction. Set 2 QC samples was prepared by spiking the same amount of lapatinib and the internal standard as Set 1 in post-extraction reconstitution solution of blank plasma. Set 3 QCs was prepared by spiking the same amount of lapatinib as Set 1 in the mobile phase. The matrix effect is expressed as the ratio of the mean peak area of an analyte spiked post-extraction (set 2) to that from neat solution (set 3). The recovery is calculated as the ratio of the mean peak area of an analyte spiked prior to extraction (set 1) to that from post-extraction solution (set 2).
2.6. Process efficiency of lapatinib from samples containing varying concentrations of plasma proteins
Process efficiency is estimated as the ratio of the peak area of an analyte spiked prior to extraction (set 1) to the peak area of the same amount of the analyte spiked in the mobile phase (set 3). Of note, process efficiency is the product of recovery and matrix factor, and hence process efficiency represents recovery when there is no apparent matrix effect (i.e., matrix factor = 1). The process efficiency of lapatinib, d3-lapatinib, and zileuton was assessed simultaneously in the samples containing varying concentrations of plasma proteins, including pooled human plasma, plasma diluted with phosphate buffer solution (PBS) at a ratio of 1:1 (50% of PBS in plasma), plasma supplemented with clinically relevant concentrations of HSA (10 or 40 mg/ml), and plasma supplemented with clinical relevant concentrations of AAG (0.5 or 3 mg/ml), as well as isolated protein solutions at clinically relevant concentrations of HSA (10, 40, or 80 mg/ml) or AAG (0.5, 1, or 3 mg/ml). Lapatinib was spiked in the above samples at a final concentration of 800 ng/ml, and each sample was made into two aliquots: one aliquot was immediately extracted with the procedure as described in section 2.3, and the other was incubated at 37°C in a shaking water bath for 4 hour and kept at −80°C overnight prior to the extraction. These two treatments determined whether the process efficiency of lapatinib in the freshly-processed samples was different from that in the incubated-frozen samples, as the calibrator standards and QC samples are routinely prepared freshly followed by immediately extraction while the patient plasma samples are usually stored at −80°C until the extraction,
2.7. Clinical application
Patient plasma samples were obtained from the patients with HER2 positive metastatic breast cancer who were treated with oral lapatinib daily alone or in combination with oral MK-2206 (an AKT inhibitor) weekly in an ongoing phase I clinical trial (NCI study #8747). The protocol was approved by the Wayne State University Institutional Review Board. All the patients provided written informed consent. To assess the potential influence of coadministration of MK-2206 on the pharmacokinetics of lapatinib, intensive pharmacokinetic sampling of lapatinib was obtained on day 1 when lapatinib was given alone and on day 29 when lapatinib was given concomitantly with MK-2206. On both days, blood samples (4 ml each) were collected at pre-dosing, 0.5, 1, 2, 3, 4, 6, 8, and 24 hours after oral administration of lapatinib (1000 or 1250 mg). The blood samples were centrifuged at 4 °C, at 3000 rpm for 10 min, and plasma samples were collected and stored at −80 °C until LC-MS/MS analysis. The plasma concentrations of lapatinib were determined using the validated LC-MS/MS method with lapatinib-d3 as the internal standard.
3. Results and discussion
3.1. Analytical performance of the LC-MS/MS method with zileuton as the internal standard
Since stable isotope-labeled internal standard for lapatinib was not available initially, zileuton was chosen as an alternative internal standard because this compound has been successfully used as the internal standard for several LC-MS/MS methods that were developed and validated in our lab [10, 11]. Our previous studies have shown that 1) zileuton is stable in aqueous and organic solvents; 2) it exhibits excellent and reproducible mass spectrometric response under the positive ionization mode; and 3) it has a good and consistent recovery from human plasma samples with ethyl acetate extraction. Hence, a LC-MS/MS method with zileuton as the internal standard was initially developed and validated for the determination of lapatinib in pooled human plasma.
The LLOQ of lapatinib in plasma for this method was 5 ng/ml, at which the signal-to-noise ratio was > 100 in all analytical runs. Selectivity test indicated no apparent interference for lapatinib and zileuton in the blank plasma samples from 6 different donors. Fig. 2 shows the representative chromatograms of a blank pooled human plasma sample, a blank plasma spiked with 5 ng/ml of lapatinib (LLOQ) and zileuton (50 ng/ml), and a patient plasma sample (which was collected at 2 hours after oral administration of lapatinib 1000 mg) spiked with zileuton (50 ng/ml).
Fig. 2.
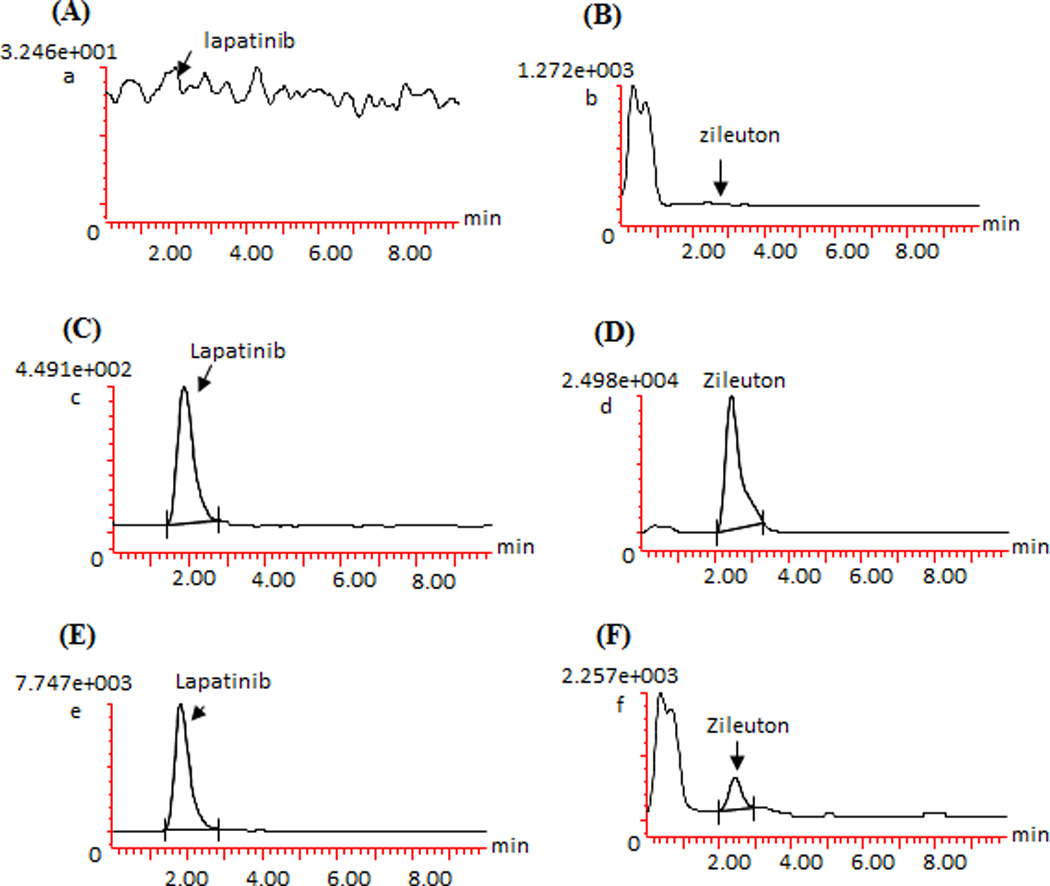
Representative chromatograms of a blank pooled human plasma (A and B), blank plasma spiked with 5 ng/ml of lapatinib (LLOQ) and zileuton (50 ng/ml) (C and D), and a patient plasma sample (which was collected at 2 hour after oral administration of lapatinib 1000 mg) spiked with zileuton (50 ng/ml) (E and F), monitored at m/z 581.40 > 364.96 for lapatinib, and m/z 237.43 > 161.09 for zileuton.
The linear calibration curves were established over the lapatinib concentration range of 5 – 5000 ng/ml in pooled human plasma. A linear correlation coefficient (R2) of > 0.99 was obtained in all analytical runs during the method validation. The inter- and intra-day accuracy and precision for all calibrator standards as well as LLOQ, low, medium, and high QC samples, were within the generally accepted validation criteria for bioanalytical method (Table 2).
Table 2.
Accuracy and intra-/inter-day precision of the lapatinib calibrator standards and quality control (QC) samples, as determined by the method with zileuton or lapatinib-d3 as the internal standard (IS).
| Nominal concentration (ng/ml) |
Zileuton as IS | Lapatinib-d3 as IS | ||||
|---|---|---|---|---|---|---|
| Average Accuracy (%) |
Intra- day (%) |
Inter-day (%) |
Average Accuracy (%) |
Intra-day (%) |
Inter-day (%) |
|
| Calibrators a | ||||||
| 5 (LLOQ) | 102.7 | 8.9 | -c | 102.7 | 10.7 | -c |
| 10 | 95.0 | 7.1 | 3.5 | 94.3 | 4.3 | 3.7 |
| 50 | 99.2 | 8.0 | 5.8 | 101.9 | 2.3 | 1.3 |
| 200 | 100.9 | 4.9 | 8.1 | 102.0 | 2.1 | 2.0 |
| 500 | 105.4 | 4.9 | 6.1 | 103.3 | 2.3 | -c |
| 1000 | 103.6 | 8.2 | -c | 100.2 | 1.9 | 4.2 |
| 2000 | 102.7 | 9.9 | -c | 100.7 | 2.5 | -c |
| 5000 | 95.3 | 8.8 | -c | 97.6 | 2.5 | 3.2 |
| QCs b | ||||||
| 5(LLOQ) | 107.1 | 2.7 | 6.3 | 102.1 | 4.6 | -c |
| 15 | 96.7 | 10.9 | 3.0 | 98.6 | 3.4 | 4.0 |
| 800 | 96.0 | 6.1 | 2.9 | 102.5 | 2.3 | 2.9 |
| 4000 | 96.7 | 7.7 | 4.9 | 98.3 | 2.4 | 3.1 |
Each calibrator standard was evaluated in duplicate on three different days.
Each QC was performed in quintuplicate on three different days.
No additional variation was observed as a result of performing assay in different days.
The matrix effect and recovery of lapatinib and zileuton were assessed in 6 different donors of plasma. No apparent matrix effects (i.e., matrix factor within 1 ± 0.15) were observed for either lapatinib or zileuton in individual donor plasma samples (Table 3). However, the recovery of lapatinib or zileuton was variable from different donors of plasma, and notably, the internal standard-normalized recovery varied up to 17-fold (range, 66 – 1136%) (Table 3), suggesting zileuton as the internal standard could not correct for the variation in lapatinib recovery but rather introduced additional variation.
Table 3.
Matrix effect and recovery of lapatinib determined in 6 different donors of plasma and in the pretreatment plasma samples from 6 cancer patients, using the method with zileuton or lapatinib-d3 as the internal standard (IS).
| Plasma samples from 6 different healthy donors |
Pretreatment plasma samples from 6 cancer patients |
|||
|---|---|---|---|---|
| Matrix factorb | Recovery (%)c | Matrix factorb | Recovery (%)c | |
| Zileuton as IS | ||||
| Lapatinib (ng/ml)a | ||||
| 15 | 0.97±0.03 | 85±22 | ND | ND |
| (0.92–1.00) | (58–108) | |||
| 800 | 1.08±0.04 | 79±22 | ND | ND |
| (1.04–1.12) | (41–101) | |||
| 4000 | 1.08±0.04 | 84±16 | ND | ND |
| (1.04–1.14) | (62–105) | |||
| Zileuton | 1.01±0.03 | 37±18 | ND | ND |
| (0.97–1.06) | (8.8–63) | |||
| IS-normalized | ||||
| 15 | 0.95±0.04 | 395±403 | ND | ND |
| (0.91–1.01) | (105–1136) | |||
| 800 | 1.09±0.04 | 346±286 | ND | ND |
| (1.03–1.14) | (66–797) | |||
| 4000 | 1.06±0.05 | 225±62 | ND | ND |
| (0.98–1.11) | (172–343) | |||
| Lapatinib-d3 as IS | ||||
| Lapatinib (ng/ml)a | ||||
| 15 | 1.12±0.07 | 43±14 | 0.85±0.02 | 30±8 |
| (1.06–1.21) | (33–70) | (0.82–0.87) | (21–39) | |
| 800 | 1.02±0.03 | 42±10 | 0.85±0.05 | 27±9 |
| (0.98–1.05) | (29–54) | (0.76–0.88) | (16–40) | |
| 4000 | 1.05±0.04 | 48±12 | 0.98±0.05 | 35±12 |
| (1.02–1.13) | (36–69) | (0.90–1.02) | (23–56) | |
| Lapatinib-d3 | 1.04±0.06 | 43±10 | 0.89±0.08 | 30±10 |
| (0.94–1.15) | (30–66) | (0.77–1.04) | (14–50) | |
| IS-normalized | ||||
| 15 | 1.03±0.06 | 108±4 | 1.02±0.03 | 91±5 |
| (0.93–1.08) | (101–113) | (0.96–1.04) | (84–95) | |
| 800 | 1.03±0.02 | 99±5 | 1.01±0.03 | 106±14 |
| (1.00–1.05) | (92–106) | (0.97–1.04) | (91–119) | |
| 4000 | 1.02±0.02 | 100±3 | 0.99±0.03 | 113±7 |
| (0.99–1.04) | (97–104) | (0.94–1.03) | (106–125) | |
The nominal concentrations of the analyte spiked in plasma before extraction (set 1). The same amounts of the analyte as in set 1 were spiked in plasma extract for set 2 and in the mobile phase for set 3.
Matrix factor is expressed as the ratio of the mean peak area of an analyte spiked postextraction (set 2) to the mean peak area of the same amount of analyte spiked in the mobile phase (set 3). Data are shown as the mean ± standard deviation (range) from six different sources of plasma.
Recovery is calculated as the ratio of the mean peak area of an analyte spiked before extraction (set 1) to the mean peak area of the same amount of the analyte spiked postextraction (set 2). Data are shown as the mean ± standard deviation (range) from six different source of plasma.
ND, not determined
Further evaluation of the method performance in two cancer patients’ plasma samples showed that the peak areas of zileuton in the cancer patient plasma samples were largely variable and significantly lower than those observed in the calibrator standards and QC samples which were prepared in pooled human plasma (Fig. 3). The reasons behind such a large variation in the recovery of zileuton between pooled human plasma and individual cancer patient plasma samples remain to be determined. Although the method with zileuton as the internal standard demonstrated acceptable specificity, accuracy, and precision for the determination of lapatinib in pooled human plasma, zileuton failed to correct for the interindividual variability in the recovery of the analyte from different sources of plasma. Recognizing this limitation, we further investigated whether the analytical performance of the method could be improved by replacing zileuton with a stable isotope-labeled internal standard, lapatinib-d3.
Fig. 3.
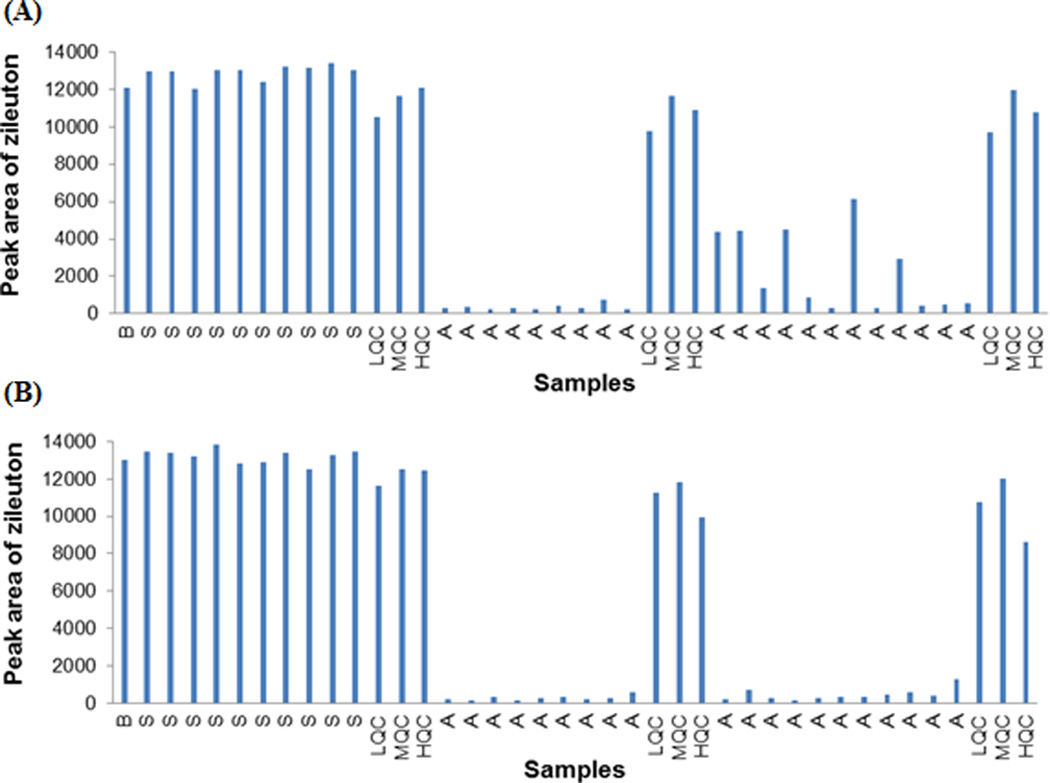
Peak areas of zileuton in the calibrator standards and quality control (QC) samples (which were prepared in pooled human plasma) as well as in the pharmacokinetic plasma samples from two cancer patients (A and B) who received lapatinib (1000 mg orally) on day 1 and the combination of lapatinib (1000 mg orally) and MK-2206 (90 mg orally) on day 29. The data are presented in the order of the analytical run sequence. Abbreviations: B, blank pooled plasma spiked with zileuton only; S, calibrator standards with lapatinib concentration ranging from 5 to 5000 ng/ml; LQC, low QC (15 ng/ml); MQC, medium QC (800 ng/ml); HQC, high QC (4000 ng/ml); A, patient pharmacokinetic plasma samples.
3.2. Analytical performance of the LC-MS/MS method with lapatinib-d3 as the internal standard
The LC-MS/MS method with lapatinib-d3 as the internal standard was developed and validated using a new batch of pooled human plasma. The LLOQ of lapatinib in plasma was 5 ng/ml, at which the signal-to-noise ratio was > 100 in all analytical runs. Selectivity test indicated no apparent interference for lapatinib and lapatinib-d3 in the blank plasma samples from 6 different donors. Fig.4 shows the representative chromatograms of a blank pooled human plasma sample, a blank plasma sample spiked with lapatinib (5 ng/ml) and lapatinib-d3 (1000 ng/ml), and a patient plasma sample (which was collected at 2 hour after the oral administration of lapatinib 1000 mg) spiked with lapatinib-d3 (1000 ng/ml).
Fig. 4.
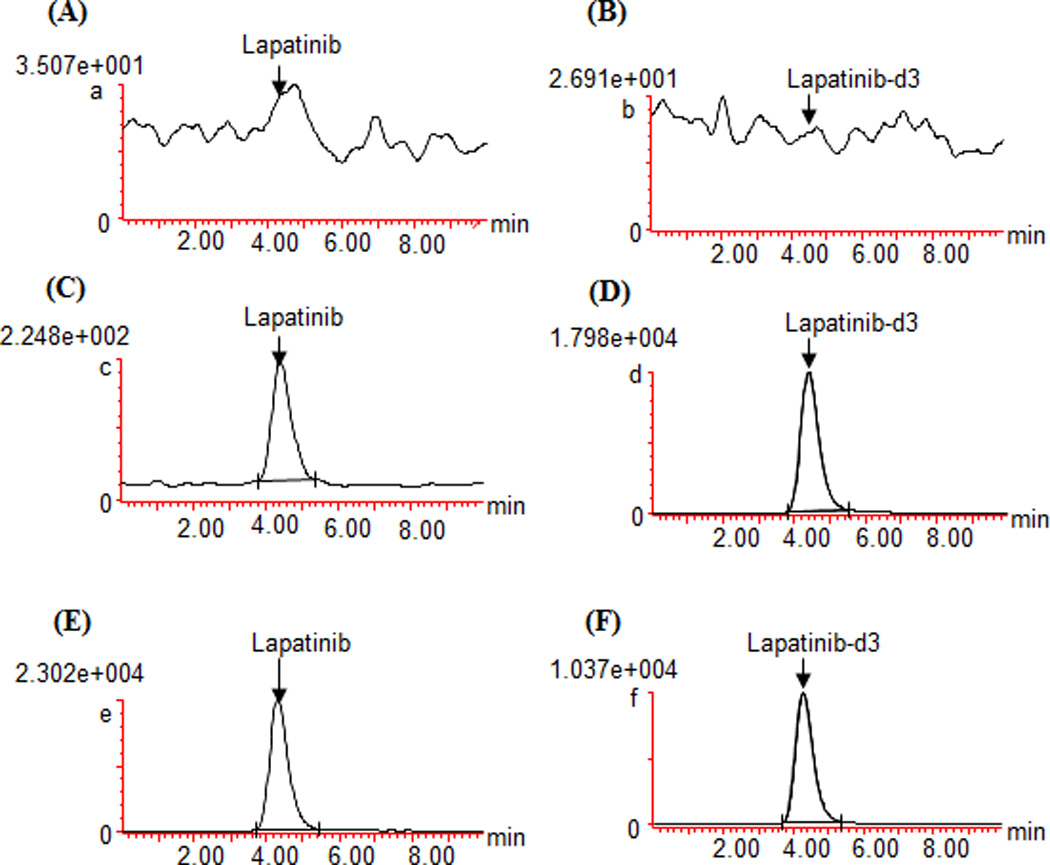
Representative chromatograms of a blank pooled human plasma (A and B), blank plasma spiked with 5 ng/ml of lapatinib (LLOQ) and lapatinib-d3 (1000 ng/ml) (C and D), and a patient plasma sample (which was collected at 2 hour after oral administration of lapatinib 1000 mg) spiked with lapatinib-d3 (1000 ng/ml) (E and F), monitored at m/z 581.40 > 364.96 for lapatinib, and m/z 584.10 > 366.00 for lapatinib-d3.
The linear calibration curves were established over the lapatinib concentration range of 5 – 5000 ng/ml. A linear correlation coefficient (R2) of > 0.99 was obtained in all analytical runs. For all lapatinib calibrator standards and QC samples (including LLOQ, low, medium, and high QCs), the intra- and inter-day accuracy and precision were within the generally accepted criteria for bioanalytical method validation (Table 2).
The matrix effect and recovery of lapatinib and lapatinib-d3 were assessed in 6 different donors of plasma and in the pretreatment plasma samples from 6 cancer patients. No apparent matrix effect (i.e., matrix factor within 1 ± 0.15) was observed for lapatinib or lapatinib-d3 in 6 different donors of plasma samples (Table 3). While a slight ion suppression (with matrix factor < 0.85) was observed in one pretreatment plasma sample from 1 of 6 cancer patients, the overall matrix factor (mean ± standard deviation) was 0.89 ± 0.08 and 0.89 ± 0.09 for lapatinib and lapatinib-d3, respectively, in the cancer patient plasma samples across three QC concentrations (Table 3). Of note, the internal standard-normalized matric factor ranged 0.94 – 1.04 (mean = 1.01) in all 6 patients’ pretreatment samples across three QC concentrations, suggesting lapatinib-d3 as the internal standard can corrected for the variation in any potential matrix effect for lapatinib.
The recovery of lapatinib was largely variable from different sources of human plasma. For example, lapatinib recovery varied up to 2.4-fold (range, 29 – 70%) in 6 different donors of plasma; the variation was even larger (varied up to 3.5-fold; range, 16 – 56%) in the pretreatment plasma samples from 6 cancer patients (Table 3). Lapatinib-d3 exhibited a similar variation in the recovery from plasma as lapatinib (Table 3). Of particular note, the internal standard-normalized recovery was within 100 ± 15% regardless of sources of plasma samples, suggesting that lapatinib-d3 can adequately correct for the interindividual variability in the recovery of lapatinib from patient plasma samples.
3.3. Stability of lapatinib
The short- and long-term stabilities of lapatinib are shown in Table 4. The bench-top stability test suggested that lapatinib was stable in both methanol (evaluated at 1 and 100 µg/ml) and in human plasma (evaluated at 15 and 4000 ng/ml) at ambient temperature (~25°C) for at least 6 hours (Table 4). The autosampler stability test suggested that lapatinib was stable in the reconstitution solution (methanol/0.45% formic acid in water, 50:50, v/v) at 4°C for at least 12 hours, allowing the assay to be performed continuously overnight for a large number of samples. The freeze-thaw stability test demonstrated that lapatinib (evaluated at 15 and 4000 ng/ml) did not degrade in human plasma through three full freeze-thaw cycles (Table 4). The long-term stability tests suggested that lapatinib was stable in methanol (at 1 mg/ml) at −20°C for at least 6 months, and it was stable in human plasma (evaluated at 15 and 4000 ng/ml) at −80°C for at least 6 months (Table 4). The long-term stability for lapatinib in methanol and in human plasma is continuously monitored.
Table 4.
Assessment of stability of lapatinib
| lapatinib plasma concentration (ng/ml)c | ||
|---|---|---|
| 15 | 4000 | |
| Bench-top stability (in plasma) (25°C) (%)a | ||
| 1.0 h | 99.5 | 102.3 |
| 2.0 h | 94.7 | 103.0 |
| 3.0 h | 95.1 | 103.7 |
| 4.0 h | 97.5 | 105.7 |
| 6.0 h | 98.1 | 106.7 |
| Auto-sampler stability (in the mobile phase) (4°C) (%)a | ||
| 1.0 h | 102.0 | 105.4 |
| 2.0 h | 108.5 | 108.7 |
| 4.0 h | 113.0 | 115.0 |
| 8.0 h | 103.4 | 104.5 |
| 12.0 h | 102.1 | 100.8 |
| Freeze-thaw stability (in plasma) (−80°C) (%)b | ||
| Cycle 1 | 90.5 | 93.1 |
| Cycle 2 | 104.0 | 100.2 |
| Cycle 3 | 109.9 | 102.3 |
| Long-term stability (in plasma) (−80°C) (%)b | ||
| 3 month | 93.1 | 98.5 |
| 6 month | 103.9 | 109.1 |
Stability data is expressed as the mean percentage of the peak area determined at certain time relative to that at time zero.
Stability data is expressed as the mean percentage of the analyte concentration determined at certain time point relative to the nominal concentration (%)
Each concentration at each time point was assessed in triplicate.
3.4 Impact of plasma protein binding on the process efficiency (or recovery) of lapatinib
Lapatinib is highly bound to plasma proteins, mainly to HSA and AAG. HSA is the most abundant protein in human blood plasma, accounting for 55–60% of the total plasma protein. The normal range of albumin is 34 – 54 mg/ml in human blood. HSA concentrations can be abnormally high (a condition called hyperalbuminemia) or low (hypoalbuminemia) in a variety of pathological conditions including advanced cancers. AAG, the second major protein in plasma, is an acute-phase protein that is synthesized in the liver. Plasma concentrations of AAG are normally around 0.28 to 0.92 mg/mL, whereas it varies considerably in pathologic or stress conditions such as chronic inflammation, myocardial infarction, and advanced cancer [12]. AAG plasma concentrations in the cancer population vary between 0.45 and 2.85 mg/ml (mean, 1.12 mg/ml) [13]. Thus, variations in plasma levels of HSA or AAG could readily account for inter- or intra-individual variability in drug plasma protein binding, particularly for highly protein-bound drugs like lapatinib.
Covalent binding to albumin has been shown to cause incomplete extraction recovery of several highly plasma protein-bound drugs from human plasma, including thymoquinone and neratinib (HKI-271) [14, 15]. Neratinib is a potent irreversible tyrosine kinase inhibitor of the ErbB family of receptors (HER1, HER2, and HER4), which covalently binds to the cytoplasmic domain of ErbB receptors, thereby preventing downstream signaling [16]. It has been shown that the incomplete recovery of nerotinib from human plasma after exhaustive extraction with organic solvent was caused by the covalent binding of the drug to the lysine residue (Lys190) of HSA via Michael addition [15]. Lys190 is located at the junction of domains I (1–188) and II (189–385) of HSA, a high-affinity region for substrate protein binding [17, 18]. Covalent binding of lapatinib to HSA has not been characterized so far and needs further investigation. Nevertheless, given the fact that lapatinib and neratinib share a similar chemical structure and both were pharmacologically designed for inhibition of the ErbB family of receptors, it is possible that lapatinib binds to HSA covalently in a similar manner as neratinib. Thus, the observed incomplete and variable recovery of lapatinib from human plasma could be attributable, at least in part, to the interindividual variability in its plasma protein binding.
The assessment of the process efficiency of lapatinib from samples containing varying concentrations of plasma proteins supported this possibility. Since no apparent matrix effect was observed for lapatinib, the process efficiency represents the recovery. As shown in Fig.5, with varying clinically relevant plasma protein concentrations, the process efficiency (or recovery) of lapatinib varied up to 3.4-fold (range, 20 – 68%) in the freshly-processed samples, and varied up to 4.5-fold (range, 16 – 71%) in the incubated-frozen samples. This degree of variation was similar to that observed in cancer patient plasma samples (Table 3). The influence of binding to HSA or AAG on the recovery of lapatinib was examined in the human plasma supplemented with clinical relevant concentrations of HSA or AAG as well as in isolated HSA or AAG solutions. The process efficiency (or recovery) of lapatinib appeared to decrease with the increase of HSA concentrations (Fig.5). On the contrary, the variation in clinical relevant concentrations of AAG (0.5 – 3 mg/ml) did not have apparent influence on the process efficiency (or recovery) of lapatinib (Fig.5). Collectively, these data suggested that the observed variation in the recovery of lapatinib from human plasma could be attributable, at least in part, to the interindividual variability in the binding to HSA (likely via covalent binding).
Fig. 5.
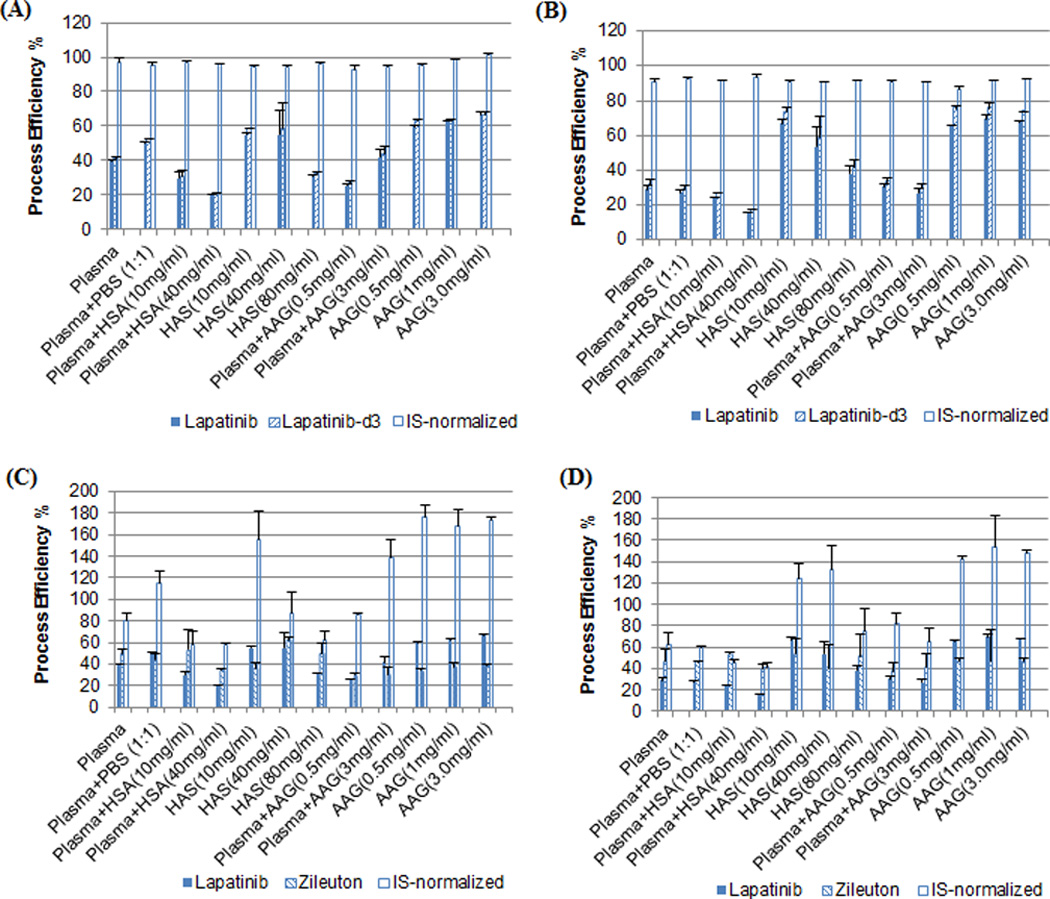
Process efficiency of lapatinib and internal standard (lapatinib-d3 or zileuton) and internal standard-normalized process efficiency in samples containing varying concentrations of plasma proteins. A and C were the results from the samples that were freshly spiked with lapatinib and processed immediately; B and D were the results from the samples that were incubated with lapatinib for 4 hours and stored at −80°C overnight prior to extraction. Process efficiency was estimated as the ratio of the peak area of the analyte spiked prior to extraction to the peak area of the same amount of the analyte spiked in the mobile phase. Data are presented as the mean ± standard deviation of triplicate measurements.
Of particular note, despite of the variation in the process efficiency (or recovery) of lapatinib, the lapatinib-d3-normalized process efficiency was within 92 – 101% (mean, 96%) and 87 – 93% (mean, 91%) in the freshly-processed and incubated-frozen samples, respectively (Fig.5). On the contrary, there was a large variation in the zileuton-normalized process efficiency, which varied from 48% to 186% and from 37% to 294% in the freshly-processed and incubated-frozen samples, respectively (Fig.5). These data further confirmed the notion that an isotope-labeled internal standard is necessary for correcting for the variation in lapatinib recovery from human plasma. Also noted was that the process efficiency of lapatinib in the freshly-processed samples was not significantly different from that in the incubated-frozen samples (paired t-test, P = 0.42) (Fig.5), suggesting that lapatinib binds to plasma proteins rapidly and thus the process efficiency (or recovery) determined from the freshly-processed calibrators or QC samples well represents that from the incurred patient samples.
3.5 Clinical application
The validated method with lapatinib-d3 as the internal standard was successfully employed to study the plasma pharmacokinetics of lapatinib when it was given alone and in combination with MK-2206 (an AKT inhibitor) in an ongoing phase I clinical trial (NCI study #8747). Fig.6 shows the plasma concentration-time profiles of lapatinib in a representative cancer patient who was treated with oral lapatinib alone (1000 mg orally) on day 1 and the combination of lapatinib (1000 mg orally) and MK-2206 (90 mg orally) on day 29. Preliminary pharmacokinetic analysis from 6 patients suggested that co-administration of MK-2206 had no apparent influence on the pharmacokinetics of lapatinib.
Fig. 6.
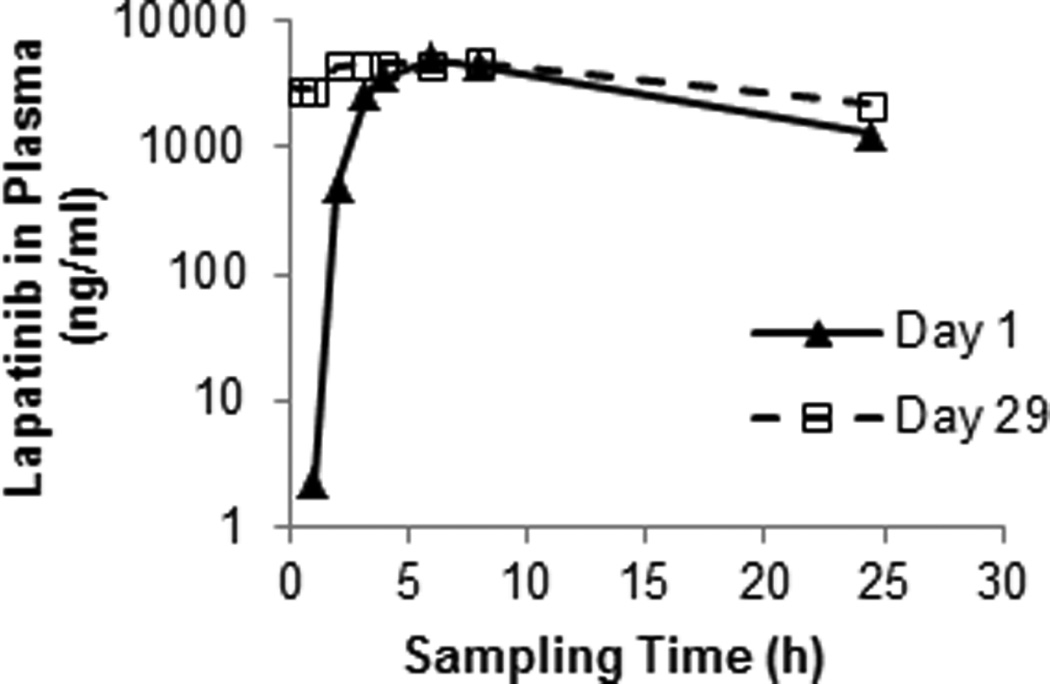
The plasma concentration-time profiles of lapatinib in a representative cancer patient treated with lapatinib alone (1000 mg orally) on day 1 and the combination of lapatinib (1000 mg orally) and MK-2206 (90 mg orally) on day 29.
4. Conclusions
Lapatinib exhibited a large variation in the recovery from different sources or individuals of plasma after exhaustive extraction with organic solvent. The observed incomplete and variable recovery could be attributable, at least in part, to the interindividual variability in the plasma protein binding of lapatinib. Because the calibrator standards and quality control samples are routinely prepared in pooled human plasma, the variation in the recovery of the analyte between different batches or individuals of plasma has the potential to cause erroneous concentration measurements in patient plasma samples. By evaluating the analytical performance of two classes of internal standards (i.e., non-isotope-labeled and isotope-labeled) in the quantitative LC-MS/MS analysis of lapatinib, we confirmed the notion that a stable isotope-labeled internal standard is indispensable for correcting for the interindividual variability in lapatinib recovery from human plasma. As inter- and intra-patient matrix variability is commonly presented in the clinical setting, this study provides an example underscoring the importance of using a stable isotope-labeled internal standard in the quantitative LC-MS/MS analysis for therapeutic drug monitoring or pharmacokinetic evaluation.
Highlights.
-
Lapatinib exhibited a large variation in the recovery from cancer patient plasma after extraction with organic solvent.
The variable recovery could be attributable, in part, to the interindividual variability in the plasma protein binding.
A stable isotope-labeled internal standard is indispensable for correcting for the variation in lapatinib recovery.
Acknowledgments
This study was supported by the United States National Institutes of Health grants: Cancer Center Support Grant P30 CA022453 and U01 CA062487.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Erp NP, Gelderblom H, Guchelaar HJ. Cancer treatment reviews. 2009;35:692–706. doi: 10.1016/j.ctrv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 2.Götze L, Hegele A, Metzelder SK, Renz H, Nockher WA. Clinica chimica acta; international journal of clinical chemistry. 2012;413:143–149. doi: 10.1016/j.cca.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 3.Roche S, McMahon G, Clynes M, O'Connor R. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2009;877:3982–3990. doi: 10.1016/j.jchromb.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 4.Bai F, Freeman BB, 3rd, Fraga CH, Fouladi M, Stewart CF. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2006;831:169–175. doi: 10.1016/j.jchromb.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 5.Bouchet S, Chauzit E, Ducint D, Castaing N, Canal-Raffin M, Moore N, Titier K, Molimard M. Clinica chimica acta; international journal of clinical chemistry. 2011;412:1060–1067. doi: 10.1016/j.cca.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 6.Haouala A, Zanolari B, Rochat B, Montemurro M, Zaman K, Duchosal MA, Ris HB, Leyvraz S, Widmer N, Decosterd LA. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2009;877:1982–1996. doi: 10.1016/j.jchromb.2009.04.045. [DOI] [PubMed] [Google Scholar]
- 7.Lankheet NA, Hillebrand MJ, Rosing H, Schellens JH, Beijnen JH, Huitema AD. Biomedical chromatography : BMC. 2013;27:466–476. doi: 10.1002/bmc.2814. [DOI] [PubMed] [Google Scholar]
- 8.Micova K, Friedecky D, Faber E, Adam T. Talanta. 2012;93:307–313. doi: 10.1016/j.talanta.2012.02.038. [DOI] [PubMed] [Google Scholar]
- 9.Hsieh S, Tobien T, Koch K, Dunn J. Rapid communications in mass spectrometry : RCM. 2004;18:285–292. doi: 10.1002/rcm.1327. [DOI] [PubMed] [Google Scholar]
- 10.Wiegand R, Wu J, Sha X, LoRusso P, Li J. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2010;878:333–339. doi: 10.1016/j.jchromb.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiegand R, Wu J, Sha X, LoRusso P, Heath E, Li J. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences. 2009;877:1460–1464. doi: 10.1016/j.jchromb.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kremer JM, Wilting J, Janssen LH. Pharmacological reviews. 1988;40:1–47. [PubMed] [Google Scholar]
- 13.Duche JC, Urien S, Simon N, Malaurie E, Monnet I, Barre J. Clinical biochemistry. 2000;33:197–202. doi: 10.1016/s0009-9120(00)00048-5. [DOI] [PubMed] [Google Scholar]
- 14.El-Najjar N, Ketola RA, Nissila T, Mauriala T, Antopolsky M, Janis J, Gali-Muhtasib H, Urtti A, Vuorela H. Journal of chemical biology. 2011;4:97–107. doi: 10.1007/s12154-010-0052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Li-Chan XX, Atherton J, Deng L, Espina R, Yu L, Horwatt P, Ross S, Lockhead S, Ahmad S, Chandrasekaran A, Oganesian A, Scatina J, Mutlib A, Talaat R. Drug metabolism and disposition: the biological fate of chemicals. 2010;38:1083–1093. doi: 10.1124/dmd.110.032292. [DOI] [PubMed] [Google Scholar]
- 16.Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, Reich MF, Shen R, Shi X, Tsou HR, Wang YF, Wissner A. Cancer research. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- 17.Yvon M, Anglade P, Wal JM. FEBS letters. 1989;247:273–278. doi: 10.1016/0014-5793(89)81351-1. [DOI] [PubMed] [Google Scholar]
- 18.Ahmed N, Dobler D, Dean M, Thornalley PJ. The Journal of biological chemistry. 2005;280:5724–5732. doi: 10.1074/jbc.M410973200. [DOI] [PubMed] [Google Scholar]