

Abstract
This review focuses on research findings in the area of diagnosis and pathogenesis of hepatitis C virus (HCV) infection over the last few decades. The information based on published literature provides an update on these two aspects of HCV. HCV infection, previously called blood transmitted non-A, non-B infection, is prevalent globally and poses a serious public health problem worldwide. The diagnosis of HCV infection has evolved from serodetection of non-specific and low avidity anti-HCV antibodies to detection of viral nucleic acid in serum using the polymerase chain reaction (PCR) technique. Current PCR assays detect viral nucleic acid with high accuracy and the exact copy number of viral particles. Moreover, multiplex assays using real-time PCR are available for identification of HCV-genotypes and their isotypes. In contrast to previous methods, the newly developed assays are not only fast and economic, but also resolve the problem of the window period as well as differentiate present from past infection. HCV is a non-cytopathic virus, thus, its pathogenesis is regulated by host immunity and metabolic changes including oxidative stress, insulin resistance and hepatic steatosis. Both innate and adaptive immunity play an important role in HCV pathogenesis. Cytotoxic lymphocytes demonstrate crucial activity during viral eradication or viral persistence and are influenced by viral proteins, HCV-quasispecies and several metabolic factors regulating liver metabolism. HCV pathogenesis is a very complex phenomenon and requires further study to determine the other factors involved.
Keywords: Hepatitis C virus, Diagnosis, Pathogenesis, Immunity, Steatosis
Core tip: This article focuses on the diagnosis and pathogenesis of hepatitis C virus infection. Both of these aspects are important in order to eradicate this endemic virus and to prevent serious liver diseases.
INTRODUCTION
Hepatitis C virus (HCV) was first characterized by Choo et al[1] and Kuo et al[2] in 1989. It was soon identified as the main causative agent of the disease previously known as post transfusion non-A, non-B hepatitis virus infection. HCV has been found to be an important cause of liver disease and remains a major public health problem worldwide. According to the World Health Organization, nearly 3% of the world population has been infected with HCV. Therefore, more than 170 million people are chronic carriers of HCV and at high risk of developing liver cirrhosis and/or hepatocellular carcinoma (HCC). Three to 4% of chronically infected individuals develop fatal HCC. Currently, HCC caused by HCV infection is considered an indication for liver transplantation[3-5].
HCV was the leading cause of post-transfusion and community-acquired non-A, non-B hepatitis until characterization of the virus in 1989 and the introduction of blood screening in 1990. The initiation of blood screening for HCV has markedly reduced its incidence. However, it still remains a significant problem in intravenous drug abusers. HCV infection is the most common cause of liver transplantation in adults. HCV and HIV-1 frequently co-infect humans and it has been estimated that as many as 18% of HIV-infected persons are also infected with HCV[4].
HCV is an enveloped RNA virus and belongs to the genus Hepacivirus of the family Flaviviridae. The HCV genome consists of 9.6-kb single-stranded RNA of positive polarity and a single open reading frame of 9033-9099 nucleotides flanked by a conserved 5’ and 3’ noncoding region (NCR) at the ends. Its genome codes for a long polyprotein of approximately 3000 amino acids[6] which is processed co-translationally and post-translationally to yield structural proteins (core, envelope E1, and E2) and non-structural (NS) proteins (NS1/p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B)[7]. The envelope proteins (E1 and E2) are the outer surface proteins of the viral particles and play important roles in virus entry into the host cell. NS5B is a variable region of the HCV genome and codes for an RNA-dependent RNA polymerase (RdRp).
RNA polymerase lacks proof reading activity and this may alter the detection, sensitivity to interferon anti-viral activity and pathogenicity of the virus (Figure 1)[8].
Figure 1.
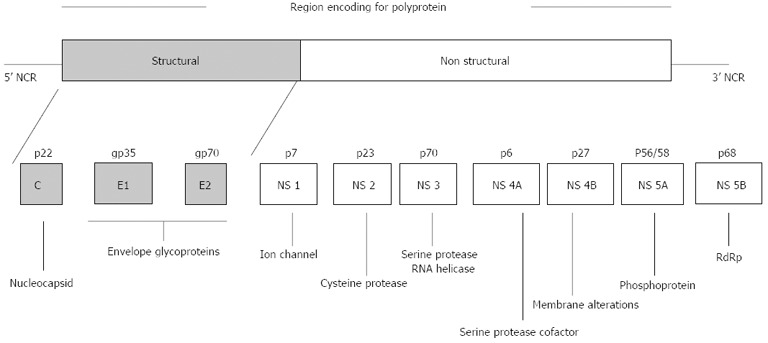
Proteins encoded by the hepatitis C virus genome. Genome organization of hepatitis C virus showing the structure of the viral genome, including the long open reading frame encoding structural and nonstructural proteins, and 5’ and 3’ non-coding regions (NCRs). [Source: Monica A et al. Expert Rev Mol Med 2003; 5].
Like several other viruses, the RNA virus has a high degree of heterogeneity[5] that varies 30%-35% among different genotypes. Based on previous studies, six major genotypes and more than 120 subtypes of HCV have been characterized to date[9]. These HCV genotypes have distinct geographic distributions, with genotype 1 and 2 frequently found worldwide[10]. In India, genotype 3 is reported to be the most prevalent, followed by genotype 1[11,12]. Different HCV genotypes have important epidemiological implications. Despite nucleotide sequence divergence between genotypes, they remain quite similar in their transmission pattern, persistence and disease development[13]. Although genetic variation is attributed to several factors, two major theories i.e., the Darwinian and Neutral evolution theories are thought to be the prominent theories in causing genetic diversity in HCV[13]. The nucleotide sequence variability is distributed throughout the viral genome. Regions encoding envelope proteins (E1, E2) and NS-1 are the most variable, whereas the 5’ NCR is the most conserved region.
HCV patients show a poor response to antiviral therapy based on the combination of pegylated interferon (IFN)-α and ribavirin. Only 40%-50% of patients infected with HCV genotype-1 and 80% of those infected with genotype-2 or 3 achieve a sustained virological response (SVR) with this regimen[14]. The recent use of direct acting anti-viral (DAA) molecules, which are active on HCV during treatment, has led to a substantial improvement in SVR rates in HCV genotype-1 infected patients. However, this may lead to the selection of resistant virus if DAA molecules are used alone[15]. Moreover, there is a high relapse rate of HCV infection after discontinuation of therapy. Recently, host genetic factors including human leukocyte antigen (HLA) and cytokine genes have been implicated in HCV infection or persistence[16]. Genetic polymorphism of cytokine genes including IFN-γ, tumor necrosis factor (TNF)-α, interleukin (IL)-10, IL-20 and SNPs in the promoter region of osteopontin gene, have been found to be crucial in determining the therapeutic outcome of HCV infection[17]. Therefore, every effort is being made to understand the pathogenesis of HCV infection to create a therapeutic model for an effective treatment against HCV. Although recent reports describe the development of in vitro replication systems leading to the production of infectious viral particles[18,19], there is currently no cell culture model suitable for synthesizing vaccines based on killed or attenuated virus. All efforts have been focused on sub-unit vaccines, composed of one or several antigens, either in the form of recombinant proteins, synthetic peptides or vectored vaccines. The earliest vaccine developed for HCV was that by the Chiron group[20]. However, very little progress was noted in this direction in subsequent years.
This article reviews the major aspects of HCV infection including the diagnosis and pathogenesis of HCV infection. Both of these aspects have a strong association with therapy, thus, newer means of accurate diagnosis and a better understanding of HCV infection pathogenesis may allow the development of a therapeutic model. This article attempts to update readers regarding the information available on these two aspects to date.
DIAGNOSIS OF HCV INFECTION
During HCV infection, every attempt is made to diagnose and differentiate acute from chronic hepatitis C infection. Acute HCV infection is typically mild. It is often not diagnosed, and the infection may be recognized only when it becomes chronic[21]. The diagnostic tests used, including the presence of anti-HCV antibodies in serum, cannot differentiate between acute and chronic HCV infection because anti-HCV IgM, used as marker of acute infection, is variable in acute infectious disease and is also detected at high rates in patients with chronic HCV infection[22,23]. The diagnostic procedures for hepatitis C virus infection used in laboratories are based on the detection of anti-HCV antibodies against recombinant HCV proteins using enzyme immunoassay (EIA) and chemiluminescence immunoassay. Non-structural and recombinant antigens are used in these assays. Four different generations of anti-HCV test kits have been developed to date. The first generation EIA detected antibodies against the nonstructural proteins (NS4) with recombinant antigen c100-3. Subsequently, the second generation assay was developed and this included antigens from the core region (c22-3), the NS3 region (c33c) and a part of c100-3 (5-1-1) from the NS4 region. The third-generation EIA included an additional antigen from the NS5 region and a reconfiguration of the core and NS3 antigens. However, all these anti-HCV assays had the disadvantages of giving high false positive results and a lack of sensitivity to detect antibodies during the window period. In addition, these antibody-based assays could not distinguish between acute, past and chronic infections. This was followed by the development of supplementary tests involving the recombinant immunoblot assay (RIBA) which was commercialized. This assay contained recombinant antigen (c33c, NS5) and synthetic peptides (5-1-1, c100 and c22). Similarly, a few other commercial assays, known as third generation immunoassays incorporated HCV antigens from the core region, E2 hypervariable region, NS3 region, NS4A, NS4B and NS5A region. All these recombinant immunoblot assays were used as supplementary tests to the anti-HCV assays. Similar to EIA, the RIBA had the disadvantages of difficulty in performance and a high percentage of indeterminate results. Therefore, these are no longer used in diagnostic laboratories. Recently, fourth generation anti-HCV assays incorporating additional nonstructural proteins are being used as screening tests[24]. These kits for anti-HCV detection target different HCV antigens and detect more than five primary antibodies to ensure the specificity and sensitivity of the detection kit.
Anti-C22c and anti-C33c may be the first HCV antibodies to appear during the acute phase of the disease, which is defined by elevated alanine aminotransferase (ALT) levels and/or clinical symptoms[25]. Anti-NS5 appears somewhat later, while anti-C100-3 is the last antibody to be detected in acute self-limited HCV infection. The diagnosis and differentiation of acute from chronic HCV infection poses another problem. Patients chronically infected with one HCV-genotype develop acute hepatitis on infection with another genotype. Multiple episodes of acute hepatitis were observed in polytransfused thalassemic children reinfected with different HCV genotypes[26,27]. Therefore, discrimination between acute and chronic infection in the same patient is sometimes very difficult. HCV RNA in the serum or liver appears to be the earliest detectable marker of acute HCV infection, preceding the appearance of anti-HCV by several weeks[25]. HCV viremia may persist despite the normalization of serum ALT levels. Thus, the use of ALT levels in the diagnosis of HCV is not helpful. However, HCV RNA in serum usually lasts for fewer than 4 mo in patients with acute self-limited HCV infection. The average time from transfusion to sero-conversion is approximately 11 to 12 wk with EIA-1 (Enzyme immunoassay-1) and 7 to 8 wk with EIA-2 (Enzyme immunoassay-2). Now attempts are being made to develop EIA assays to differentiate HCV sub-types[28]. Patients with post-transfusion chronic non-A, non-B hepatitis develop anti-HCV antibodies in the majority of cases. Anti-HCV antibodies are not neutralizing, especially with HCV envelope proteins E1 and E2[29]. High levels of anti-C100-3 were correlated with high titers of circulating HCV in chimpanzees[30]. Therefore, the development and persistence of diagnostic antibodies to HCV seem to reflect concomitant virus replication and consequently a high potential for infectivity.
HCV RNA is frequently detected in patients with chronic hepatitis C and in patients carrying anti-HCV antibodies. A study carried out in Hong Kong demonstrated that 83% of anti-HCV positive patients were viremic when HCV RNA was determined using polymerase chain reaction (PCR) with two different sets of primers for noncoding regions[27]. Similarly, in another study, 98 of 100 patients with chronic non-A, non-B liver disease were positive for antibodies by EIA-2, but all 100 patients were positive for HCV RNA by PCR. With the currently available EIA systems, chronic HCV infection can readily be identified in most patients. Measurement of HCV RNA by PCR does not substantially increase the numbers of patients found to have chronic HCV infection[31]. Following the introduction and wider use of real-time PCR, it is now easier to diagnose and monitor the progress of HCV viremia in a very short time period[32]. In addition, the use of multiplex PCR by real time is another advancement in the detection of possible hepatitis viral co-infections in single attempt analysis[33].
Based on published information regarding various aspects of HCV infection including the currently available diagnostic assays and therapeutic regimens, the American Association for the Study of Liver Diseases and Centers for Disease Control and Prevention, United States have approved a document as “practice guidelines” for use in the diagnosis and treatment of HCV infection. This is an important document and describes details of the guidelines to be followed for laboratory diagnosis of acute/chronic HCV infection[34].
PATHOGENESIS OF HCV INFECTION
HCV is a non-cytopathic virus[35] that enters the liver cell and undergoes replication simultaneously causing cell necrosis by several mechanisms including immune-mediated cytolysis in addition to various other phenomena such as hepatic steatosis, oxidative stress and insulin resistance. The proteins/peptides encoded by different sub-genomic regions of the HCV genome and their quasispecies influence the above mechanism, and thus, have a significant role in HCV pathogenesis and disease causation. A brief description of HCV pathogenesis in the light of these factors is given in the following section (Figure 2).
Figure 2.
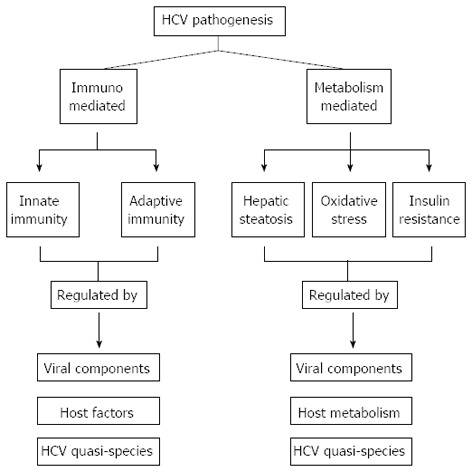
Regulation of hepatitis C virus pathogenesis by host immunity and metabolic factors. HCV: Hepatitis C virus.
Viral entry
HCV is a blood-transmitted virus that reaches the liver via circulation. The entry of HCV isolates requires at least 4 host-derived factors including scavenger receptor class B type I, Occludin, Claudin-I (CLDNI) and CD81. In addition, CLDN6 and CLDN9 have been shown to substitute for CLDN1 as HCV entry factors in human non-liver cells[36]. The CD81 molecule on host cell surfaces acts as a viral receptor, which binds with the viral particle and facilitates its entry in the liver cell[37,38]. CD81 is expressed on the surface of almost all nucleated cells and complexes with a variety of other cell-surface receptors such as CD19 and CD21 on B cells, and sends a costimulatory signal to the cells[39]. The viral envelop protein, E2, binds to the major extracellular loop of CD8[40]. HCV shows multi-site binding and can also bind to several other molecules such as the receptor for low-density lipoprotein, the dendritic cell (DC)-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN), and its liver counterpart[41,42]. E2 is the most variable viral protein, and therefore, its interactions with CD81 have been reported to be strain-specific[43]. It has two hyper variable regions, HVR-1 and HVR-2 which undergo frequent mutations, possibly due to virus-neutralizing antibodies and HCV-specific cytolytic T lymphocytes (CTLs). HCV also has a high mutation rate due to the lack of proofreading ability of its RNA-dependent RNA polymerase. Therefore, HCV exists in several distinct, but closely related virus species within an infected individual. These species are called HCV quasispecies.
HOST IMMUNITY
Innate immunity
Innate immunity presents a first line defense for the control of HCV infection as it does for several other viral infections. During HCV infection, cells produce Type 1 IFN which prepares and induces the cells to resist infection, check viral replication, promote adaptive immunity and activate natural killer (NK) cells, DCs and Kupffer cells etc. Once inside the cell, the innate immunity vs HCV is triggered through host recognition of viral macromolecular motifs, known as pathogen-associated molecular patterns (PAMPs), as non-self by cellular pathogen recognition receptors. These receptors includes toll-like receptors (TLRs) and retinoic acid-inducible gene-I (RIG-I) like receptors (RLRs)[44]. RIG-I binds PAMP on HCV-RNA and activates interferon regulatory factor-3 (IRF-3) for expression of IFN-α/β and anti-viral/interferon stimulated genes (ISGs)[45]. The secreted IFN and cytokines then activate NKs, DCs and Kupffer cells etc. These cells also play a significant role in mounting T/B cell-based immunity[46]. The PAMP region lies on the 3’ untranslated region (UTR) of HCV and induces RIG-1 signaling[47] that results in a RIG-1 interaction with IFN-β promoter stimulator (IPS-1) which causes activation of IRF-3 and nuclear factor κB (NFκB).
HCV can effectively evade innate immunity resulting in persistent viral infection. This occurs because HCV has evolved to counteract the RIG-1 pathway[48] and thus evade the immune challenge. This phenomenon is the reason for chronicity in the majority of HCV infected patients. For this, the non-structural proteins of HCV i.e., NS3 and NS4A form a complex which activates the NS protease domain to target cleavage of IPS-1. After cleavage, IPS-1 can no longer signal downstream to activate IRF-3 and NFκB and the infected cells no longer produce IFN-β or express ISGs[49].
NK cells, a major arm of innate immunity, play an important role in eradication of HCV. The liver is enriched in NK cells that are usually activated in an early phase of HCV infection. The activated NK cells recruit virus-specific T cells and induce antiviral immunity in the liver. They also eliminate virus-infected hepatocytes directly by cytolytic mechanisms and indirectly by secreting cytokines including IFN-γ and TNF-α. These cytokines induce an antiviral state in host cells. Surprisingly, HCV has evolved multiple strategies to counter the host’s NK cell response. It is interesting that activated NK cells contribute toward liver injury, while inactive or compromised NK cells permit the virus to continue invasion[50].
Adaptive immunity
After entry and replication of the virus inside liver cells, the viral molecules are transported to the endoplasmic reticulum and associate with major histocompatibility complex (MHC) molecules, which are finally transported to the cell surface. These molecules on the cell surface are recognized by T cells for their immune action. The majority of CTLs are CD8+ and recognize antigens presented on MHC class I molecules. Approximately 10% of CTLs are CD4+ which recognize antigens presented on MHC II molecules. These CTLs eliminate cells infected with virus. However, HCV is reported to have evolved mechanisms to avoid recognition by CTLs. They either reduce the expression of MHC molecules or prevent the viral peptide from presentation at the cell surface. Thus, CTLs play a major role in viral eradication[51] and immunopathogenesis of HCV infection[52].
In another pathway of the disease mechanism, the destruction of HCV-infected hepatocytes release HCV fragments that are taken up by myeloid DCs. These DCs migrate to the draining lymph nodes and express HCV antigens on HLA class II molecules. Subsequently, they increase expression of costimulatory molecules (CD80, CD86) which interact with and activate antigen-specific helper T (Th) cells[53]. These activated Th cells promote the maturation of DCs and increase the expression of CD40 ligand and TNF-α. The mature DCs induce T-cell activation by overexpression of their surface molecules. They also enhance antigen presentation capacity via HLA-I and production of cytokines that stimulate T-cell activation. IL-12 has been shown to play an important role in stimulating IFN-γ production from activated T cells[54,55], and thus, induces development of the type 1 (Th1) immune response characteristic of CTL activation. The effector CTLs release perforin, granzyme, and TNF-α, or express Fas ligand, and initiate a direct attack on HCV-infected hepatocytes[56,57].
The hepatocytes infected with HCV and DCs produce Type I IFNs which suppress viral replication by inducing enzymes such as 2’-5’ oligoadenylate synthetase (OAS) and RNA-dependent protein kinase (PKR) in hepatocytes[58]. The plasmacytoid DC recognizes HCV-related markers through TLR-7, which interacts with single-stranded RNA[59]. The TLR-signaling up-regulates PDC-triggering receptor expressed on myeloid cells (PDC-TREM) which induce further production of IFN-α[60]. Activated OAS destroys viral RNAs, whereas PKR inhibits forming polysomes of viral mRNA[58]. When HCV-specific CTL responses are not strong enough to eradicate the virus this leads to persistent infection[61].
Successful clearance of HCV during acute HCV infection depends on the rise, vigor and persistence of the Th1 immune response[62,63]. Patients who developed a strong Th1 response showed efficient viral clearance and a self-limited disease course. In contrast, those who lacked IL-12 and IFN-γ production invariably developed chronic persistence of the virus. The majority of patients fail to control the infection and develop a chronic infection with a variable degree of hepatitis and viremia[64,65]. Experimental studies have also demonstrated that HCV components induce an antigen processing mechanism and IFN-stimulated genes in infected livers[66-68]. Impaired function of DCs, as antigen-presenting cells in inducing immunity, may be responsible for the impaired immune responses. Various studies have reported that viral proteins including HCV core, E1, and NS3 inhibit DC maturation[69,70]. HCV infects DCs through the binding of HCV E2 protein and thereby suppress DC function in promoting an antiviral effect[41,71].
CTLs activated by viral proteins, not only kill virus-infected cells, but also contribute to virus control through a noncytolytic mechanism by secreting cytokines, e.g., IFN-γ, IFN-α/β and TNF-α. These cytokines induce an antiviral state in host cells. This also renders uninfected cells resistant to infection and prevents viral replication. The progression of the majority of infected persons to chronic infection suggests inability of the antiviral immunity to contain this infection. There may be several reasons for this failure, including the emergence of escape variants as a result of a high rate of virus mutations, decreased production of antiviral cytokines or “stunning” of HCV-specific CTLs, a compromised cytolytic potential of the CTLs and antagonistic peptides[72].
It is important to note here that the HCV genome in a single host is a dynamic population of different, but closely related genomes, designated quasispecies. The generation of quasispecies is usually ascribed to high variation in hyper variable region-1 (HVR-1) during viral replication[73]. In acute resolving hepatitis, HVR-1 shows very little variation, as compared to that in chronic hepatitis[74]. HVR-1 induces anti-HCV neutralizing antibodies[75,76] and HVR-1 specific CD4+ and CD8+ T cells[77,78]. Using the responding host cellular immune response differentially, HVR-1 favors viral escape[79,80]. HVR-1 variations result from the action of a continuous immune-driven positive selection[81,82]. Thus, HVR-1 complexity helps in the virus adaptive strategy to escape the immune onset. HCV clearance is associated with a vigorous HCV specific CD4+ and CD8+ T cell response in the acute phase of infection. In contrast, viral persistence is associated with a weak and dysfunctional virus specific T cell response[79-83]. T cell failure and HCV immune evasion have been explained in several reports[84-86].
Role of T regulatory cells in adaptive immunity
Recent studies have suggested a possible role for different regulatory T cell populations in HCV persistence. These studies showed a higher frequency of CD4+CD25+ regulatory T cells in the blood and CD4+FoxP3+ T cells in the liver of chronically HCV infected patients[87-89]. CD4+CD25+ regulatory T cells suppress HCV specific CD8+ T cell and CD4+ T cell proliferation as well as CD8+ T cell IFN-γ secretion[87,90-92]. After HCV antigen stimulation, Treg cells secrete IL-10 and transforming growth factor-β (TGF-β) which suppress virus specific T cell responses[91-93]. CD4+CD25+ Treg cells obtained from chronically HCV infected patients demonstrated greater suppressive activity against HCV specific CD8+ T cells compared to Treg cells isolated from acute HCV infected patients. However, the suppressive effect observed in patients who successfully cleared the virus was still significant[90]. Another study showed that the frequency of CD4+CD25+FoxP3+ Treg cells and their suppressive capacity against virus specific T cell responses were as high in HCV recovered chimpanzees as those in persistently HCV infected chimpanzees[94]. This observation requires further in-depth studies to explore the actual suppressive effect of Treg cells during HCV infection. Induction of Treg cells by HCV antigens was first demonstrated by the response of CD4+ T cell to HCV core protein. HCV-specific IL-10 secreting T cells were detected in the blood of chronic HCV infected persons[95]. Regulatory CD8+ T cells may play an important role in chronic HCV infection. HCV-specific CD8+CD25+FoxP3+ T cells from the blood of chronically infected patients suppress HCV-specific T cell responses via TGF-β secretion. The blockade of TGF-β markedly enhanced HCV specific IFN-γ secretion by CD4+ and CD8+ T cells[96].
Few other studies have shown that chronic HCV infection results in exhaustion or impairment of HCV-specific CD8+ T cells. During chronic HCV infection, CD8+ T cells fail to proliferate or secrete antiviral cytokines including IFN-γ. This phenomenon is promoted by a lack of CD4+ T cells and the expression of immunomodulatory cytokines such as IL-10[97]. The major cause of HCV-specific CD8+ T cell impairment is ascribed to the expression of inhibitory receptors such as Programmed death-1, lymphocyte-activation gene-3 (a protein related to CD4), CTLA-4 (a member of the CD28 receptor family), T-cell immunoglobulin mucin-3 and 2B4 on HCV-specific CD8+ T cells in blood and liver[98]. Expression of these inhibitory receptors is associated with low levels of CD127 expression and impaired proliferation and differentiation of T cells. Thus, different mechanisms contribute to the dysfunction of HCV-specific CD8+ T cells in chronic HCV infection.
In addition to cytotoxic T lymphocytes, humoral immune response against viral and cellular components during HCV infection is also present. Patients positive for HCV RNA and/or anti-HCV antibodies have type I anti-liver kidney microsome antibodies, which also recognize cytochrome P450 (CYP) 2D6. The patient’s liver is infiltrated with auto reactive mononuclear cells, which recognize CYP2D6. It is interesting that the viral core protein residues 178-187 bear sequence homology with human cytochrome P450 (CYP2A6 and CYP2A7) residues 8-17[96]. Although HCV is a hepatotropic virus and infects hepatocytes, viral genome and its replicative intermediates are frequently present in peripheral blood mononuclear cells and lymphoid tissues of chronically infected persons. The viral glycoprotein E2 has been implicated in the oligoclonal expansion of several lymphoma cells[99]. The most common rheumatic and cutaneomucous symptoms in HCV-infected patients include fatigue, arthralgia, paraestheisa, myalgia, pruritus, and the sicca syndrome[100].
ROLE OF VIRAL PROTEINS AND GENOTYPES
The role of structural and non-structural components of the HCV virion has been explained by variation in their interactions with metabolites affecting pathogenic pathways leading to liver damage. HCV-core protein has a prominent role in all these interactions as compared to envelope and non-structural proteins. Moreover, when the mechanism of this interaction was studied in relation to various HCV genotypes, it was observed that different genotypes behave differently to regulate all these pathogenic pathways.
The role of NS5A and E2 region was found be important. NS5A has a role in viral replication, inactivating PKR[101-104], blocking the apoptotic pathway, binding of growth factor receptor-bound protein 2[105,106] and induction of anti-inflammatory interleukin secretion[107,108]. Similarly, E2 protein inhibits PKR[109,110]. The region of NS5A which interacts with PKR, shows clustering of amino acid changes during IFN treatment and plays an important role in the evasion mechanism[111]. Furthermore, this association varies with genotype and thus, alters their sensitivity to IFN treatment. NS5A remains under strong immune selection, has T- and B-cell epitopes and possibly, in combination with individuals’ HLA, selects immune cells to produce sensitivity/resistance to IFN therapy[112]. The functional activity of NS5A towards immune selection is clearly governed by the HCV-genotypes and varies accordingly. The response of genotype 2 and 3 to IFN treatment may be due to individuals recognizing the NS5A protein immunologically[13].
Binding of HCV E2 protein to DCs induces their maturation. Several HCV viral proteins, including core, NS3, NS5A and NS5B proteins, have been shown to inhibit DC functions[69]. Consequently, the functions of both CD4+ Th cells and CD8+ CTLs are impaired in chronic HCV patients. This has been suggested to be one of the mechanisms that HCV utilizes to weaken host immune responses and spread the infection. Indeed, many clinical studies have shown that in chronic HCV patients, not only the functions of DCs are impaired[113,114], the functions of both CD4+ and CD8+ T cells are also impaired[115]. A similar inductive effect of E2 protein was also reported in other cell types, including T cells, B cells[116], hepatocytes[117] and hepatic stellate cells[118].
The role of HCV genotypes in the progression of liver disease is one of the most controversial areas of HCV research. In patients with chronic HCV, infection with genotype-1b is reportedly associated with a more severe liver disease and a more aggressive course than the infection with other HCV genotypes. Similarly, it was found that HCV genotype-1b was significantly more prevalent among patients with liver cirrhosis and those with decompensated liver disease requiring liver transplantation than among those with chronic active hepatitis C[119-121]. Although this is indirect evidence, it suggests an association between HCV genotype-1b and the development of these complications. HCV genotype-1b is a marker for more severe HCV associated liver disease, because it reflects a longer time of infection than a mere aggressive form of hepatitis C.
METABOLIC CONDITIONS AFFECTING HCV PATHOGENESIS
In addition to immune mediated HCV pathogenesis, there are several other clinical and metabolic conditions that have a strong association with HCV pathogenesis. These include HCV-induced insulin resistance, oxidative stress and hepatic steatosis. The following is a brief description of the conditions affecting HCV pathogens:
HCV-induced insulin resistance
HCV infection influences overall metabolism leading to increased steatosis, fibrosis, inflammation, apoptosis and insulin resistance (IR)[122,123] during the course of the disease. The resulting IR shows a modulating impact on liver pathogenesis by HCV infection[124]. IR increases the de novo lipogenesis i.e., fatty acid (FA) synthesis via overexpression and maturation of SREBP-1c. This in turn, increases the activities of lipogenic enzymes including Acetyl CoA carboxylase and FA synthase. At the same time, intermediates of triglyceride biosynthesis also activate inhibitors of insulin signaling. For example, activation of protein kinase C-E by phosphorylating insulin receptor substrate, and thus inhibiting phosphatidyl inositol-3,4,5-triphosphate[125], inhibits Akt translocation by ceramides etc.[126]. HCV-core protein, either directly or via increased secretion of TNF-α, causes IR[127,128]. The HCV core can activate inhibitors of insulin signaling including mammalian target of rapamycin[129] and suppressor of cytokine signaling (SOCS)-3 and C-Jun N-terminal kinase (JNK)[130,131]. The activation of JNK by HCV core may follow a direct or indirect proinflammatory cytokine-mediated mechanism.
HCV-associated oxidative stress
Oxidative stress is reported to be an important part of HCV-induced liver damage. Previous studies investigated the role of different molecular components of HCV structure in modulating oxidative stress during HCV infection. HCV-core protein present within the outer membrane of mitochondria induces oxidation of glutathione and promotes Ca2+ uptake into mitochondria. Clément et al[96] explained the molecular mechanism and demonstrated that following glutathione oxidation, there is increased reactive oxygen species (ROS) production by mitochondrial electron transport complex I and III. The HCV non-structural protein, NS5A, promotes ROS production in the membrane of endoplasmic reticulum (ER) by activating the release of Ca2+ from ER, thereby inducing oxidative stress[97]. NS3 protein induces ROS production by activation of NADPH oxidase[97]. Increased ROS production and consequent oxidative stress is evident by the presence of markers of increased oxidative stress in the blood. Levels of 8-hydroxy deoxyguanosine and 4-hydroxy-2-nonenol are increased in HCV infection[132,133]. Similarly, few studies have shown reduced levels of glutathione during HCV infection. Another study showed that the serum level of thioredoxin, a marker of oxidative stress, was significantly reduced in HCV infection[134-136].
The presence of oxidative stress has been noted in different types of hepatitis including hepatitis B. However, there is a marked increase in oxidative stress (OS) in HCV infection[132]. Several studies have shown that structural components of HCV induce effective OS[132]. HCV-core and non-structural components, NS3 and NS5A proteins, directly induce OS[137-139]. Core protein is involved in OS generation via oxidation of mitochondrial glutathione and uptake of Ca2+ into mitochondria[139,140] thus, changing the permeability of its membrane[141]. Electron transport complex I increases production of ROS and redistributes cytochrome from mitochondria to the cytosolic fraction[93]. NS5A is associated with the ER membrane[142] and activates signal transducer transcription and NFκB[107]. These activations lead to inflammation, immune response and apoptosis[143]. Similarly, NS3 triggers ROS by activating NADPH oxidase 2 in mononuclear and polymorphonuclear phagocytes[144] which increase apoptosis of hepatocytes[144]. All these reports conclude that the structural and non-structural components of HCV induce a significant increase in OS that results in liver damage during HCV infection.
HCV-induced steatosis
HCV infection is reported to have a strong association with hepatic steatosis. There are several other factors also responsible for steatosis, which include alcohol consumption, obesity, and diabetes[145-147]. Studies on steatosis in relation to hepatotropic viruses demonstrated that HCV infection directly causes steatosis in some patients[148]. Studies in experimental animals have shown that HCV-core protein promotes liver steatosis[149,150]. Furthermore, when steatosis was studied in relation to HCV-genotypes, it was noted that although steatosis is induced by all HCV-genotypes, it appears more prominent and frequent with HCV-genotype 3 infection[151-153]. In patients carrying genotype-3 infection, there was a good correlation between the level of steatosis and HCV replication[153,154] and the presence of HCV-core in the liver. In addition, steatosis resolves in patient with genotype-3 when treated successfully with anti-viral therapy as compared to those with non-genotype-3 who remain steatotic[155,156]. Steatosis reappears with relapse of infection[155]. This clearly demonstrates that some HCV-genotypes have more steatogenic potential. Subsequent studies[157] indicated that genotype-3 interferes with very low-density lipoprotein (VLDL) secretion. Core protein, which promotes lipid accumulation in hepatocytes[158,159], was more efficient from genotype-3 compared to core protein from genotype-1.
All these reports concluded that HCV causes steatosis in three different ways: (1) Impaired secretion of lipids from hepatocytes; (2) Increased de novo synthesis of free fatty acids (FFAs); and (3) Impaired FA degradation. The first aspect of HCV-induced steatosis was proposed due to the impaired secretion of VLDL. To substantiate this, reports from different studies demonstrated a decreased level of apolipoprotein B and cholesterol in chronic HCV infected patients[159,160]. These low levels pointed to HCV disturbing the assembly and secretion of VLDL from the liver[161]. Another important aspect in this relationship was increased de novo synthesis of FFAs in the presence of HCV infection. In this context, it is suggested that HCV upregulated the sterol regulatory element binding protein-1c (SREBP-1c) signaling pathway[158] with NS2 and NS4B proteins inducing SREBP at the transcriptional level[162,163]. SREBP was also induced by expression of HCV core protein. Studies in chimpanzees infected with HCV also demonstrated that HCV increased the activity of lipogenic enzymes such as ATP citrate lyase[164]. HCV-core, in particular, activates and helps in cellular lipid synthesis[164], possibly via its binding with retinoid receptor.
HCV-induced steatosis is also due to impaired FA degradation by HCV. Expression of HCV-core protein is reported to reduce the expression of peroxisome proliferation activated receptor-α (PPARα), a nuclear receptor involved in FA degradation and down-regulation of mitochondria β-oxidation[165]. Genotype-3 shows significant down-regulation of PPARα as compared to genotype-1[166,167]. HCV-core protein also down-regulates PPARα and therefore, is more effective when from genotype-3 as compared to genotype-1. The core protein from genotype-3 also down-regulated the PPARγ and up-regulated SOCS-7 in human hepatoma cells[167]. These data clearly show that HCV-core protein may modulate the expression of various genes responsible for FA degradation via down-regulation of PPARs.
CONCLUSION
HCV infection, previously known as blood borne non-A, non-B infection, is a serious public health problem worldwide. The diagnosis of HCV is based on the detection of anti-HCV antibodies and/or viral nucleic acid in serum. Studies over the last few years have developed assays not only for the accurate serodiagnosis of infection, but also identification of HCV serotypes. The pathogenesis of HCV infection is quite complex and regulated by host immunity as well as several metabolic activities influencing liver function. Whereas both innate and adaptive immunity are involved in the pathogenic action of HCV, the cytotoxic lymphocytes are crucial in deciding the eradication or persistence of viral particles. Moreover, the persistence of HCV infection is also affected by viral proteins, HCV isotypes and liver metabolism. In order to understand HCV pathogenesis further investigations are needed.
ACKNOWLEDGMENTS
We thank and appreciate the financial aid provided by ICMR, New Delhi, India to conduct this study. We are also thankful to Mrs. Suman Rawat for preparing this manuscript.
Footnotes
P- Reviewer: Wang Y S- Editor: Ma YJ L- Editor: Webster JR E- Editor: Zhang DN
References
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, Miyamura T, Dienstag JL, Alter MJ, Stevens CE. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 3.Seeff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36:S35–S46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 4.Pawlotsky JM. The nature of interferon-alpha resistance in hepatitis C virus infection. Curr Opin Infect Dis. 2003;16:587–592. doi: 10.1097/00001432-200312000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Testino G, Sumberaz A, Leone S, Borro P. Recurrent hepatitis C and non-alcoholic fatty liver disease in transplanted patients: a review. Minerva Med. 2013;104:225–232. [PubMed] [Google Scholar]
- 6.Lindenbach BD, Rice CM. Flaviviridae: the viruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology. Philadelphia: Lippubcott Williams and Wilkins; 2001. p. 991–1041. [Google Scholar]
- 7.Simmonds P. Variability of hepatitis C virus. Hepatology. 1995;21:570–583. doi: 10.1002/hep.1840210243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamane D, McGivern DR, Masaki T, Lemon SM. Liver injury and disease pathogenesis in chronic hepatitis C. Curr Top Microbiol Immunol. 2013;369:263–288. doi: 10.1007/978-3-642-27340-7_11. [DOI] [PubMed] [Google Scholar]
- 9.Irshad M, Ansari MA, Singh A, Nag P, Raghvendra L, Singh S, Badhal SS. HCV-genotypes: a review on their origin, global status, assay system, pathogenecity and response to treatment. Hepatogastroenterology. 2010;57:1529–1538. [PubMed] [Google Scholar]
- 10.Das BR, Kundu B, Khandapkar R, Sahni S. Geographical distribution of hepatitis C virus genotypes in India. Indian J Pathol Microbiol. 2002;45:323–328. [PubMed] [Google Scholar]
- 11.Hissar SS, Goyal A, Kumar M, Pandey C, Suneetha PV, Sood A, Midha V, Sakhuja P, Malhotra V, Sarin SK. Hepatitis C virus genotype 3 predominates in North and Central India and is associated with significant histopathologic liver disease. J Med Virol. 2006;78:452–458. doi: 10.1002/jmv.20561. [DOI] [PubMed] [Google Scholar]
- 12.Irshad M, Acharya SK, Joshi YK. Prevalence of hepatitis C virus antibodies in the general population & amp; in selected groups of patients in Delhi. Indian J Med Res. 1995;102:162–164. [PubMed] [Google Scholar]
- 13.Okamoto H, Kojima M, Okada S, Yoshizawa H, Iizuka H, Tanaka T, Muchmore EE, Peterson DA, Ito Y, Mishiro S. Genetic drift of hepatitis C virus during an 8.2-year infection in a chimpanzee: variability and stability. Virology. 1992;190:894–899. doi: 10.1016/0042-6822(92)90933-g. [DOI] [PubMed] [Google Scholar]
- 14.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Häussinger D, Diago M, Carosi G, Dhumeaux D, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 15.Pawlotsky JM. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology. 2011;53:1742–1751. doi: 10.1002/hep.24262. [DOI] [PubMed] [Google Scholar]
- 16.Huang Y, Yang H, Borg BB, Su X, Rhodes SL, Yang K, Tong X, Tang G, Howell CD, Rosen HR, et al. A functional SNP of interferon-gamma gene is important for interferon-alpha-induced and spontaneous recovery from hepatitis C virus infection. Proc Natl Acad Sci USA. 2007;104:985–990. doi: 10.1073/pnas.0609954104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naito M, Matsui A, Inao M, Nagoshi S, Nagano M, Ito N, Egashira T, Hashimoto M, Mishiro S, Mochida S, et al. SNPs in the promoter region of the osteopontin gene as a marker predicting the efficacy of interferon-based therapies in patients with chronic hepatitis C. J Gastroenterol. 2005;40:381–388. doi: 10.1007/s00535-005-1558-3. [DOI] [PubMed] [Google Scholar]
- 18.Lauer GM. Immune responses to hepatitis C virus (HCV) infection and the prospects for an effective HCV vaccine or immunotherapies. J Infect Dis. 2013;207 Suppl 1:S7–S12. doi: 10.1093/infdis/jis762. [DOI] [PubMed] [Google Scholar]
- 19.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 20.Abstracts of the 12th International Symposium on Viral Hepatitis and Liver Disease, Paris, France, July 1-5, 2006. J Clin Virol. 2006;36 Suppl 2:S1–218. [PubMed] [Google Scholar]
- 21.Ndimbie OK, Nedjar S, Kingsley L, Riddle P, Rinaldo C. Long-term serologic follow-up of hepatitis C virus-seropositive homosexual men. Clin Diagn Lab Immunol. 1995;2:219–224. doi: 10.1128/cdli.2.2.219-224.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farci P, Alter HJ, Govindarajan S, Wong DC, Engle R, Lesniewski RR, Mushahwar IK, Desai SM, Miller RH, Ogata N. Lack of protective immunity against reinfection with hepatitis C virus. Science. 1992;258:135–140. doi: 10.1126/science.1279801. [DOI] [PubMed] [Google Scholar]
- 23.Yuki N, Hayashi N, Ohkawa K, Hagiwara H, Oshita M, Katayama K, Sasaki Y, Kasahara A, Fusamoto H, Kamada T. The significance of immunoglobulin M antibody response to hepatitis C virus core protein in patients with chronic hepatitis C. Hepatology. 1995;22:402–406. [PubMed] [Google Scholar]
- 24.Kesli R. An Overview of the Laboratory Assay Systems and Reactives Used in the Diagnosis of Hepatitis C Virus (HCV) Infections. In: Abuelzein E, editor. Trends in Immunolabelled and Related Techniques. Rijeka: InTech; 2012. pp. 340–350. [Google Scholar]
- 25.Kato N, Yokosuka O, Hosoda K, Ito Y, Ohto M, Omata M. Detection of hepatitis C virus RNA in acute non-A, non-B hepatitis as an early diagnostic tool. Biochem Biophys Res Commun. 1993;192:800–807. doi: 10.1006/bbrc.1993.1485. [DOI] [PubMed] [Google Scholar]
- 26.Kao JH, Chen PJ, Lai MY, Chen DS. Superinfection of heterologous hepatitis C virus in a patient with chronic type C hepatitis. Gastroenterology. 1993;105:583–587. doi: 10.1016/0016-5085(93)90737-w. [DOI] [PubMed] [Google Scholar]
- 27.Lai ME, Mazzoleni AP, Argiolu F, De Virgilis S, Balestrieri A, Purcell RH, Cao A, Farci P. Hepatitis C virus in multiple episodes of acute hepatitis in polytransfused thalassaemic children. Lancet. 1994;343:388–390. doi: 10.1016/s0140-6736(94)91224-6. [DOI] [PubMed] [Google Scholar]
- 28.Ansari MA, Irshad M, Agarwal SK, Chosdol K. Expression of the full-length HCV core subgenome from HCV gentoype-1a and genotype-3a and evaluation of the antigenicity of translational products. Eur J Gastroenterol Hepatol. 2013;25:806–813. doi: 10.1097/MEG.0b013e32835eb9b9. [DOI] [PubMed] [Google Scholar]
- 29.Alter HJ, Purcell RH, Shih JW, Melpolder JC, Houghton M, Choo QL, Kuo G. Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A, non-B hepatitis. N Engl J Med. 1989;321:1494–1500. doi: 10.1056/NEJM198911303212202. [DOI] [PubMed] [Google Scholar]
- 30.Chien DY, Choo QL, Ralston R, Spaete R, Tong M, Houghton M, Kuo G. Persistence of HCV despite antibodies to both putative envelope glycoproteins. Lancet. 1993;342:933. doi: 10.1016/0140-6736(93)91983-s. [DOI] [PubMed] [Google Scholar]
- 31.Chemello L, Cavalletto D, Pontisso P, Bortolotti F, Donada C, Donadon V, Frezza M, Casarin P, Alberti A. Patterns of antibodies to hepatitis C virus in patients with chronic non-A, non-B hepatitis and their relationship to viral replication and liver disease. Hepatology. 1993;17:179–182. [PubMed] [Google Scholar]
- 32.Irshad M, Ansari MA, I K, L R. A Novel Single Step Multiplex Real Time Pcr Assay for Simultaneous Quantification of Hepatitis Virus A, B, C & E in Serum. J Gastroenterol Hepatol. 2013:Epub ahead of print. doi: 10.1111/jgh.12302. [DOI] [PubMed] [Google Scholar]
- 33.Yang JH, Lai JP, Douglas SD, Metzger D, Zhu XH, Ho WZ. Real-time RT-PCR for quantitation of hepatitis C virus RNA. J Virol Methods. 2002;102:119–128. doi: 10.1016/s0166-0934(02)00007-1. [DOI] [PubMed] [Google Scholar]
- 34.Ghany MG, Strader DB, Thomas DL, Seeff LB; American Association for the Study of Liver Diseases. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology. 2009;49:1335–1374. doi: 10.1002/hep.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irshad M, Dhar I. Hepatitis C virus core protein: an update on its molecular biology, cellular functions and clinical implications. Med Princ Pract. 2006;15:405–416. doi: 10.1159/000095485. [DOI] [PubMed] [Google Scholar]
- 36.Haid S, Grethe C, Dill MT, Heim M, Kaderali L, Pietschmann T. Isolate-dependent use of Claudins for cell entry by hepatitis C virus. Hepatology. 2013:Epub ahead of print. doi: 10.1002/hep.26567. [DOI] [PubMed] [Google Scholar]
- 37.Masciopinto F, Freer G, Burgio VL, Levy S, Galli-Stampino L, Bendinelli M, Houghton M, Abrignani S, Uematsu Y. Expression of human CD81 in transgenic mice does not confer susceptibility to hepatitis C virus infection. Virology. 2002;304:187–196. doi: 10.1006/viro.2002.1631. [DOI] [PubMed] [Google Scholar]
- 38.Zeisel MB, Felmlee DJ, Baumert TF. Hepatitis C virus entry. Curr Top Microbiol Immunol. 2013;369:87–112. doi: 10.1007/978-3-642-27340-7_4. [DOI] [PubMed] [Google Scholar]
- 39.Maecker HT, Todd SC, Levy S. The tetraspanin superfamily: molecular facilitators. FASEB J. 1997;11:428–442. [PubMed] [Google Scholar]
- 40.Flint M, Maidens C, Loomis-Price LD, Shotton C, Dubuisson J, Monk P, Higginbottom A, Levy S, McKeating JA. Characterization of hepatitis C virus E2 glycoprotein interaction with a putative cellular receptor, CD81. J Virol. 1999;73:6235–6244. doi: 10.1128/jvi.73.8.6235-6244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lozach PY, Lortat-Jacob H, de Lacroix de Lavalette A, Staropoli I, Foung S, Amara A, Houles C, Fieschi F, Schwartz O, Virelizier JL, et al. DC-SIGN and L-SIGN are high affinity binding receptors for hepatitis C virus glycoprotein E2. J Biol Chem. 2003;278:20358–20366. doi: 10.1074/jbc.M301284200. [DOI] [PubMed] [Google Scholar]
- 42.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 2002;21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roccasecca R, Ansuini H, Vitelli A, Meola A, Scarselli E, Acali S, Pezzanera M, Ercole BB, McKeating J, Yagnik A, et al. Binding of the hepatitis C virus E2 glycoprotein to CD81 is strain specific and is modulated by a complex interplay between hypervariable regions 1 and 2. J Virol. 2003;77:1856–1867. doi: 10.1128/JVI.77.3.1856-1867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu HM, Gale M. Hepatitis C Virus Evasion from RIG-I-Dependent Hepatic Innate Immunity. Gastroenterol Res Pract. 2010;2010:548390. doi: 10.1155/2010/548390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saito T, Gale M. Regulation of innate immunity against hepatitis C virus infection. Hepatol Res. 2008;38:115–122. doi: 10.1111/j.1872-034X.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 47.Saito T, Gale M. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J Exp Med. 2008;205:1523–1527. doi: 10.1084/jem.20081210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schoggins JW, Rice CM. Innate immune responses to hepatitis C virus. Curr Top Microbiol Immunol. 2013;369:219–242. doi: 10.1007/978-3-642-27340-7_9. [DOI] [PubMed] [Google Scholar]
- 49.Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci USA. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Golden-Mason L, Rosen HR. Natural killer cells: multifaceted players with key roles in hepatitis C immunity. Immunol Rev. 2013;255:68–81. doi: 10.1111/imr.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zinkernagel RM, Haenseler E, Leist T, Cerny A, Hengartner H, Althage A. T cell-mediated hepatitis in mice infected with lymphocytic choriomeningitis virus. Liver cell destruction by H-2 class I-restricted virus-specific cytotoxic T cells as a physiological correlate of the 51Cr-release assay? J Exp Med. 1986;164:1075–1092. doi: 10.1084/jem.164.4.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neumann-Haefelin C, Thimme R. Adaptive immune responses in hepatitis C virus infection. Curr Top Microbiol Immunol. 2013;369:243–262. doi: 10.1007/978-3-642-27340-7_10. [DOI] [PubMed] [Google Scholar]
- 53.Malta FM, Bruno FR, Carvalho KI, Nastri AC, Kalil J, Carrilho FJ, Kallas EG, Pinho JR. HCV viremia drives an increment of CD86 expression by myeloid dendritic cells. J Med Virol. 2013;85:1919–1924. doi: 10.1002/jmv.23692. [DOI] [PubMed] [Google Scholar]
- 54.Jaime-Ramirez AC, Mundy-Bosse BL, Kondadasula S, Jones NB, Roda JM, Mani A, Parihar R, Karpa V, Papenfuss TL, LaPerle KM, et al. IL-12 enhances the antitumor actions of trastuzumab via NK cell IFN-γ production. J Immunol. 2011;186:3401–3409. doi: 10.4049/jimmunol.1000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heufler C, Koch F, Stanzl U, Topar G, Wysocka M, Trinchieri G, Enk A, Steinman RM, Romani N, Schuler G. Interleukin-12 is produced by dendritic cells and mediates T helper 1 development as well as interferon-gamma production by T helper 1 cells. Eur J Immunol. 1996;26:659–668. doi: 10.1002/eji.1830260323. [DOI] [PubMed] [Google Scholar]
- 56.Holder KA, Stapleton SN, Gallant ME, Russell RS, Grant MD. Hepatitis C virus-infected cells downregulate NKp30 and inhibit ex vivo NK cell functions. J Immunol. 2013;191:3308–3318. doi: 10.4049/jimmunol.1300164. [DOI] [PubMed] [Google Scholar]
- 57.Zhang S, Saha B, Kodys K, Szabo G. IFN-γ production by human natural killer cells in response to HCV-infected hepatoma cells is dependent on accessory cells. J Hepatol. 2013;59:442–449. doi: 10.1016/j.jhep.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809, table of contents. doi: 10.1128/CMR.14.4.778-809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu YJ, Kanzler H, Soumelis V, Gilliet M. Dendritic cell lineage, plasticity and cross-regulation. Nat Immunol. 2001;2:585–589. doi: 10.1038/89726. [DOI] [PubMed] [Google Scholar]
- 60.Watarai H, Sekine E, Inoue S, Nakagawa R, Kaisho T, Taniguchi M. PDC-TREM, a plasmacytoid dendritic cell-specific receptor, is responsible for augmented production of type I interferon. Proc Natl Acad Sci USA. 2008;105:2993–2998. doi: 10.1073/pnas.0710351105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pasetto A, Aleman S, Chen M. Functional Attributes of Responding T Cells in HCV Infection: The Recent Advances in Engineering Functional Antiviral T Cells. Arch Immunol Ther Exp (Warsz) 2013:Epub ahead of print. doi: 10.1007/s00005-013-0248-8. [DOI] [PubMed] [Google Scholar]
- 62.Aberle JH, Formann E, Steindl-Munda P, Weseslindtner L, Gurguta C, Perstinger G, Grilnberger E, Laferl H, Dienes HP, Popow-Kraupp T, et al. Prospective study of viral clearance and CD4(+) T-cell response in acute hepatitis C primary infection and reinfection. J Clin Virol. 2006;36:24–31. doi: 10.1016/j.jcv.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 63.Fahey S, Dempsey E, Long A. The role of chemokines in acute and chronic hepatitis C infection. Cell Mol Immunol. 2013:Epub ahead of print. doi: 10.1038/cmi.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 65.Valiante NM, D’Andrea A, Crotta S, Lechner F, Klenerman P, Nuti S, Wack A, Abrignani S. Life, activation and death of intrahepatic lymphocytes in chronic hepatitis C. Immunol Rev. 2000;174:77–89. doi: 10.1034/j.1600-0528.2002.017417.x. [DOI] [PubMed] [Google Scholar]
- 66.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, et al. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci USA. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koziel MJ. The role of immune responses in the pathogenesis of hepatitis C virus infection. J Viral Hepat. 1997;4 Suppl 2:31–41. doi: 10.1111/j.1365-2893.1997.tb00178.x. [DOI] [PubMed] [Google Scholar]
- 68.Chisari FV. Cytotoxic T cells and viral hepatitis. J Clin Invest. 1997;99:1472–1477. doi: 10.1172/JCI119308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sarobe P, Lasarte JJ, Zabaleta A, Arribillaga L, Arina A, Melero I, Borrás-Cuesta F, Prieto J. Hepatitis C virus structural proteins impair dendritic cell maturation and inhibit in vivo induction of cellular immune responses. J Virol. 2003;77:10862–10871. doi: 10.1128/JVI.77.20.10862-10871.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szabo G, Dolganiuc A. Subversion of plasmacytoid and myeloid dendritic cell functions in chronic HCV infection. Immunobiology. 2005;210:237–247. doi: 10.1016/j.imbio.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 71.Pöhlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, McKeating JA. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol. 2003;77:4070–4080. doi: 10.1128/JVI.77.7.4070-4080.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Irshad M, Khushboo I, Singh S, Singh S. Hepatitis C virus (HCV): a review of immunological aspects. Int Rev Immunol. 2008;27:497–517. doi: 10.1080/08830180802432178. [DOI] [PubMed] [Google Scholar]
- 73.Weiner AJ, Brauer MJ, Rosenblatt J, Richman KH, Tung J, Crawford K, Bonino F, Saracco G, Choo QL, Houghton M. Variable and hypervariable domains are found in the regions of HCV corresponding to the flavivirus envelope and NS1 proteins and the pestivirus envelope glycoproteins. Virology. 1991;180:842–848. doi: 10.1016/0042-6822(91)90104-j. [DOI] [PubMed] [Google Scholar]
- 74.Farci P, Shimoda A, Coiana A, Diaz G, Peddis G, Melpolder JC, Strazzera A, Chien DY, Munoz SJ, Balestrieri A, et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science. 2000;288:339–344. doi: 10.1126/science.288.5464.339. [DOI] [PubMed] [Google Scholar]
- 75.Farci P, Alter HJ, Wong DC, Miller RH, Govindarajan S, Engle R, Shapiro M, Purcell RH. Prevention of hepatitis C virus infection in chimpanzees after antibody-mediated in vitro neutralization. Proc Natl Acad Sci USA. 1994;91:7792–7796. doi: 10.1073/pnas.91.16.7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shimizu YK, Hijikata M, Iwamoto A, Alter HJ, Purcell RH, Yoshikura H. Neutralizing antibodies against hepatitis C virus and the emergence of neutralization escape mutant viruses. J Virol. 1994;68:1494–1500. doi: 10.1128/jvi.68.3.1494-1500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Del Porto P, Puntoriero G, Scottà C, Nicosia A, Piccolella E. High prevalence of hypervariable region 1-specific and -cross-reactive CD4(+) T cells in HCV-infected individuals responsive to IFN-alpha treatment. Virology. 2000;269:313–324. doi: 10.1006/viro.2000.0238. [DOI] [PubMed] [Google Scholar]
- 78.Tsai SL, Chen YM, Chen MH, Huang CY, Sheen IS, Yeh CT, Huang JH, Kuo GC, Liaw YF. Hepatitis C virus variants circumventing cytotoxic T lymphocyte activity as a mechanism of chronicity. Gastroenterology. 1998;115:954–965. doi: 10.1016/s0016-5085(98)70268-9. [DOI] [PubMed] [Google Scholar]
- 79.Frasca L, Scottà C, Del Porto P, Nicosia A, Pasquazzi C, Versace I, Masci AM, Racioppi L, Piccolella E. Antibody-selected mimics of hepatitis C virus hypervariable region 1 activate both primary and memory Th lymphocytes. Hepatology. 2003;38:653–663. doi: 10.1053/jhep.2003.50387. [DOI] [PubMed] [Google Scholar]
- 80.Grakoui A, Shoukry NH, Woollard DJ, Han JH, Hanson HL, Ghrayeb J, Murthy KK, Rice CM, Walker CM. HCV persistence and immune evasion in the absence of memory T cell help. Science. 2003;302:659–662. doi: 10.1126/science.1088774. [DOI] [PubMed] [Google Scholar]
- 81.Manzin A, Solforosi L, Petrelli E, Macarri G, Tosone G, Piazza M, Clementi M. Evolution of hypervariable region 1 of hepatitis C virus in primary infection. J Virol. 1998;72:6271–6276. doi: 10.1128/jvi.72.7.6271-6276.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ray SC, Wang YM, Laeyendecker O, Ticehurst JR, Villano SA, Thomas DL. Acute hepatitis C virus structural gene sequences as predictors of persistent viremia: hypervariable region 1 as a decoy. J Virol. 1999;73:2938–2946. doi: 10.1128/jvi.73.4.2938-2946.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dustin LB, Rice CM. Flying under the radar: the immunobiology of hepatitis C. Annu Rev Immunol. 2007;25:71–99. doi: 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- 84.Shoukry NH, Cawthon AG, Walker CM. Cell-mediated immunity and the outcome of hepatitis C virus infection. Annu Rev Microbiol. 2004;58:391–424. doi: 10.1146/annurev.micro.58.030603.123836. [DOI] [PubMed] [Google Scholar]
- 85.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–952. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 86.Kim HS, Lee JK, Yang IH, Ahn JK, Oh YI, Kim CJ, Kim YS, Lee CK. Identification of hepatitis C virus core domain inducing suppression of allostimulatory capacity of dendritic cells. Arch Pharm Res. 2002;25:364–369. doi: 10.1007/BF02976640. [DOI] [PubMed] [Google Scholar]
- 87.Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang KM. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology. 2003;38:1437–1448. doi: 10.1016/j.hep.2003.09.026. [DOI] [PubMed] [Google Scholar]
- 88.Rushbrook SM, Ward SM, Unitt E, Vowler SL, Lucas M, Klenerman P, Alexander GJ. Regulatory T cells suppress in vitro proliferation of virus-specific CD8+ T cells during persistent hepatitis C virus infection. J Virol. 2005;79:7852–7859. doi: 10.1128/JVI.79.12.7852-7859.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ward SM, Fox BC, Brown PJ, Worthington J, Fox SB, Chapman RW, Fleming KA, Banham AH, Klenerman P. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J Hepatol. 2007;47:316–324. doi: 10.1016/j.jhep.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 90.Thimme R, Lohmann V, Weber F. A target on the move: innate and adaptive immune escape strategies of hepatitis C virus. Antiviral Res. 2006;69:129–141. doi: 10.1016/j.antiviral.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 91.Boettler T, Spangenberg HC, Neumann-Haefelin C, Panther E, Urbani S, Ferrari C, Blum HE, von Weizsäcker F, Thimme R. T cells with a CD4+CD25+ regulatory phenotype suppress in vitro proliferation of virus-specific CD8+ T cells during chronic hepatitis C virus infection. J Virol. 2005;79:7860–7867. doi: 10.1128/JVI.79.12.7860-7867.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bolacchi F, Sinistro A, Ciaprini C, Demin F, Capozzi M, Carducci FC, Drapeau CM, Rocchi G, Bergamini A. Increased hepatitis C virus (HCV)-specific CD4+CD25+ regulatory T lymphocytes and reduced HCV-specific CD4+ T cell response in HCV-infected patients with normal versus abnormal alanine aminotransferase levels. Clin Exp Immunol. 2006;144:188–196. doi: 10.1111/j.1365-2249.2006.03048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Haseda F, Imagawa A, Murase-Mishiba Y, Terasaki J, Hanafusa T. CD4+ CD45RA- FoxP3high activated regulatory T cells are functionally impaired and related to residual insulin-secreting capacity in patients with type 1 diabetes. Clin Exp Immunol. 2013;173:207–216. doi: 10.1111/cei.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Manigold T, Shin EC, Mizukoshi E, Mihalik K, Murthy KK, Rice CM, Piccirillo CA, Rehermann B. Foxp3+CD4+CD25+ T cells control virus-specific memory T cells in chimpanzees that recovered from hepatitis C. Blood. 2006;107:4424–4432. doi: 10.1182/blood-2005-09-3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.MacDonald AJ, Duffy M, Brady MT, McKiernan S, Hall W, Hegarty J, Curry M, Mills KH. CD4 T helper type 1 and regulatory T cells induced against the same epitopes on the core protein in hepatitis C virus-infected persons. J Infect Dis. 2002;185:720–727. doi: 10.1086/339340. [DOI] [PubMed] [Google Scholar]
- 96.Clément S, Pascarella S, Negro F. Hepatitis C virus infection: molecular pathways to steatosis, insulin resistance and oxidative stress. Viruses. 2009;1:126–143. doi: 10.3390/v1020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, Pircher H, Thimme R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6:e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Agnello V, Chung RT, Kaplan LM. A role for hepatitis C virus infection in type II cryoglobulinemia. N Engl J Med. 1992;327:1490–1495. doi: 10.1056/NEJM199211193272104. [DOI] [PubMed] [Google Scholar]
- 100.Ivanovski M, Silvestri F, Pozzato G, Anand S, Mazzaro C, Burrone OR, Efremov DG. Somatic hypermutation, clonal diversity, and preferential expression of the VH 51p1/VL kv325 immunoglobulin gene combination in hepatitis C virus-associated immunocytomas. Blood. 1998;91:2433–2442. [PubMed] [Google Scholar]
- 101.Tan SL, Katze MG. How hepatitis C virus counteracts the interferon response: the jury is still out on NS5A. Virology. 2001;284:1–12. doi: 10.1006/viro.2001.0885. [DOI] [PubMed] [Google Scholar]
- 102.Reyes GR. The nonstructural NS5A protein of hepatitis C virus: an expanding, multifunctional role in enhancing hepatitis C virus pathogenesis. J Biomed Sci. 2002;9:187–197. doi: 10.1007/BF02256065. [DOI] [PubMed] [Google Scholar]
- 103.Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol. 2004;85:2485–2502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 104.Fournier C, Helle F, Descamps V, Morel V, François C, Dedeurwaerder S, Wychowski C, Duverlie G, Castelain S. Natural selection of adaptive mutations in non-structural genes increases trans-encapsidation of hepatitis C virus replicons lacking envelope protein genes. J Gen Virol. 2013;94:996–1008. doi: 10.1099/vir.0.049676-0. [DOI] [PubMed] [Google Scholar]
- 105.Gong G, Waris G, Tanveer R, Siddiqui A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-kappa B. Proc Natl Acad Sci USA. 2001;98:9599–9604. doi: 10.1073/pnas.171311298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Majumder M, Ghosh AK, Steele R, Ray R, Ray RB. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J Virol. 2001;75:1401–1407. doi: 10.1128/JVI.75.3.1401-1407.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Polyak SJ, Khabar KS, Paschal DM, Ezelle HJ, Duverlie G, Barber GN, Levy DE, Mukaida N, Gretch DR. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J Virol. 2001;75:6095–6106. doi: 10.1128/JVI.75.13.6095-6106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Foy E, Li K, Wang C, Sumpter R, Ikeda M, Lemon SM, Gale M. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300:1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 109.Pavio N, Taylor DR, Lai MM. Detection of a novel unglycosylated form of hepatitis C virus E2 envelope protein that is located in the cytosol and interacts with PKR. J Virol. 2002;76:1265–1272. doi: 10.1128/JVI.76.3.1265-1272.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- 111.Shanmugam S, Yi M. Efficiency of E2-p7 processing modulates production of infectious hepatitis C virus. J Virol. 2013;87:11255–11266. doi: 10.1128/JVI.01807-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sarrazin C, Herrmann E, Bruch K, Zeuzem S. Hepatitis C virus nonstructural 5A protein and interferon resistance: a new model for testing the reliability of mutational analyses. J Virol. 2002;76:11079–11090. doi: 10.1128/JVI.76.21.11079-11090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tu Z, Zhang P, Li H, Niu J, Jin X, Su L. Cross-linking of CD81 by HCV-E2 protein inhibits human intrahepatic plasmacytoid dendritic cells response to CpG-ODN. Cell Immunol. 2013;284:98–103. doi: 10.1016/j.cellimm.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Díaz-Valdés N, Manterola L, Belsúe V, Riezu-Boj JI, Larrea E, Echeverria I, Llópiz D, López-Sagaseta J, Lerat H, Pawlotsky JM, et al. Improved dendritic cell-based immunization against hepatitis C virus using peptide inhibitors of interleukin 10. Hepatology. 2011;53:23–31. doi: 10.1002/hep.23980. [DOI] [PubMed] [Google Scholar]
- 115.Arnaud C, Pradat P, Spaziante M, Berthillon P, Maynard M, Taliani G, Chemin I, Trépo C, Petit MA. Pretreatment predictive factors for hepatitis C therapy outcome: relevance of anti-E1E2 antibodies compared to IP-10 and IL28B genotypes. Antivir Ther. 2013:Epub ahead of print. doi: 10.3851/IMP2671. [DOI] [PubMed] [Google Scholar]
- 116.Rosa D, Saletti G, De Gregorio E, Zorat F, Comar C, D’Oro U, Nuti S, Houghton M, Barnaba V, Pozzato G, et al. Activation of naïve B lymphocytes via CD81, a pathogenetic mechanism for hepatitis C virus-associated B lymphocyte disorders. Proc Natl Acad Sci USA. 2005;102:18544–18549. doi: 10.1073/pnas.0509402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao LJ, Wang L, Ren H, Cao J, Li L, Ke JS, Qi ZT. Hepatitis C virus E2 protein promotes human hepatoma cell proliferation through the MAPK/ERK signaling pathway via cellular receptors. Exp Cell Res. 2005;305:23–32. doi: 10.1016/j.yexcr.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 118.Mazzocca A, Sciammetta SC, Carloni V, Cosmi L, Annunziato F, Harada T, Abrignani S, Pinzani M. Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J Biol Chem. 2005;280:11329–11339. doi: 10.1074/jbc.M410161200. [DOI] [PubMed] [Google Scholar]
- 119.Silini E, Bono F, Cividini A, Cerino A, Bruno S, Rossi S, Belloni G, Brugnetti B, Civardi E, Salvaneschi L. Differential distribution of hepatitis C virus genotypes in patients with and without liver function abnormalities. Hepatology. 1995;21:285–290. [PubMed] [Google Scholar]
- 120.Zein NN. Clinical significance of hepatitis C virus genotypes. Clin Microbiol Rev. 2000;13:223–235. doi: 10.1128/cmr.13.2.223-235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zein NN, Rakela J, Krawitt EL, Reddy KR, Tominaga T, Persing DH. Hepatitis C virus genotypes in the United States: epidemiology, pathogenicity, and response to interferon therapy. Collaborative Study Group. Ann Intern Med. 1996;125:634–639. doi: 10.7326/0003-4819-125-8-199610150-00002. [DOI] [PubMed] [Google Scholar]
- 122.Arrese M, Riquelme A, Soza A. Insulin resistance, hepatic steatosis and hepatitis C: a complex relationship with relevant clinical implications. Ann Hepatol. 2010;9 Suppl:112–118. [PubMed] [Google Scholar]
- 123.Fartoux L, Poujol-Robert A, Guéchot J, Wendum D, Poupon R, Serfaty L. Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut. 2005;54:1003–1008. doi: 10.1136/gut.2004.050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bièche I, Asselah T, Laurendeau I, Vidaud D, Degot C, Paradis V, Bedossa P, Valla DC, Marcellin P, Vidaud M. Molecular profiling of early stage liver fibrosis in patients with chronic hepatitis C virus infection. Virology. 2005;332:130–144. doi: 10.1016/j.virol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 125.Foster DA. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007;67:1–4. doi: 10.1158/0008-5472.CAN-06-3016. [DOI] [PubMed] [Google Scholar]
- 126.Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev. 2008;29:381–402. doi: 10.1210/er.2007-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shintani Y, Fujie H, Miyoshi H, Tsutsumi T, Tsukamoto K, Kimura S, Moriya K, Koike K. Hepatitis C virus infection and diabetes: direct involvement of the virus in the development of insulin resistance. Gastroenterology. 2004;126:840–848. doi: 10.1053/j.gastro.2003.11.056. [DOI] [PubMed] [Google Scholar]
- 128.Pazienza V, Clément S, Pugnale P, Conzelman S, Foti M, Mangia A, Negro F. The hepatitis C virus core protein of genotypes 3a and 1b downregulates insulin receptor substrate 1 through genotype-specific mechanisms. Hepatology. 2007;45:1164–1171. doi: 10.1002/hep.21634. [DOI] [PubMed] [Google Scholar]
- 129.Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, Taniguchi E, Kumemura H, Hanada S, Maeyama M, et al. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165:1499–1508. doi: 10.1016/S0002-9440(10)63408-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bernsmeier C, Duong FH, Christen V, Pugnale P, Negro F, Terracciano L, Heim MH. Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J Hepatol. 2008;49:429–440. doi: 10.1016/j.jhep.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 131.Banerjee S, Saito K, Ait-Goughoulte M, Meyer K, Ray RB, Ray R. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream akt/protein kinase B signaling pathway for insulin resistance. J Virol. 2008;82:2606–2612. doi: 10.1128/JVI.01672-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fujita N, Sugimoto R, Ma N, Tanaka H, Iwasa M, Kobayashi Y, Kawanishi S, Watanabe S, Kaito M, Takei Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J Viral Hepat. 2008;15:498–507. doi: 10.1111/j.1365-2893.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- 133.Romero MJ, Bosch-Morell F, Romero B, Rodrigo JM, Serra MA, Romero FJ. Serum malondialdehyde: possible use for the clinical management of chronic hepatitis C patients. Free Radic Biol Med. 1998;25:993–997. doi: 10.1016/s0891-5849(98)00118-x. [DOI] [PubMed] [Google Scholar]
- 134.Mitsuyoshi H, Itoh Y, Sumida Y, Minami M, Yasui K, Nakashima T, Okanoue T. Evidence of oxidative stress as a cofactor in the development of insulin resistance in patients with chronic hepatitis C. Hepatol Res. 2008;38:348–353. doi: 10.1111/j.1872-034X.2007.00280.x. [DOI] [PubMed] [Google Scholar]
- 135.Houglum K, Venkataramani A, Lyche K, Chojkier M. A pilot study of the effects of d-alpha-tocopherol on hepatic stellate cell activation in chronic hepatitis C. Gastroenterology. 1997;113:1069–1073. doi: 10.1053/gast.1997.v113.pm9322499. [DOI] [PubMed] [Google Scholar]
- 136.Gabbay E, Zigmond E, Pappo O, Hemed N, Rowe M, Zabrecky G, Cohen R, Ilan Y. Antioxidant therapy for chronic hepatitis C after failure of interferon: results of phase II randomized, double-blind placebo controlled clinical trial. World J Gastroenterol. 2007;13:5317–5323. doi: 10.3748/wjg.v13.i40.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- 138.Abdalla MY, Ahmad IM, Spitz DR, Schmidt WN, Britigan BE. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J Med Virol. 2005;76:489–497. doi: 10.1002/jmv.20388. [DOI] [PubMed] [Google Scholar]
- 139.Dionisio N, Garcia-Mediavilla MV, Sanchez-Campos S, Majano PL, Benedicto I, Rosado JA, Salido GM, Gonzalez-Gallego J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J Hepatol. 2009;50:872–882. doi: 10.1016/j.jhep.2008.12.026. [DOI] [PubMed] [Google Scholar]
- 140.Li Y, Boehning DF, Qian T, Popov VL, Weinman SA. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007;21:2474–2485. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- 141.Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MM. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J Virol. 2006;80:7199–7207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 143.Joyce MA, Walters KA, Lamb SE, Yeh MM, Zhu LF, Kneteman N, Doyle JS, Katze MG, Tyrrell DL. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009;5:e1000291. doi: 10.1371/journal.ppat.1000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Thorén F, Romero A, Lindh M, Dahlgren C, Hellstrand K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: role of NADPH oxidase-derived oxygen radicals. J Leukoc Biol. 2004;76:1180–1186. doi: 10.1189/jlb.0704387. [DOI] [PubMed] [Google Scholar]
- 145.Asselah T, Rubbia-Brandt L, Marcellin P, Negro F. Steatosis in chronic hepatitis C: why does it really matter? Gut. 2006;55:123–130. doi: 10.1136/gut.2005.069757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Khan M, Jahan S, Khaliq S, Ijaz B, Ahmad W, Samreen B, Hassan S. Interaction of the hepatitis C virus (HCV) core with cellular genes in the development of HCV-induced steatosis. Arch Virol. 2010;155:1735–1753. doi: 10.1007/s00705-010-0797-7. [DOI] [PubMed] [Google Scholar]
- 147.Hwang SJ, Lee SD. Hepatic steatosis and hepatitis C: Still unhappy bedfellows? J Gastroenterol Hepatol. 2011;26 Suppl 1:96–101. doi: 10.1111/j.1440-1746.2010.06542.x. [DOI] [PubMed] [Google Scholar]
- 148.Rubbia-Brandt L, Giostra E, Mentha G, Quadri R, Negro F. Expression of liver steatosis in hepatitis C virus infection and pattern of response to alpha-interferon. J Hepatol. 2001;35:307. doi: 10.1016/s0168-8278(01)00087-3. [DOI] [PubMed] [Google Scholar]
- 149.Rubbia-Brandt L, Fabris P, Paganin S, Leandro G, Male PJ, Giostra E, Carlotto A, Bozzola L, Smedile A, Negro F. Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut. 2004;53:406–412. doi: 10.1136/gut.2003.018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roingeard P. Hepatitis C virus diversity and hepatic steatosis. J Viral Hepat. 2013;20:77–84. doi: 10.1111/jvh.12035. [DOI] [PubMed] [Google Scholar]
- 151.Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology. 2001;33:1358–1364. doi: 10.1053/jhep.2001.24432. [DOI] [PubMed] [Google Scholar]
- 152.Hui JM, Kench J, Farrell GC, Lin R, Samarasinghe D, Liddle C, Byth K, George J. Genotype-specific mechanisms for hepatic steatosis in chronic hepatitis C infection. J Gastroenterol Hepatol. 2002;17:873–881. doi: 10.1046/j.1440-1746.2002.02813.x. [DOI] [PubMed] [Google Scholar]
- 153.Abid K, Pazienza V, de Gottardi A, Rubbia-Brandt L, Conne B, Pugnale P, Rossi C, Mangia A, Negro F. An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J Hepatol. 2005;42:744–751. doi: 10.1016/j.jhep.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 154.Adinolfi LE, Restivo L, Marrone A. The predictive value of steatosis in hepatitis C virus infection. Expert Rev Gastroenterol Hepatol. 2013;7:205–213. doi: 10.1586/egh.13.7. [DOI] [PubMed] [Google Scholar]
- 155.Kumar D, Farrell GC, Fung C, George J. Hepatitis C virus genotype 3 is cytopathic to hepatocytes: Reversal of hepatic steatosis after sustained therapeutic response. Hepatology. 2002;36:1266–1272. doi: 10.1053/jhep.2002.36370. [DOI] [PubMed] [Google Scholar]
- 156.Poynard T, Ratziu V, McHutchison J, Manns M, Goodman Z, Zeuzem S, Younossi Z, Albrecht J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology. 2003;38:75–85. doi: 10.1053/jhep.2003.50267. [DOI] [PubMed] [Google Scholar]
- 157.Hofer H, Bankl HC, Wrba F, Steindl-Munda P, Peck-Radosavljevic M, Osterreicher C, Mueller C, Gangl A, Ferenci P. Hepatocellular fat accumulation and low serum cholesterol in patients infected with HCV-3a. Am J Gastroenterol. 2002;97:2880–2885. doi: 10.1111/j.1572-0241.2002.07056.x. [DOI] [PubMed] [Google Scholar]
- 158.Oem JK, Jackel-Cram C, Li YP, Zhou Y, Zhong J, Shimano H, Babiuk LA, Liu Q. Activation of sterol regulatory element-binding protein 1c and fatty acid synthase transcription by hepatitis C virus non-structural protein 2. J Gen Virol. 2008;89:1225–1230. doi: 10.1099/vir.0.83491-0. [DOI] [PubMed] [Google Scholar]
- 159.Shi ST, Polyak SJ, Tu H, Taylor DR, Gretch DR, Lai MM. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology. 2002;292:198–210. doi: 10.1006/viro.2001.1225. [DOI] [PubMed] [Google Scholar]
- 160.Serfaty L, Andreani T, Giral P, Carbonell N, Chazouillères O, Poupon R. Hepatitis C virus induced hypobetalipoproteinemia: a possible mechanism for steatosis in chronic hepatitis C. J Hepatol. 2001;34:428–434. doi: 10.1016/s0168-8278(00)00036-2. [DOI] [PubMed] [Google Scholar]
- 161.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chrétien Y, Koike K, Pessayre D, Chapman J, Barba G, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB J. 2002;16:185–194. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 162.Park CY, Jun HJ, Wakita T, Cheong JH, Hwang SB. Hepatitis C virus nonstructural 4B protein modulates sterol regulatory element-binding protein signaling via the AKT pathway. J Biol Chem. 2009;284:9237–9246. doi: 10.1074/jbc.M808773200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jackel-Cram C, Babiuk LA, Liu Q. Up-regulation of fatty acid synthase promoter by hepatitis C virus core protein: genotype-3a core has a stronger effect than genotype-1b core. J Hepatol. 2007;46:999–1008. doi: 10.1016/j.jhep.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 164.Tsutsumi T, Suzuki T, Shimoike T, Suzuki R, Moriya K, Shintani Y, Fujie H, Matsuura Y, Koike K, Miyamura T. Interaction of hepatitis C virus core protein with retinoid X receptor alpha modulates its transcriptional activity. Hepatology. 2002;35:937–946. doi: 10.1053/jhep.2002.32470. [DOI] [PubMed] [Google Scholar]
- 165.Cheng Y, Dharancy S, Malapel M, Desreumaux P. Hepatitis C virus infection down-regulates the expression of peroxisome proliferator-activated receptor alpha and carnitine palmitoyl acyl-CoA transferase 1A. World J Gastroenterol. 2005;11:7591–7596. doi: 10.3748/wjg.v11.i48.7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.de Gottardi A, Pazienza V, Pugnale P, Bruttin F, Rubbia-Brandt L, Juge-Aubry CE, Meier CA, Hadengue A, Negro F. Peroxisome proliferator-activated receptor-alpha and -gamma mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Aliment Pharmacol Ther. 2006;23:107–114. doi: 10.1111/j.1365-2036.2006.02729.x. [DOI] [PubMed] [Google Scholar]
- 167.Dharancy S, Malapel M, Perlemuter G, Roskams T, Cheng Y, Dubuquoy L, Podevin P, Conti F, Canva V, Philippe D, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology. 2005;128:334–342. doi: 10.1053/j.gastro.2004.11.016. [DOI] [PubMed] [Google Scholar]