

Abstract
Cellular DNA damage response is critical to preserving genomic integrity following exposure to genotoxic stress. A complex series of networks and signaling pathways become activated after DNA damage and trigger the appropriate cellular response, including cell cycle arrest, DNA repair, and apoptosis. The response elicited is dependent upon the type and extent of damage sustained, with the ultimate goal of preventing propagation of the damaged DNA. A major focus of our studies is to determine the cellular pathways involved in processing damage induced by altered helical structures, specifically triplexes. Our lab has demonstrated that the TFIIH factor XPD occupies a central role in triggering apoptosis in response to triplex-induced DNA strand breaks. We have shown that XPD co-localizes with γH2AX, and its presence is required for the phosphorylation of H2AX tyrosine142, which stimulates the signaling pathway to recruit pro-apoptotic factors to the damage site. Herein, we examine the cellular pathways activated in response to triplex formation and discuss our finding that suggests that XPD-dependent apoptosis plays a role in preserving genomic integrity in the presence of excessive structurally induced DNA damage.
Keywords: triplex DNA, H-DNA, DNA repair, apoptosis, XPD, H2AX, genomic instability, nucleotide excision repair, triplex-forming oligonucleotides
Introduction
Humans are faced with the fundamental problem that the form in which their genetic material is stored is not chemically inert. As a result, DNA integrity is challenged daily by the damaging effects of numerous chemical and physical agents. These events result in various DNA lesions, including double strand breaks (DSBs), mispaired bases, and helix-distorting lesions. If left unrepaired, these lesions can lead to mutations and eventually result in hereditary disease or cancer [1]. To counteract the potentially devastating effects of endogenous and exogenous genotoxins on genomic integrity, all organisms have evolved an intricate balance between two essential pathways, DNA repair and apoptosis [2]. As the first line of defense against genotoxic insult, a network of DNA repair systems is in place to process specific types of DNA damage. These major repair pathways are categorized as homologous recombinational repair (HRR), non-homologous end joining (NHEJ), nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR) [3-5]. The importance of functional repair systems is demonstrated in the number of human diseases resulting from defects in DNA repair pathways, such as Fanconi anemia (FA), Cockayne syndrome (CS), Xeroderma pigmentosa (XP), and Bloom syndrome (BS) [6,7].
Alternatively, if too much damage has been sustained, apoptosis plays an important role in maintaining genomic integrity by providing a mechanism by which a cell can actively control its own death [8-10]. This process offers the cell a “fail-safe” mechanism that can be activated if the repair networks become overwhelmed by significant levels of DNA damage and thus inefficient at repair. However, a key issue exists as to how the cell triggers the switch from repair to apoptosis in the presence of excessive DNA damage. This mechanism is critical in avoiding progression to cancer, because it prevents expansion of cells that contain potential disease initiating mutations. In this overview, we focus on the DNA damage response to helix-distorting lesions and discuss our studies aimed at identifying proteins that not only contribute to cell survival by participating in the repair of altered helical structures but also occupy a central position in stimulating the cell to activate apoptosis in the presence of excessive structurally induced DNA damage.
Endogenous Triplex DNA and Genomic Instability
The human genome consists of sequence patterns that can adopt a variety of alternative structures in addition to the double-helical B-conformation described by Watson and Crick in 1953 [11]. For example, H-DNA (triplex) formation is favored by sequences that contain mirror repeat symmetry and occurs at purine/pyrimidine tracts [12-14]. These sequences can assume an H-DNA conformation when the polypurine tract becomes disrupted into single strands as a result of the energy provided from supercoiling. One of the dissociated single strands can fold backward and associate in a parallel orientation with the purine-rich strand in the remaining duplex. This results in the formation of a structure that has an unpaired single-stranded region and a three-stranded helical region via non-Watson Crick interactions.
Over the past several years, research has established that 1 in every 50,000 base pairs within the mammalian genome contains a mirror repeat sequence with the potential to generate an H-DNA structure [15]. Formation of these structures causes severe genomic alterations and represents an endogenous source of genomic instability [16-18]. These sequences are typically found in promoters and exons and are believed to be involved in the regulation of expression of several disease-linked genes [19-22]. For instance, the translocation and overexpression of the c-myc gene observed in some tumors has been attributed in part to a H-DNA forming sequence located in the promoter region of the gene [19]. By using the endogenous H-DNA forming sequences found in the human c-myc promoter, Wang et al. have shown that these sequences are intrinsically mutagenic in mammalian cells [18]. In addition, DSBs were detected near the H-DNA locus, suggesting that this structure is a source of genetic instability. Further supporting this hypothesis, many breakpoints on the translocated gene are clustered around the triplex-forming sequences in the promoter region of Burkitt’s lymphoma [23]. These findings suggest that the H-DNA structures result in fragile sites or mutation hot spots, which can lead to DSBs and subsequent translocation of the gene. Combined, these findings demonstrate that naturally occurring DNA sequences capable of forming triplex structures represent an endogenous source of genomic instability.
Triplex-Forming Oligonucleotides
In 1957, Felsenfeld and Rich revealed that nucleic acids can also interact to form triplex structures [24]. Synthetic triplex DNA can be generated at polypurine/polypyrimidine sequences when oligonucleotides serve as the third strand and bind to duplex DNA through specific major groove interactions [25]. These structures occur oriented either parallel or anti-parallel to the purine strand of the duplex. In the parallel motif, a pyrimidine-rich oligonucleotide binds parallel to the purine strand of duplex DNA via Hoogsteen bonds with the canonocial triplets being T.A:T and C.G:C. Because protonation at the N3 position of cytosine is required for proper Hoogsteen binding with N7 guanine, pyrimidine oligonucleotides do not usually bind to DNA at physiological pH without further modification. In the anti-parallel motif, a homopurine oligonucleotide binds anti-parallel to the purine strand in the duplex via reverse Hoogsteen hydrogen bonds where the triplets are A.A:T and G.G:C. The canonical base triplets formed by either Hoogsteen or reverse Hoogsteen hydrogen bonds provides sequence-specificity between the target duplex and the triplex-forming oligonucleotide (TFO).
The formation of triplex structures via TFOs induces significant structural distortions in the target duplex [26-31]. Crytallographic studies on triplex structures reveal large changes in the phosphate backbone torsion angles of the purine-rich strand [32]. In addition, lower twists and higher opening angles of the Watson and Crick pairs at the duplex-triplex junction indicate that the transition from duplex to triplex results in partial unwinding of the DNA [32]. These triplex-induced changes in the parameters of the DNA double helix likely mediate induction of several cellular DNA metabolic responses, such as repair. Because the triplex region found in H-DNA is similar in structure to intermolecular triplexes formed by TFOs, they represent an excellent model to study naturally occurring non-canonical structures and advance our understanding of the mechanisms activated in response to structurally induced DNA damage.
Triplex-Induced Double Strand Breaks
Endogenous triplex structures are speculated to be responsible for some genomic breakpoints in common chromosomal translocations observed in cancers such as leukemia, lymphoma, and sarcoma [33-35]. Our lab has now determined that triplex formation via TFOs can also act as a fragile site resulting in DSBs [36]. Neutral comet assays determined that TFOs were capable of inducing significantly more DSBs in cells containing multiple triplex target sites compared to a control oligonucleotide, which cannot bind as a third strand. Given this result, we initiated several studies that utilized γH2AX as a quantitative marker for DSBs and confirmed the presence of DSBs after triplex formation by Western blot analysis and immunofluorescence microscopy [36]. Flow cytometry studies staining for γH2AX in the presence of propidium iodine ensured that γH2AX foci formation was indeed a hallmark of DSBs and was not generated in the course of DNA fragmentation during apoptosis. Although triplex-induced γH2AX expression was determined to be cell cycle-independent, persistent γH2AX signal for up to 24 hours after TFO treatment demonstrated that many sites marked by γH2AX foci remained unrepaired. These results suggest that formation of multiple triplex structures can generate substantial DSBs, which can overwhelm the repair capacity of the cell [36].
We have extended these studies in vivo and have explored the potential for triplex structures to induce DSBs in animals [36]. A transgenic mouse model with multiple copies of the triplex target site chromosomally integrated into its genome was used to evaluate damage generated by triplexes [37]. Immunohistochemistry staining for γH2AX in spleen samples harvested from TFO-treated mice determined that there was an increase in the percentage of cells positive for γH2AX compared to mice treated with PBS or a control oligonucleotide, implicating the presence of triplex-induced DSBs. In relevant work, Wang et al. have developed a mouse model that incorporates the naturally occurring H-DNA sequence found at the breakage hotspot in the human c-myc promoter [38]. This group had previously reported that this naturally occurring sequence was highly mutagenic and induced double strand breaks in mammalian cells [18]. Large-scale chromosomal deletions and translocations were observed in regions of the H-DNA sequence in approximately 8 percent of the animals carrying the c-myc sequence, thus implicating DNA structure in chromosome breakage [38].
Nucleotide Excision Repair and Triplex Structures
Studies have shown that the more a lesion distorts the normal helical structure, the more efficiently it is repaired [39]. The ability of TFOs to bind to undamaged duplex DNA and stimulate repair was demonstrated using an assay to measure the induction of repair synthesis [40,41]. Research has determined that a TFO bound to its target site on a supercoiled plasmid creates a helical distortion that strongly provokes DNA repair synthesis in HeLa extracts supplemented with α-32P dCTP and an ATP regenerating system [40,41]. Increased α-32P dCTP incorporation was detected following incubation of the TFO with the target plasmid compared to a control plasmid, which lacks the target site, indicating TFO-induced DNA repair synthesis.
Detection of TFO-induced repair synthesis prompted investigation into which repair pathway was responsible for recognition and repair of this type of lesion. The nucleotide excision repair (NER) pathway occupies an important position in repairing a wide array of helix-distorting lesions [39,42]. The common denominator in NER-recognized lesions can be attributed to significant chemical or physical distortion of the DNA helix. As a result, we hypothesized that triplex structures were identified as a lesion by the NER pathway. In order to determine whether NER participated in the repair of triplex structures, we repeated the DNA repair assay using XPA-depleted HeLa extracts. As a major contributor to the NER pathway, the XPA protein is responsible for validating the altered DNA structure and recruiting the remaining NER proteins to the damage site. Immunodepletion of XPA from the extracts resulted in a substantial reduction in the ability of the TFOs to induce repair synthesis [41]. These results were verified by conducting experiments, which complemented the XPA-depleted extracts with purified XPA protein and observing a restoration in repair activity [41]. Additional studies from other laboratories have also demonstrated by gel shift analysis that the NER proteins XPA and RPA are involved in the recognition of triplex lesions [43].
Subsequent studies have also investigated the role of transcription-coupled NER (TC-NER) in the processing of triplex structures [44]. Triplex formation has been previously determined to specifically inhibit gene transcription on a plasmid in HeLa nuclear extracts. Additional studies demonstrated that TC-NER was important for the repair of the triplex structure and restoration of gene transcription. In fact, the degree of TFO-induced TC-NER repair activity coincided with the level of transcription [44]. When the extracts lacked individual transcription components such as TFIIB, TFIIH, or RNA Pol II, inhibition of repair was evident, thus suggesting that triplex-induced repair synthesis was related to transcription. Additional experiments also indicate that the ability to repair TFO-induced structures was altered in nuclear extracts deficient in the transcription factor CSA (defective in TC-NER), implying the importance of TC-NER. In addition to TC-NER, evidence for global genome NER (GG-NER) participating in resolving TFO-associated lesions provided evidence that repair of triplex structures takes place outside of the actively transcribed regions. XPC, the primary recognition factor of the GG-NER sub-pathway, was determined to be required for the repair of TFO-induced triplex structures. Given these results, the conclusion can be drawn that both TC-NER and GG-NER subpathways are involved in the repair of triplex structures.
Triplex Formation Activates Apoptosis
TFOs create helical distortions upon binding to duplex DNA that strongly provoke NER-dependent repair [40,41]. Our current work has now established that these structures also form a severe enough alteration in the DNA double helix to induce enough DNA damage and activate apoptosis [36]. Studies conducted in our laboratory have demonstrated a decrease in cell survival that was proportional to an increase in the formation of triplex structures, indicating that excessive DNA damage induced by helical distortions can lead to cell death. Using quantification of Annexin V binding to exposed phosphatidylserine residues in the cell membrane as an early marker for apoptosis, we determined that the observed cell death resulted from activation of an apoptotic pathway. Our research also confirmed that the observed apoptosis can be attributed to the generation of triplex structures and not to nonspecific toxicity created by the oligonucleotide itself, since apoptosis was not observed with a non-triplex-forming control oligonucleotide [36]. Importantly, we also demonstrated that in vivo triplex formation could also elicit an apoptotic response. Immunohistochemistry staining of activated caspase 3 can be utilized as an apoptotic marker. Examination of spleen tissue samples obtained from TFO-treated mice revealed that 26 percent of their spleen cells were positive for activated caspase 3, while tissue samples obtained from control animals, which lack the triplex target site, were void of activated caspase 3 [36].
NER Proteins Involved in Apoptosis
Based on the finding that triplex-induced DNA damage can activate an apoptotic response, we proceeded to investigate which proteins were responsible for maintaining a balance between NER-dependent repair and apoptosis after triplex formation. Cellular DNA-repair pathways involve proteins that have roles in other DNA metabolic processes in addition to their function in DNA damage removal [2]. Key proteins that contribute to cell survival by acting in DNA repair can become executioners when faced with extreme DNA damage. Consequently, we hypothesized that a dual role NER protein like XPD, which contributes to repair through its 5′→3′ helicase activity and to apoptosis via binding to p53, may be pivotal in maintaining the balance [45-48]. Mechanistically, our work has identified a role for XPD in the activation of apoptosis following excessive triplex-induced DNA damage. We found that XPD-knockdown resulted in a decrease in apoptosis, although the same level of triplex-induced DNA damage was measured in the XPD-deficient cells compared with XPD-proficient cells (as determined by neutral comet assay and γH2AX foci formation). Our results support a mechanism where XPD is important for activation of apoptosis but not required for the formation of triplex-induced DSBs [36].
Signaling Pathways Activated Following Triplex-Induced DNA Damage
As mentioned above, tumor suppressor p53, a central component of apoptosis, can bind to and inhibit the helicase activity of XPD. We observed by Western blot that an increase in p53 protein levels corresponded to an increase in triplex-induced apoptosis. Additionally, triplex-induced damage increased p53 phosphorylation at serine 15, which participates in signaling activation of apoptosis. Conversely, we have demonstrated a reduction in p53 (serine 15) phosphorylation after triplex formation in XPD-depleted cells, further lending support to our finding that XPD is required for triplex-induced apoptosis [36].
In earlier work, Xiao et al. [49] and Cook et al. [50] have independently discovered that the phosphorylation status of H2AX at tyrosine 142 (Y142) is crucial in determining the relative recruitment of either DNA repair or pro-apoptotic factors to the DSBs site. When repair is possible following damage, Y142 is dephosphorylated and phosphorylation of serine 139 (γH2AX) recruits repair factors. However, in cases of excessive damage, Y142 is phosphorylated in the presence of serine 139 phosphorylation, which results in the mobilization of apoptotic factors. We have determined that H2AX Y142 is phosphorylated in response to triplex-induced DSBs, resulting in apoptosis [36]. In further studies, we have found that XPD is required for phosphorylation of Y142 in response to triplex-induced DNA damage. Taking the next step, we have detected by confocal microscopy and co-immunoprecipitation that XPD colocalizes with γH2AX at triplex-induced DSBs. These results would suggest that an absence of XPD disrupts the signaling pathway used to trigger apoptosis following formation of multiple triplex structures (Figure 1).
Figure 1.
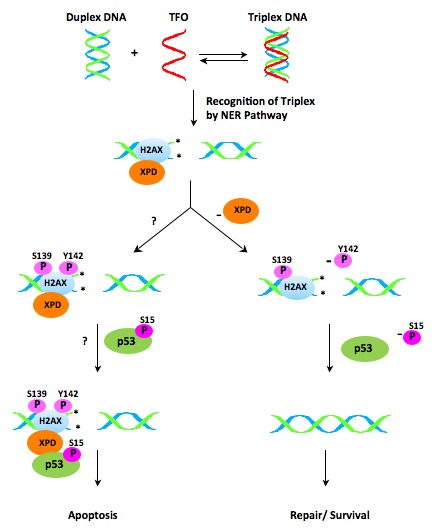
Proposed model of XPD-dependent activation of apoptosis in response to triplex-induced DNA damage. The formation of triplex structures can result in DNA double strand breaks. XPD is recruited to the triplex-induced double strand break and co-localizes with γH2AX at the damage site. H2AX tyrosine142 (Y142) phosphorylation is an important post-translational modification that differentiates between activation of apoptosis and initiation of DNA repair. XPD is required for H2AX Y142 phosphorylation and subsequent recruitment of pro-apoptotic factors to γH2AX in response to triplex-induced damage.
Conclusions and Outlook
Overall, our work has determined that triplex formation in addition to stimulating NER-dependent repair creates a helical distortion that can activate apoptotic pathways. Apoptosis is important for maintaining genomic integrity, especially under conditions where the repair network is unable to efficiently process DNA damage. Several groups have established that triplex formation generated by endogenous sequences or TFOs can induce mutations both in vitro and in vivo [51,52]. Our experiments indicate that the ability to activate apoptosis in response to triplex-induced DNA damage was altered in cells defective in XPD, resulting in an increase in TFO-induce mutations [36]. The thrust of our findings positions apoptosis as an important pathway in triplex-induced DNA damage response and solidifies its importance in preserving genomic integrity. Moreover, we have begun to identify the potential players involved in maintaining a balance between NER-dependent repair and apoptosis. We expect that further characterization of triplex-induced DNA damage response will provide insight into the driving forces responsible for making the ultimate choice between DNA repair and apoptotic cell death in response to genotoxic stress.
Acknowledgments
This work is based on a presentation by F.A.R. at the 50 Years of DNA Repair Symposium held by the Department of Therapeutic Radiology at Yale. It was supported by a grant from the NIH (K22 CA120049, K22 CA120049-03S1 to F.A.R.).
Abbreviations
- DSBs
double strand breaks
- TFO
triplex-forming oligonucleotides
- NER
nucleotide excision repair
- HRR
homologous recombinational repair
- NHEJ
non-homologous end joining
- BER
base excision repair
- MMR
mismatch repair
- FA
Fanconi anemia
- CS
Cockayne syndrome
- XP
Xeroderma pigmentosal
- BS
Bloom syndrome
- GG-NER
global genome NER
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein C, Bernstein H, Payne CM, Garewal H. DNA repair/pro-apoptotic dual-role proteins in five major DNA repair pathways: fail-safe protection against carcinogenesis. Mutat Res. 2002;511(2):145–178. doi: 10.1016/s1383-5742(02)00009-1. [DOI] [PubMed] [Google Scholar]
- Hansen WK, Kelley MR. Review of mammalian DNA repair and translational implications. J Pharmacol Exp Ther. 2000;295(1):1–9. [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Wood RD. DNA repair in eukaryotes. Annu Rev Biochem. 1996;65:135–167. doi: 10.1146/annurev.bi.65.070196.001031. [DOI] [PubMed] [Google Scholar]
- O’Driscoll M. Diseases associated with defective responses to DNA damage. Cold Spring Harb Perspect Biol. 2012;4(12) doi: 10.1101/cshperspect.a012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses RE. DNA damage processing defects and disease. Annu Rev Genomics Hum Genet. 2001;2:41–68. doi: 10.1146/annurev.genom.2.1.41. [DOI] [PubMed] [Google Scholar]
- Payne CM, Bernstein C, Bernstein H. Apoptosis overview emphasizing the role of oxidative stress, DNA damage and signal-transduction pathways. Leuk Lymphoma. 1995;19(1-2):43–93. doi: 10.3109/10428199509059662. [DOI] [PubMed] [Google Scholar]
- Searle J, Kerr JF, Bishop CJ. Necrosis and apoptosis: distinct modes of cell death with fundamentally different significance. Pathol Annu. 1982;17 Pt 2:229–259. [PubMed] [Google Scholar]
- Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4(2):139–163. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- Htun H, Dahlberg JE. Single strands, triple strands, and kinks in H-DNA. Science. 1988;241(4874):1791–1796. doi: 10.1126/science.3175620. [DOI] [PubMed] [Google Scholar]
- Voloshin ON, Mirkin SM, Lyamichev VI, Belotserkovskii BP, Frank-Kamenetskii MD. Chemical probing of homopurine-homopyrimidine mirror repeats in supercoiled DNA. Nature. 1988;333(6172):475–476. doi: 10.1038/333475a0. [DOI] [PubMed] [Google Scholar]
- Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, Frank-Kamenetskii MC. DNA H form requires a homopurine-homopyrimidine mirror repeat. Nature. 1987;330(6147):495–497. doi: 10.1038/330495a0. [DOI] [PubMed] [Google Scholar]
- Schroth GP, Ho PS. Occurrence of potential cruciform and H-DNA forming sequences in genomic DNA. Nucleic Acids Res. 1995;23(11):1977–1983. doi: 10.1093/nar/23.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DN, Bacolla A, Ferec C, Vasquez KM, Kehrer-Sawatzki H, Chen JM. On the sequence-directed nature of human gene mutation: the role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease. Hum Mutat. 2011;32(10):1075–1099. doi: 10.1002/humu.21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Shen Y, Zhang F, Chiang C, Pillalamarri V, Blumenthal I. et al. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. Am J Hum Genet. 2013;92(3):375–386. doi: 10.1016/j.ajhg.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Vasquez KM. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc Natl Acad Sci USA. 2004;101(37):13448–13453. doi: 10.1073/pnas.0405116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinniburgh AJ. A cis-acting transcription element of the c-myc gene can assume an H-DNA conformation. Nucleic Acids Res. 1989;17(19):7771–7778. doi: 10.1093/nar/17.19.7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestov DG, Dayn A, Siyanova E, George DL, Mirkin SM. H-DNA and Z-DNA in the mouse c-Ki-ras promoter. Nucleic Acids Res. 1991;19(23):6527–6532. doi: 10.1093/nar/19.23.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Jaworski A, Connors TD, Wells RD. Pkd1 unusual DNA conformations are recognized by nucleotide excision repair. J Biol Chem. 2001;276(21):18597–18604. doi: 10.1074/jbc.M100845200. [DOI] [PubMed] [Google Scholar]
- Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J Biol Chem. 2007;282(44):32433–32441. doi: 10.1074/jbc.M704618200. [DOI] [PubMed] [Google Scholar]
- Saglio G, Grazia Borrello M, Guerrasio A, Sozzi G, Serra A, di Celle PF. et al. Preferential clustering of chromosomal breakpoints in Burkitt's lymphomas and L3 type acute lymphoblastic leukemias with a t(8;14) translocation. Genes Chromosomes Cancer. 1993;8(1):1–7. doi: 10.1002/gcc.2870080102. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G, Rich A. Studies on the formation of two- and three-stranded polyribonucleotides. Biochim Biophys Acta. 1957;26(3):457–468. doi: 10.1016/0006-3002(57)90091-4. [DOI] [PubMed] [Google Scholar]
- Francois JC, Saison-Behmoaras T, Helene C. Sequence-specific recognition of the major groove of DNA by oligodeoxynucleotides via triple helix formation. Footprinting studies. Nucleic Acids Res. 1988;16(24):11431–11440. doi: 10.1093/nar/16.24.11431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XG, Cao EH, He YJ, Qin JF. Spectroscopic comparison of different DNA structures formed by oligonucleotides. J Biomol Struct Dyn. 1999;16(4):863–872. doi: 10.1080/07391102.1999.10508298. [DOI] [PubMed] [Google Scholar]
- Asensio JL, Dosanjh HS, Jenkins TC, Lane AN. Thermodynamic, kinetic, and conformational properties of a parallel intermolecular DNA triplex containing 5' and 3' junctions. Biochemistry. 1998;37(43):15188–15198. doi: 10.1021/bi980057m. [DOI] [PubMed] [Google Scholar]
- Han ZJ, Rhee S, Liu K, Miles HT, Davies DR. Crystallization and preliminary crystallographic study of triple-helical DNA. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 1):104–105. doi: 10.1107/s0907444999012895. [DOI] [PubMed] [Google Scholar]
- He Y, Scaria PV, Shafer RH. Studies on formation and stability of the d[G(AG)5]* d[G(AG)5]. d[C(TC)5] and d[G(TG)5]* d[G(AG)5]. d[C(TC)5] triple helices. Biopolymers. 1997;41(4):431–441. doi: 10.1002/(SICI)1097-0282(19970405)41:4<431::AID-BIP7>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Kan LS, Callahan DE, Trapane TL, Miller PS, Ts’o PO, Huang DH. Proton NMR and optical spectroscopic studies on the DNA triplex formed by d-A-(G-A)7-G and d-C-(T-C)7-T. J Biomol Struct Dyn. 1991;8(5):911–933. doi: 10.1080/07391102.1991.10507857. [DOI] [PubMed] [Google Scholar]
- Hartman DA, Kuo SR, Broker TR, Chow LT, Wells RD. Intermolecular triplex formation distorts the DNA duplex in the regulatory region of human papillomavirus type-11. J Biol Chem. 1992;267(8):5488–5494. [PubMed] [Google Scholar]
- Rhee S, Han Z, Liu K, Miles HT, Davies DR. Structure of a triple helical DNA with a triplex-duplex junction. Biochemistry. 1999;38(51):16810–16815. doi: 10.1021/bi991811m. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH. Chromosomal translocations in human cancer. Nature. 1994;372(6502):143–149. doi: 10.1038/372143a0. [DOI] [PubMed] [Google Scholar]
- Bacolla A, Jaworski A, Larson JE, Jakupciak JP, Chuzhanova N, Abeysinghe SS. et al. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc Natl Acad Sci USA. 2004;101(39):14162–14167. doi: 10.1073/pnas.0405974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Rowley JD. Chromatin structural elements and chromosomal translocations in leukemia. DNA Repair (Amst) 2006;5(9-10):1282–1297. doi: 10.1016/j.dnarep.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Kaushik Tiwari M, Rogers FA. XPD-dependent activation of apoptosis in response to triplex-induced DNA damage. Nucleic Acids Res. 2013 Aug 2; doi: 10.1093/nar/gkt670. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers FA, Lin SS, Hegan DC, Krause DS, Glazer PM. Targeted gene modification of hematopoietic progenitor cells in mice following systemic administration of a PNA-peptide conjugate. Mol Ther. 2012;20(1):109–118. doi: 10.1038/mt.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Carbajal S, Vijg J, DiGiovanni J, Vasquez KM. DNA structure-induced genomic instability in vivo. J Natl Cancer Inst. 2008;100(24):1815–1817. doi: 10.1093/jnci/djn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batty DP, Wood RD. Damage recognition in nucleotide excision repair of DNA. Gene. 2000;241(2):193–204. doi: 10.1016/s0378-1119(99)00489-8. [DOI] [PubMed] [Google Scholar]
- Wang G, Seidman MM, Glazer PM. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science. 1996;271(5250):802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- Rogers FA, Vasquez KM, Egholm M, Glazer PM. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc Natl Acad Sci USA. 2002;99(26):16695–16700. doi: 10.1073/pnas.262556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunz D, Hess MT, Naegeli H. Recognition of DNA adducts by human nucleotide excision repair. Evidence for a thermodynamic probing mechanism. J Biol Chem. 1996;271(41):25089–25098. doi: 10.1074/jbc.271.41.25089. [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Christensen J, Li L, Finch RA, Glazer PM. Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc Natl Acad Sci USA. 2002;99(9):5848–5853. doi: 10.1073/pnas.082193799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Chen Z, Zhang S, Wilson GL, Jing K. Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclear extracts. Nucleic Acids Res. 2001;29(8):1801–1807. doi: 10.1093/nar/29.8.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13(7):768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- Wang XW, Vermeulen W, Coursen JD, Gibson M, Lupold SE, Forrester K. et al. The XPB and XPD DNA helicases are components of the p53-mediated apoptosis pathway. Genes Dev. 1996;10(10):1219–1232. doi: 10.1101/gad.10.10.1219. [DOI] [PubMed] [Google Scholar]
- Robles AI, Wang XW, Harris CC. Drug-induced apoptosis is delayed and reduced in XPD lymphoblastoid cell lines: possible role of TFIIH in p53-mediated apoptotic cell death. Oncogene. 1999;18(33):4681–4688. doi: 10.1038/sj.onc.1202862. [DOI] [PubMed] [Google Scholar]
- Wang XW, Yeh H, Schaeffer L, Roy R, Moncollin V, Egly JM. et al. p53 modulation of TFIIH-associated nucleotide excision repair activity. Nat Genet. 1995;10(2):188–195. doi: 10.1038/ng0695-188. [DOI] [PubMed] [Google Scholar]
- Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H. et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457(7225):57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458(7238):591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez KM, Narayanan L, Glazer PM. Specific mutations induced by triplex-forming oligonucleotides in mice. Science. 2000;290(5491):530–533. doi: 10.1126/science.290.5491.530. [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Wang G, Havre PA, Glazer PM. Chromosomal mutations induced by triplex-forming oligonucleotides in mammalian cells. Nucleic Acids Res. 1999;27(4):1176–1181. doi: 10.1093/nar/27.4.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]