

Abstract
AIM: To characterize the nuclear import of hepatitis B virus (HBV) polymerase (P) and its relevance for the viral life cycle.
METHODS: Sequence analysis was performed to predict functional motives within P. Phosphorylation of P was analyzed by in vitro phosphorylation. Phosphorylation site and nuclear localization signal (NLS) were destroyed by site directed mutagenesis. Functionality of the identified NLS was analyzed by confocal fluorescence microscopy and characterizing the karyopherin binding. Relevance of the structural motives for viral life cycle was studied by infection of primary Tupaia hepatocytes with HBV.
RESULTS: We identified by sequence alignment and functional experiments a conserved bipartite NLS containing a casein kinase II (CKII) phosphorylation site located within the terminal protein domain (TP) of the HBV polymerase. Inhibition of CKII impairs the functionality of this NLS and thereby prevents the nuclear import of the polymerase. Binding of the import factor karyopherin-α2 to the polymerase depends on its CKII-mediated phosphorylation of the bipartite NLS. In HBV-infected primary Tupaia hepatocytes CKII inhibition in the early phase (post entry phase) of the infection process prevents the establishment of the infection.
CONCLUSION: Based on these data it is suggested that during HBV infection the final import of the genome complex into the nucleus is mediated by a novel bipartite NLS localized in the TP domain of HBV polymerase.
Keywords: Hepatitis B virus, Nuclear localization signal, Casein kinase II, Trafficking, Replication
Core tip: The mechanism mediating import of the hepatitis B virus (HBV) genome into the nucleus is still not fully understood. We describe the identification and characterization of a bipartite nuclear localization signal (NLS) in the HBV polymerase that harbours a phosphorylation site for casein kinase II (CKII). Integrity of the phosphorylation site is crucial for the functionality of the NLS. Moreover, inhibition of CKII prevents karyopherin-α2 from binding to the polymerase and thereby the import of the polymerase is impaired. Analysing the viral life cycle we observed that inhibition of CKII blocks the import of the genome into the nucleus resulting in impaired cccDNA formation and so the establishment of the viral infection is prevented.
INTRODUCTION
Infection with human hepatitis B virus (HBV) can cause acute or chronic inflammation of the liver. At present there are about 400 million chronically infected people worldwide. Moreover, persistently infected individuals have an increased risk of developing primary hepatocellular carcinoma (HCC)[1-6]. HBV is the prototype member of the hepadnaviridae family, which encompasses members isolated from woodchucks, ground squirrels and avian viruses isolated from e.g., pekin duck, grey heron and stork.
The HBV polymerase (P) has four domains (Figure 1A). The terminal protein domain (TP) contains the tyrosine residue that primes DNA synthesis and covalently links P to the viral DNA[7-9]. The spacer domain has no known function other than to connect the terminal protein domain with the rest of P. The spacer domain, however, harbors at aa position 320 a thrombin cleavage site. This generates the possibility that not a full length protein, but a truncated polymerase is linked to the HBV genome due to intracellular proteolytic processing[10]. The reverse transcriptase domain and the RNase H domain contain the two enzymatic active sites catalyzing the reverse transcription of the RNA template to DNA and degradation of the RNA template.
Figure 1.
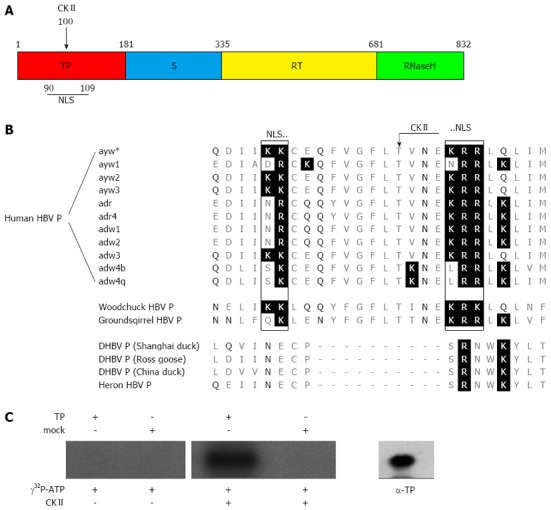
Sequence alignment of the hepatitis B virus polymerase from various virus subtypes and species. A: Scheme of the hepatitis B virus (HBV) polymerase showing the different domains. The terminal protein domain (TP) is shown in red, the spacer domain (S) in blue, the reverse transcriptase domain (RT) in yellow and the RNase H domain in green. The numbers designate the amino acids referred to HBV genotype D. The positions of the casein kinase II (CKII) phosphorylation site and of the bipartite nuclear localization signal (NLS) identified in this study (see Figure 1B) are indicated; B: This figure shows amino acid alignment of Q86-M111 referred to the sequence of subtype ayw*, which was used in this study. Basic amino acids are highlighted by a black background and polar (δ+) amino acids are highlighted by black letters. A conserved protein kinase CKII recognition site (arrow) was identified in orthohepadnaviruses at Thr100 (protein kinase CKII: T/S-X-X-E/D; X = any amino acid). A bipartite nuclear localization signal was identified in orthohepadnaviruses, which is flanking the CKII recognition site by its two basic amino acid clusters (rectangles). All identified putative motifs were not found in the aligned P proteins of the compared avihepadnaviruses; C: Purified TP domain and mock purified proteins (empty vector products) were incubated with [γ-32P] ATP and recombinant protein kinase CKII. To control auto-phosphorylation TP domain was incubated with [γ-32P] ATP in absence of the kinase. The protein specificity was verified by Western blotting using a TP specific antibody (α-TP) on a separate lane. All experiments were performed in triplicate. One representative is shown.
Regarding the life cycle of HBV one open question concerns the fate of the viral nucleocapsid after the virus has entered the cell. There is evidence that the virus enters the cell by receptor mediated endocytosis and at the end of this process the nucleocapsid is released into the cytoplasm[11,12] and transported towards the nuclear pore complex[13,14]. Productive viral infection requires the transport of the HBV genome into the nucleus where the conversion into cccDNA occurs[15].
The phase of nuclear entry is not fully understood. It is discussed that the intact viral capsid shuttles the genome-polymerase complex into the nuclear basket of the nuclear pore complex[16-18] where a partial disassembly of the DNA-loaded nucleocapsid leads to a release of the polymerase-linked genome[13,19-21]. The polymerase-genome complex is too big for free diffusion through the nuclear pore complex.
Due to the facts that that although P is too big for free diffusion through the nuclear pore complex the polymerase-genome complex is imported into the nucleus and that a fraction of P protein is found within the nucleus[22-24] we examined the P protein for conserved motifs that could play a role for nuclear import and aimed to characterize the nuclear import of HBV polymerase-genome complex.
MATERIALS AND METHODS
Cell lines and culture conditions
The human hepatoblastoma cell lines HuH-7[25] and HBV producing cell line HepG2.2.15[26] were cultured in D-MEM medium containing 10% (v/v) fetal calf serum (FCS), 500 U/L penicillin and 100 mg/L streptomycin (PAA, Pasching, Austria). Inducible HBV producing cell line HepAD38[27] were cultivated like HepG2.2.15 but with 400 mg/L G418, 50 µmol/L hydrocortisone and 2.5 mg/L insulin (Sigma-Aldrich, Sleeze, Germany), additionally.
Subcellular fractionation
Subcellular fractionation was performed as described[28,29].
Infection of primary hepatocytes
Primary Tupaia belangeri hepatocytes were isolated, cultivated and infected as described[30,31]. Trypsin treatment for removal of attached viral particles was performed as described[12,31-33]. HBeAg and HBsAg synthesis were analysed 120 h after infection.
Generation of expression constructs
Plasmids were sub-cloned in Escherichia coli strain DH5α. The relevant mutations in the listed primer sequences are highlighted, restriction sites underlined and the corresponding backward primer sequences of mutation primers are reverse complementary to the forward primer if not citied otherwise.
The 1.2 fold HBV genome pJO19 (subtype ayw, genotype D) was derived from plasmid pSM2[26] by a stepwise truncation of the plasmid with BseAI and AatII. Mutant versions of the wild type genome with alterations in the polymerase coding sequence were generated based on pJO19: The CKII recognition site deficient genome pJO19[Τ100Ι] was generated using primer ∆CKII_fw (5’-CAg TTT gTA ggC CCA CTC ATA gTT AAT gAg AAA AgA AgA TTg CAA TTA ATT ATg CCT gCC), the pseudophosphorylated genome pJO19[T100D] was generated with primer *CKII_fw (5’-CAg TTT gTA ggC CCA CTCg ACg TTA ATg AgA AAA gAA gAT TgC AAT TAA TTA TgC CTg CC), the genome with an inactivated NLS pJO19[K105D,K106S] was generated with forward primer ∆NLS_fw (5’-ggC CCA CTC ACA gTT AAT gAg CAg TCT AgA TTg CAA TTg ATT ATg CCT g). GFP wild type expression was obtained by pEGFP-N1 (BD, Heidelberg, Germany). The N-terminal fusions of NLS signals to GFP were generated from a modified version of pEGFP-N1 (pJO21). In pJO21 the first base of the transcriptional START of the GFP reading frame was deleted by site directed mutagenesis to prevent wild type GFP expression using primer GFP∆start_fw (5’-CCA CCg gTC gCC ACC Tgg TgA gCA Agg gCg Agg). The plasmid pNLSNP-GFP was generated by amplifying human nucleoplasmin from a cDNA library. The polymerase chain reaction (PCR) product was flanked by terminal BglII sites, which were generated by primer N-NLS-3fw (5’-TTT AgA TCT gTT CAg ggC CAg TgC) and primer N-NLS-3bw (5’-TTT AgA TCT TTT TAC TTT TTT CTg Tgg). The PCR product was ligated to the BamHI cut pJO21. An engineered BglII site followed by an optimized Kozak sequence[34] with transcriptional START was inserted immediately upstream of the NLS sequence by site directed mutagenesis using forward primer N-NLS-4fw (5’-ggA TgT gAA ACT CTT AAg TAg ATC TCg CCA CCA Tgg gAA AgC ggT CTg CCC CTg g). The dispensable upstream sequence was removed by a BglII digest. Analogue to pNLSNP-GFP, the plasmid pNLSTP-GFP was generated by amplifying the putative NLS of the TP domain from pJO19 using forward primer TP-NLS-3fw (5’-CCC ggA TCC ATg CCC CTA TCC TAT CAA CAC) and backward primer TP-NLS-3bw (5’-TTT AgA TCT TCT TTT CTC ATT AAC Tg). The upstream BglII site, the Kozak signal and the transcriptional START was inserted using primer TP-NLS-4fw (5’-CCT AAT ATA CAT TTA CAC CAA gAC AgA TCT CgC CAC CAT ggT gAA AAA ATg TgA ACA gTT TgT Agg C).
The P-expression constructs were generated based on pJo19 or the corresponding mutants by PCR and subcloned in the pCDNA.3 eukaryotic expression vector.
Site-directed mutagenesis
was performed as described[35] by amplification of the whole plasmid using Pfu Turbo Hotstart DNA-Polymerase (Invitrogen, Karlsruhe, Germany). All synthetic oligonucleotides are purchased by Tib-Molbiol, Berlin, Germany.
Purification of recombinant proteins
The coding sequence for the TP domain (amino acid 1-181) of HBV polymerase was amplified by PCR and inserted into the eubacterial expression vector pQE60 (Qiagen, Hilden, Germany), which encodes a C-terminal His-tag. Expression was performed at room temperature to reduce the formation of inclusion bodies. The soluble fraction of recombinant TP was purified by affinity chromatography on a Ni-NTA column under native conditions as described recently[36]. TP protein inclusion bodies were solved using 6 mol/L guanidine hydrochloride. Ni-NTA affinity purification under denaturing conditions was performed as described[37]. For further purification the TP containing fractions were pooled, dialyzed to buffer AMS (6 mol/L urea, 20 mmol/L sodium acetate, 2% (v/v) ethanol, pH 5.5) and polished by cationic exchange chromatography using a pre-packed Tricorn MonoS column (GE Healthcare, Freiburg, Germany). The elution was performed by a linear gradient over 20 column volumes (cv) between buffer AMS and AMS containing 1 mol/L sodium chloride.
In vitro phosphorylation
experiments were performed using highly purified E. coli produced terminal protein domain dialyzed against kinase buffer (25 mmol/L Tris-HCl, 25 mmol/L beta-glycerophosphate, 10 mmol/L MgCl2, 1 mmol/L DTT, pH 7.5). Phosphorylation was started by addition of 10 µCi [γ-32P] ATP and recombinant human CKII (Merck, Darmstadt, Germany). After 30 min incubation at 30 °C the reaction was stopped by addition of SDS sample buffer and heat treatment (5 min, 95 °C). Proteins were separated by 12% (v/v) SDS-PAGE and detected by autoradiography.
On column phosphorylation of was performed using polished, denatured TP from the cationic exchange chromatography. After addition of 20 mmol/L 2-mercaptoethanol and 100 mmol/L Tris, pH 8 the TP containing fraction was incubated for 1 h at room temperature with 2 cv Ni-NTA agarose, which was pre-washed with buffer AD (6 mol/L urea, 100 mmol/L Tris, pH 8.0). The coupling efficiency was 90%, which was determined by optical density at 280 nm. Equal amounts of TP-agarose were loaded on two empty chromatography columns. A controlled refolding of TP was initiated by a 30 cv linear gradient of buffer AD to buffer R (20 mmol/L Tris, 134 mmol/L sodium chloride, 10% (v/v) glycerol, 10% (v/v) sucrose, 20 mmol/L 2-mercaptoethanol, 0.1% (v/v) Tween-20, pH 7.5). The buffer was changed by a 10 cv linear gradient to buffer K (20 mmol/L Tris, 50 mmol/L potassium chloride, 10 mmol/L imidazole, 20 mmol/L 2-mercaptoethanol, 20 mmol/L beta-glycerol phosphate, 0.1 mmol/L sodium ortho-vanadate, 0.1% (v/v) Tween-20, pH 7.5). 2500 U recombinant protein kinase CKII (Merck, Darmstadt, Germany) was injected together with 200 µmol/L GTP in buffer K and the column was incubated for 3 h at 28 °C. The reaction was stopped by washing the column with 5 cv buffer K.
Binding partner fishing
Six confluent grown 175 cm² culture flasks of HuH-7 cells were lysed by sonification in TBS buffer including protease inhibitor cocktail (1 mmol/L PMSF, 5 mg/L aprotinin, 1 mg/L pepstatin, 4 mmol/L leupeptin, 1 mmol/L EDTA). The crude lysate was cleared by centrifugation at 20000 rpm in a TST41 rotor. The lipid content of the supernatant was reduced by precipitation of the proteins at 75% (w/v) ammonium sulfate. The protein pellet was resolved in TBS buffer and desalted by gel filtration using a HiTrap Desalting column (GE Healthcare, Freiburg, Germany). The desalted 75% (w/v) ammonium sulfate fraction of HuH-7 cell lysate was diluted in buffer B (20 mmol/L Tris, 25 mmol/L beta-glycerol phosphate, 1 mmol/L ortho-vanadate, 20 mmol/L 2-mercaptoethanol, 0.1% (v/v) Tween-20, pH 7.5). Equal amounts of this protein solution were injected to the two terminal protein bound columns and to a blank column only loaded with nickel-agarose. After washing the three columns for 5 cv with buffer B the binding partners were eluted by 1 mol/L sodium chloride and analyzed by western blotting using antibody karyopherin-α2 (C-20) purchased from Santa Cruz, Heidelberg, Germany.
Kinase inhibitor experiments
HepG2.2.15 cells were seeded in 6-well plates with an initial density of 5 × 105 cells/well. Three days after cell seeding CKII inhibitor DMAT (Merck, Darmstadt, Germany) was added to the consumed cell culture medium for 2.5 h. After washing with phosphate buffered saline the cells were incubated for additional 18 h with consumed cell culture medium from the non-HBV producing cell line HuH-7, supplemented with the same concentration of DMAT as already pre-treated. The concentration of the solvent dimethyl sulfoxide (DMSO) was kept at 0.7% (v/v) in all investigated samples.
HBV quantification
Virus genomes were extracted from cell culture supernatant using High Pure Viral Nucleic Acid Kit and determined by LightCyler PCR (Roche, Mannheim, Germany) using a HBx specific probe[38]. cccDNA was extracted from HBV infected Tupaia hepatocytes according standard protocols for genomic DNA extraction with phenol/chloroform[39]. Southern blotting of HBV DNA using a HBV specific 32P labeled probe was performed as described[31].
Endogenous polymerase reaction
HuH-7 cells (3 × 105) were seeded in 6-well plates and transfected with 2 µg HBV DNA using Fugene 6 (Roche, Mannheim, Germany). The enveloped viral particles were precipitated 5 d after transfection by sheep anti-HBs polyclonal serum (kindly gift from Klaus-H. Heermann, University Goettingen, Dept. Virology, Germany) and swollen protein-A sepharose beads (Sigma-Aldrich, Sleeze, Germany) from the cell culture supernatant. The endogenous polymerase reaction (EPR) reaction was performed as described[40] using 10 µCi [α-32P] dCTP (GE Healthcare, Freiburg, Germany) for the labeling.
Confocal laser scanning microscopy
HuH-7 cells (5 × 104) were grown on cover slides in 24-well plates and fixed with 4% formaldehyde/PBS for 30 min at 25 °C. For visualization of actin filaments, the cells were stained with FITC-labelled phalloidin (Sigma, Munich, Germany). Staining was performed as described[41,42]. Rabbit-derived polyclonal TP-specific or spacer domain-specific sera were used for detection of P. Confocal laser scanning microscopy (CLSM) immunofluorescence was performed using the Zeiss LSM 510 microscope (Zeiss, 20 × and 63 × objectives).
RESULTS
Identification of conserved motifs on HBV polymerase
Previous data of our lab based on cell permeable HBV capsids[13] and studies by M Kann’s lab[21] argue against the concept that intact viral capsids[17] or HBcAg shuttles the genome-polymerase complex into the nucleus[16,20]. Based on the data from the TLM-nucleocapsid model system it can be assumed that a partial disassembly of the capsid occurs within the nuclear pore complex or in a perinuclear domain that leads to a release of the genome complex. This raises the question about the final import of the polymerase linked genome into the nucleus.
Sequence analysis of HBV polymerase subtype ayw predicted the existence of a bipartite nuclear localization signal within the terminal domain (TP) (amino acid K90-K91, K104-R106) (Figure 1A). Moreover, a phosphorylation site for CKII (T100) was found within the putative NLS (Figure 1B). The family hepadnaviridae encompasses two genera: orthohepadnavirus and avihepadnavirus. Further analysis revealed that the bipartite NLS and the enclosed CKII phosphorylation site are conserved within the orthohepadna viruses but not within the avihepadna viruses (Figure 1C). The existence of a functional NLS would enable the transfer of the genome-polymerase complex through the nuclear pore complex into the nucleus.
TP domain is phosphorylated by CKII in vitro
To control experimentally whether the predicted phosphorylation site indeed can be phosphorylated by CKII in vitro phosphorylation was performed. Thereto, highly purified recombinant TP domain was incubated with [γ-32P] ATP in the presence of CKII. To exclude any phosphorylation by contaminating kinases the purified TP domain was incubated as described above, but CKII was omitted. As an additional control a mutated TP domain was used in which the predicted CKII phosphorylation site was destroyed by a T to A conversion at aa position 100. Figure 1B shows a significant specific phosphorylation of the TP domain only if CKII is present. In case of the controls no significant phosphorylation was observed. To confirm the identity of the phosphorylated species with the TP domain Western blotting analysis was performed (Figure 1C, right panel). This indicates that the predicted kinase recognition site on the terminal protein is indeed accessible for phosphorylation.
P protein harbors a functional bipartite NLS, which depends on phosphorylation
To study the functionality of the TP-derived putative bipartite NLS HuH-7 cells were transfected with an expression plasmid encoding for a fusion protein of the putative NLS and GFP (NLSP-GFP). As a positive control served a 17 aa long prototype NLS (K142 to K158) derived from human nucleoplasmin (gi114762) fused to the amino terminus of GFP (NLSNP-GFP). The intracellular distribution of the GFP fluorescence was quantified by confocal laser scan microscopy in living cells. Compared to wild type GFP expression, which was found evenly distributed within the cell (Figure 2A), the level of NLSP-GFP was approximately 30% higher in the nucleus then in the cytosol (Figure 2B). In case of the positive control (NLSNP-GFP) an about 75% elevated level of GFP specific fluorescence in the nucleus was observed (Figure 2C). This confirms that the predicted sequence indeed acts as a functional NLS. To analyze a putative relevance of CKII-dependent phosphorylation for the functionality of the TP-derived NLS, NLSP-GFP producing cells were incubated for 2 h with CKII inhibitor DMAT prior analysis by confocal microscopy. The quantification of GFP fluorescence revealed that presence of the CKII inhibitor prevented the directed nuclear enrichment of the NLSP-GFP (Figure 2D). Comparable results were obtained for cells expression the NLSP-GFP(T100A) mutant. An equal distribution comparable to GFP was observed (data not shown).
Figure 2.
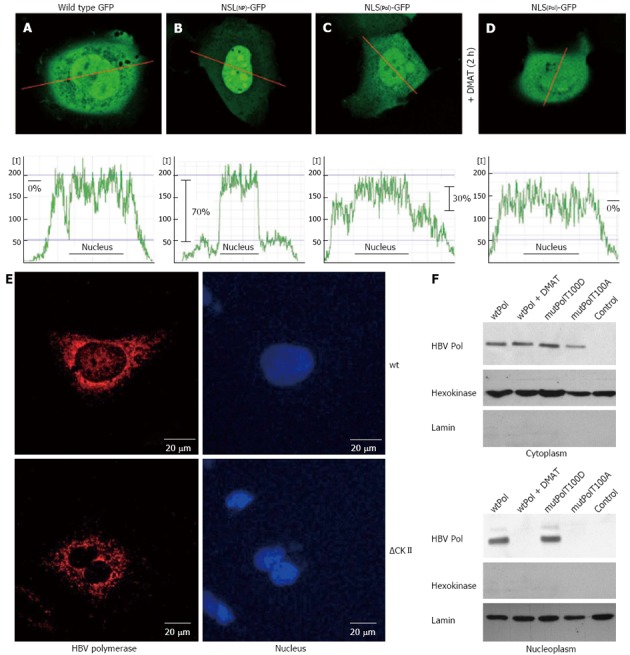
Hepatitis B virus polymerase harbors a functional nuclear localization signal in the terminal protein domain. A-D: HuH-7 cells were transfected with (A) the negative control wild type green fluorescent protein (GFP) (pEGFP-N1), (B) a positive control: GFP fused to a prototype bipartite nuclear localization signal (NLS) of human nucleoplasmin, (C) GFP fused to the putative bipartite NLS of hepatitis B virus (HBV) P protein, (D) the same as (C) but cells were treated with 10 × IC50 of casein kinase II (CKII) inhibitor 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) 2 h prior analysis. The fluorescence was measured in living cells by confocal laser scan analysis. The central layer (out of 6) was quantitated along the red indicated line and displayed in the corresponding graph of the lower panel as relative fluorescence intensity [I]. Differences of mean fluorescence intensities in the cytoplasm and within the nucleus (indicated as black line in the graph) were calculated and are indicated in percent. One representative cell for each fusion protein is shown; E: Confocal immunofluorescence microscopy of HuH-7 cells transfected with an expression vector encoding wt P or the mutant ∆CKII (= T100I) that is not phosphorylated by CKII. For detection of P (red) a rabbit-derived spacer domain-specific serum was used. Nuclei were stained with DAPI (blue); F: HuH-7 cells were transfect with the indicated expression vectors and left untreated or treated with 10× IC50 of CKII inhibitor DMAT 5 h prior analysis. Transfection with pCDNA.3 served as control. Cells were lysed, the cytosolic and nuclear fraction were isolated by differential centrifugation and analyzed by western blotting. For detection of P a TP-domain specific serum was used. Detection of hexokinase and of lamin served as loading control and as control for the purity of the subcellular fractions. All experiments were performed in triplicate. One representative is shown.
To study the relevance of the identified bipartite NLS for the subcellular distribution of the HBV polymerase cells were transfected with an expression construct encoding for HBV polymerase and analyzed by confocal immunofluorescence microscopy or subjected to cell fractionation. The immunofluorescence microscopy shows that in HBV P overproducing cells a fraction was found within the nucleus. However, in cells overexpressing the T100A mutant that destroys the CKII phosphorylation site the nuclear-localized fraction disappeared and the P was exclusively found in the cytoplasm (Figure 2E). Western blotting analysis of the cytoplasmic and of the nuclear fraction confirmed that in addition to the cytosolic fraction a significant amount of P was detectable in the nucleus. However in cells treated with DMAT or overexpressing the T100A mutant of the putative CKII phosphorylation site no P-specific signal was detectable in the nuclear fraction (Figure 2F).
Taken together these results indicate that the HBV polymerase harbors a bipartite nuclear localization signal which functionality is dependent on CKII-mediated phosphorylation.
Binding of karyopherin-α2 to TP depends on CKII mediated phosphorylation
Karyopherin-α2 is an essential factor for NLS-mediated nuclear import. Therefore, it was investigated whether the data described above are reflected by an increased binding of karyopherin-α2 to in vitro phosphorylated TP as compared to unphosphorylated TP. Equal amounts of terminal protein domain were immobilized on two columns. One column was in vitro phosphorylated by CKII and its substrate GTP the other and a blank control column were treated equally without the addition of the kinase. The pretreated columns were equilibrated and loaded with the desalted 75% (w/v) ammonium sulfate fraction of HuH-7 cell lysate. Finally, binding partners were eluted by sodium chloride and the eluted fractions were analyzed by Western blotting using a karyopherin-α2 specific antibody. Interestingly, karyopherin-α2 was only found in the eluate of the column with CKII pre-treated TP (Figure 3). This indicates clearly that in vitro binding of karyopherin-α2, the key enzyme for nuclear import, is dependent on CKII phosphorylation of the terminal protein.
Figure 3.
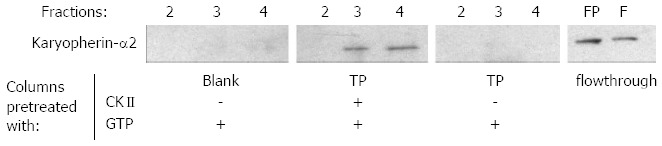
Binding of karyopherin-α2 to terminal protein depends on casein kinase II mediated phosphorylation. The interaction of soluble protein fraction of HuH-7 cells to immobilized terminal protein (TP) domain was investigated by western blotting of the sodium chloride eluted fractions 2-4. Karyopherin-α2 bound only to the casein kinase II (CKII) pre-treated TP column (upper panel). FP: Flow through of the CKII treated TP column; F: Flow trough of the untreated TP column.
Inhibition of CKII impairs HBV replication in primary Tupaia hepatocytes
To study the relevance of the NLS and of the CKII phosphorylation site for the HBV life cycle the HBV P protein was mutated based on a recombinant 1.2 fold HBV genome (subtype ayw) by site directed mutagenesis. The changes in the DNA sequence did not affect other reading frames or regulatory elements.
In the NLS-deficient mutant (∆NLS) the NLS is inactivated by manipulating the basicity of the downstream cluster to K105D and K106S. The putative CKII recognition site on the P protein is destroyed by a T100I substitution (∆CKII), whereas a pseudo-phosphorylated mutant was generated by a T100D conversion (CKII*). However, the attempt to produce mutant virus by transfection of HepG2 or HuH-7 cells with the respective 1.2 fold genomes failed in case of ∆NLS and of the ∆CKII mutant (Figure 4A). In both cases significant less virus was produced (about hundredfold) as compared to the wt genome or the mutant encoding the CKII* mutant. In a transfection experiment the nuclear import is not the limiting step. It can be concluded that mutations affecting the integrity of the NLS motive or of the CKII phosphorylation site cause secondary effects affecting the functionality of the polymerase. Therefore a direct analysis of the relevance of the NLS and of the CKII substrate domain based on mutated virus was not possible.
Figure 4.
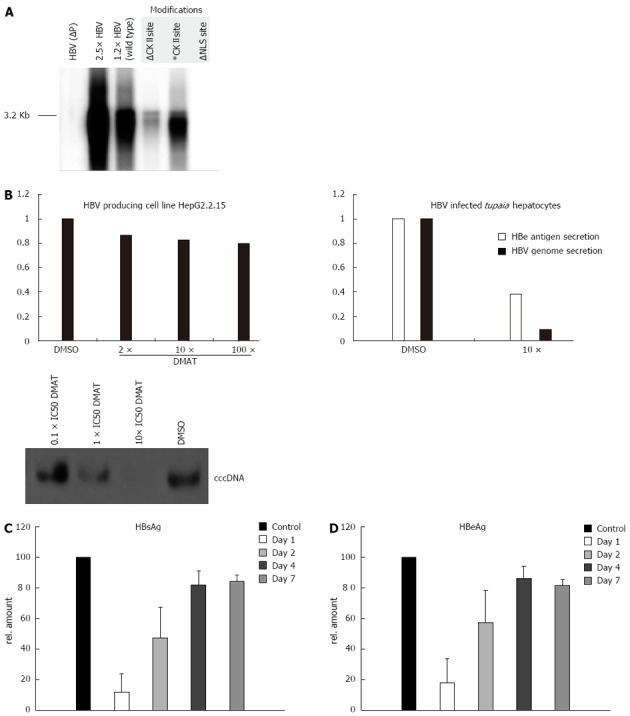
Casein kinase II inhibition impairs hepatitis B virus replication in infected primary Tupaia hepatocytes. A: HuH-7 cells were transfected with mutant versions of a 1.2 fold hepatitis B virus (HBV) genome. After 5 d the secreted viral particles were precipitated with a HBs specific antibody and the containing 3.2 kb HBV genomes were visualized by the radioactive tracer [α-32P] dCTP incorporated using the endogenous polymerase activity. The unphosphorylated form of the casein kinase II (CKII) recognition site in the P protein was simulated by a T100I substitution (∆) and the pseudo-phosphorylation was simulated by a T100D substitution (*) in the 1.2 fold HBV wild type genome. The nuclear localization signal (NLS) was inactivated by mutating the downstream basic cluster (K105D and K106S) on the P protein. A 2.5 × HBV genome and a P deficient genome [HBV (P-)] served as controls; B: Primary Tupaia hepatocytes were infected with HepAD38 derived HBV and post infection treated for 36 h with solvent DMSO or 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole (DMAT) (10 × IC50). Twelve days after infection the HBV genome secretion was measured by Lightcycler polymerase chain reaction. The cccDNA content of infected Tupaia hepatocytes that were incubated for 36 h with the indicated amounts of DMAT was visualized by Southern blot using a HBV specific probe. The specificity of DMAT incubation was analyzed by 2 h inhibitor pre-treatment of the stably HBV transfected cell line HepG2.2.15 followed by treatment of 36 h with 10 × IC50 CKII inhibitor DMAT (IC50 in rat liver = 150 nmol/L) and genome secretion was compared to the solvent control DMSO measured by Lightcycler PCR. All experiments were performed in triplicate. The figure shows one representative experiment; C, D: Primary Tupaia hepatocytes were infected with HepAD38-derived HBV and at day 1, day 2, day 4 and day 7 treated for 36 h DMAT (10 × IC50). Twelve days after infection the HBV replication was measured by HBeAg or HBsAg-specific enzyme linked immunosorbent assay. The bars represent the standard deviation.
The in vitro data described above had shown that the functionality of the NLS identified in the TP-domain depends on the CKII-dependent phosphorylation (Figure 2). To study the relevance of CKII-dependent phosphorylation of the TP domain for HBV life cycle primary Tupaia hepatocytes were infected with wtHBV particles for 8 h. Adherent HBV particles were removed by trypsin treatment as described above. After infection the cells were grown for 36 h in the presence of the cell permeable small molecular CKII inhibitor DMAT and the virus replication was analyzed by quantification of HBsAg and HBeAg secretion (Figure 4B). Moreover secreted viral particles were quantified by real time PCR. Both approaches revealed that inhibition of CKII by DMAT caused a strong and significant reduction of virus replication. This was further confirmed by analysis of cccDNA formation in infected PTHs. Inhibition of CKII by increasing concentrations of DMAT abolishes cccDNA formation (Figure 4B).
For a more detailed analysis primary Tupaia hepatocytes were infected as described above. One, two, four and seven days after infection the hepatocytes were grown for 36 h in the presence of the cell permeable small molecular CKII inhibitor DMAT and the virus replication was analyzed by quantification of HBsAg and HBeAg secretion (Figure 4C and D). Both approaches revealed that inhibition of CKII by DMAT at 1 and 2 d after infection caused a strong and significant reduction of virus replication, while inhibition after 4 and 7 d pi resulted only in a small reduction of virus replication.
To control the specificity of the observed effect the constitutively HBV expressing cell line HepG2.2.15 were instrumental. This cell line harbors a stably integrated HBV genome. Due to the stable integration of the genome the re-import of de novo synthesized genomes plays a minor role for maintaining the pool of transcriptional templates. Therefore, inhibition of polymerase import should exert a small effect. HepG2.2.15 cells were treated with DMAT, HBeAg and HBsAg secretion were analyzed by ELISA and virus secretion was quantified by LightCycler PCR (Figure 4B). Under these conditions inhibition of CKII with DMAT did slightly but not significantly reduce virus secretion as compared to the solvent control (Figure 4B). Comparable results were obtained for the cell line HepAD38 (data not shown).
Taken together these data provide indirect evidence that the functionality of the NLS in the TP domain of P is required for the import of the viral genome into the nucleus.
DISCUSSION
It is well as established that HBV replicates its genome inside the nucleus (recent reviews[18,43,44]). This raises the question about the post entry transport of the genome complex to and about its final import into the nucleus. Previous reports discussed whether the final import of the assembled nucleocapsid in the nucleus harboring the genome complex can occur[17]. More recent reports provide evidence that the mature nucleocapsid disassembles in the nuclear pore complex and the final import of the genome complex could be mediated by an association with HBcAg oligomers that harbor in their C-terminal domain a NLS[20,21,44-47]. For DHBV it was described that completition of plus-strand DNA synthesis triggers genomic DNA deproteinization and conformational changes of the nucleocapsid. This could lead to the exposure of a NLS within the core and thereby could enable the import of the rcDNA[48]. In this context it is interesting to mention that the presence of the identified NLS is not conserved for the genus avihepadnavirus.
Recently we developed cell permeable HBV nucleocapsids as a vehicle for gene transfer (Brandenburg et al[13]) Based on this system it was observed that neither HBcAg dimers nor nucleocapsids were visible in the nucleus of TLM-nucleocapsid treated cells although an efficient expression of the packaged, P-linked genome occurred suggesting that neither the nucleocapsid nor HBcAg dimers mediate the final import of the genome complex. Therefore the question arose about an alternative import mechanism: with about 90 kDa the covalent complex of HBV polymerase and genome clearly exceeds the size that freely can pass the nuclear pore complex.
In this context it is interesting that previous in vitro experiments have shown that the HBV genome complex can be efficiently imported into the nucleus. However if the complex is deproteinized, the naked genome fails to enter efficiently the nucleus[23]. These data suggest that the genome-linked polymerase could be relevant for the nuclear entry process.
The bipartite NLS identified in the TP domain of P could mediate the entry of the genome complex into the nucleus. The functional analysis of the genome complex revealed that the predicted NLS indeed has the potential to act as a nuclear localization signal. However, compared to other nuclear localization signals the TP-derived NLS is not a strong signal. This might reflect the different functions of P[18]. On the one hand P recognizes in the cytoplasm the de novo-synthesized 3.5 kb mRNA[43,49] and on the other hand the genome associated Pol is assumed to mediate by its NLS the entry of the genome complex into the nucleus. It is obvious that a too strong NLS signal might counteract the RNA-recognizing function in the cytoplasm. Yet, it was described that in duck HBV replicating cells in addition to the encapsidated polymerase non-encapsidated polymerase exists[24]. The major fraction of the non-encapsidated duck HBV polymerase is found in the cytoplasm a smaller fraction however can be detected within the nucleus[24]. Interestingly, in cells overexpressing HBV polymerase in the absence of other viral proteins a fraction of HBV polymerase is found within the nucleus, co-localized with the p11 protein of PML bodies[22].
A further interesting feature is the CKII phosphorylation site localized in the center of the bipartite NLS. The functionality of the HBV polymerase-derived NLS depends on the CKII-mediated phosphorylation. CKII is not a very tightly regulated kinase[50]. It can be assumed that CKII exerts a housekeeping phosphorylation function[51]. If subcellular localization and function of HBV polymerase is subjected to a tight control, it is not likely that CKII exerts this function. It is tempting to speculate that phosphatases could play an important role to regulate the subcellular localization and thereby function of HBV polymerase. CKII phosphorylation is reported to influence subcellular localization of various nuclear proteins[52]. For example CKII phosphorylation upstream of the NLS of simian virus 40 T-antigen enhances its nuclear import up to 40 fold[53]. But immediate phosphorylation one or two amino acid upstream of the crucial amino acid of classical monopartite NLS seems to have inhibitory effects on karyopherin binding due to a disturbance of the NLS basicity[54]. In case of bipartite nuclear localization signals this correlation is not evident. For example the spacer of the functional bipartite NLS of the Agrobacterium tumefaciens protein nopaline contains four negative charged aspartates, one even immediate located ate the downstream basic cluster[55]. On the other hand an increase of the hydrophobicity of the 10-12 amino acid spacer seems to decrease its functionality[56].
In transfection experiments it was found that destruction of the NLS or of the CKII-site almost completely abolishes HBV replication. Under these experimental conditions the import of the viral genome into the nucleus does not represent the limiting step. The plasmid DNA freely moves into the nucleus. However this observation suggests that perturbation of this domain has further effects on the polymerase function. Since the ∆NLS and ∆CKII-mutants were replication deficient it was not possible to study the replication of the respective mutant viruses. The in vitro data however have shown that the functionality of the TP-derived NLS requires the functionality of CKII. Based on this it could be shown that inhibition of CKII in the early phase of the infection abolished the establishment of HBV infection while inhibition of CKII in a later phase of the infection or in a stable system had no effect on HBV replication.
In conclusion we demonstrate the presence of a bipartite NLS within the TP domain of P and provide evidence for a novel model describing the import of the HBV genome into the nucleus.
ACKNOWLEDGMENTS
We thank Sabine Mac Nelly for excellent preparation of the Tupaia primary hepatocytes.
COMMENTS
Background
Human hepatitis B virus (HBV) enters the cell by receptor mediated endocytosis and at the end of this process the nucleocapsid that harbours the viral genome is released into the cytoplasm and transported towards the nuclear pore complex. Establishment of a productive viral infection requires the transport of the HBV genome that is covalently linked to the polymerase into the nucleus. In the nucleus the partial- double stranded DNA genome is converted to covalently closed circular (ccc) double stranded DNA.
Research frontiers
The phase of nuclear entry is not fully understood. It is discussed that the intact viral capsid shuttles the genome-polymerase complex into the nuclear basket of the nuclear pore complex. Here, after a partial disassembly of the nucleocapsid the polymerase-linked genome is released. The polymerase-genome complex however is too big to pass freely through the nuclear pore complex. This raises the question about the import mechanism.
Innovations and breakthroughs
The identification and characterization of a bipartite NLS in the HBV polymerase that harbours a phosphorylation site for casein kinase II (CKII) is described in this manuscript. The integrity of the phosphorylation site is crucial for the functionality of the NLS. Moreover, inhibition of CKII prevents karyopherin α2 from binding to the polymerase. Thereby the import of the polymerase is impaired resulting in inhibited cccDNA formation that prevents the establishment of the viral infection. The data identify novel structural and functional prerequisites for the establishment of HBV infection.
Applications
The data describe a potential novel target for antiviral that could block the establishment of a HBV-infection.
Peer review
In this work, they identified a novel NLS located in the terminal protein domain of HBV polymerase and defined a CKII phosphorylation site (threonine) which is adjacent to the NLS. This paper is well written.
Footnotes
Supported by A Grant from DZIF to Hildt E
P- Reviewer: Lo SJ S- Editor: Wen LL L- Editor: A E- Editor: Zhang DN
References
- 1.Buendia MA. Hepatitis B viruses and cancerogenesis. Biomed Pharmacother. 1998;52:34–43. doi: 10.1016/s0753-3322(97)86239-7. [DOI] [PubMed] [Google Scholar]
- 2.Lupberger J, Hildt E. Hepatitis B virus-induced oncogenesis. World J Gastroenterol. 2007;13:74–81. doi: 10.3748/wjg.v13.i1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prange AN, Bartsch M, Meiners J, Serek M, Winkelmann T. Interspecific somatic hybrids between Cyclamen persicum and C. coum, two sexually incompatible species. Plant Cell Rep. 2012;31:723–735. doi: 10.1007/s00299-011-1190-z. [DOI] [PubMed] [Google Scholar]
- 4.Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19:859–868. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tan YJ. Hepatitis B virus infection and the risk of hepatocellular carcinoma. World J Gastroenterol. 2011;17:4853–4857. doi: 10.3748/wjg.v17.i44.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivière L, Ducroux A, Buendia MA. The oncogenic role of hepatitis B virus. Recent Results Cancer Res. 2014;193:59–74. doi: 10.1007/978-3-642-38965-8_4. [DOI] [PubMed] [Google Scholar]
- 7.Shin YC, Ko C, Ryu WS. Hydrophobic residues of terminal protein domain of hepatitis B virus polymerase contribute to distinct steps in viral genome replication. FEBS Lett. 2011;585:3964–3968. doi: 10.1016/j.febslet.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Wang YX, Xu X, Luo C, Ma ZM, Jiang HL, Ding JP, Wen YM. A putative new domain target for anti-hepatitis B virus: residues flanking hepatitis B virus reverse transcriptase residue 306 (rtP306) J Med Virol. 2007;79:676–682. doi: 10.1002/jmv.20835. [DOI] [PubMed] [Google Scholar]
- 9.Beck J, Nassal M. A Tyr residue in the reverse transcriptase domain can mimic the protein-priming Tyr residue in the terminal protein domain of a hepadnavirus P protein. J Virol. 2011;85:7742–7753. doi: 10.1128/JVI.00482-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin CG, Yang SJ, Hwang WL, Su TS, Lo SJ. Demonstration of the presence of protease-cutting site in the spacer of hepatitis B viral Pol protein. J Virol Methods. 1995;51:61–73. doi: 10.1016/0166-0934(94)00118-z. [DOI] [PubMed] [Google Scholar]
- 11.Köck J, Theilmann L, Galle P, Schlicht HJ. Hepatitis B virus nucleic acids associated with human peripheral blood mononuclear cells do not originate from replicating virus. Hepatology. 1996;23:405–413. doi: 10.1002/hep.510230303. [DOI] [PubMed] [Google Scholar]
- 12.Stoeckl L, Funk A, Kopitzki A, Brandenburg B, Oess S, Will H, Sirma H, Hildt E. Identification of a structural motif crucial for infectivity of hepatitis B viruses. Proc Natl Acad Sci USA. 2006;103:6730–6734. doi: 10.1073/pnas.0509765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brandenburg B, Stockl L, Gutzeit C, Roos M, Lupberger J, Schwartlander R, Gelderblom H, Sauer IM, Hofschneider PH, Hildt E. A novel system for efficient gene transfer into primary human hepatocytes via cell-permeable hepatitis B virus-like particle. Hepatology. 2005;42:1300–1309. doi: 10.1002/hep.20950. [DOI] [PubMed] [Google Scholar]
- 14.Rabe B, Glebe D, Kann M. Lipid-mediated introduction of hepatitis B virus capsids into nonsusceptible cells allows highly efficient replication and facilitates the study of early infection events. J Virol. 2006;80:5465–5473. doi: 10.1128/JVI.02303-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nassal M. Hepatitis B virus replication: novel roles for virus-host interactions. Intervirology. 1999;42:100–116. doi: 10.1159/000024970. [DOI] [PubMed] [Google Scholar]
- 16.Rabe B, Vlachou A, Panté N, Helenius A, Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci USA. 2003;100:9849–9854. doi: 10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panté N, Kann M. Nuclear pore complex is able to transport macromolecules with diameters of about 39 nm. Mol Biol Cell. 2002;13:425–434. doi: 10.1091/mbc.01-06-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schädler S, Hildt E. HBV life cycle: entry and morphogenesis. Viruses. 2009;1:185–209. doi: 10.3390/v1020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol. 1999;145:45–55. doi: 10.1083/jcb.145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rabe B, Delaleau M, Bischof A, Foss M, Sominskaya I, Pumpens P, Cazenave C, Castroviejo M, Kann M. Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog. 2009;5:e1000563. doi: 10.1371/journal.ppat.1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitz A, Schwarz A, Foss M, Zhou L, Rabe B, Hoellenriegel J, Stoeber M, Panté N, Kann M. Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog. 2010;6:e1000741. doi: 10.1371/journal.ppat.1000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi J, Chang JS, Song MS, Ahn BY, Park Y, Lim DS, Han YS. Association of hepatitis B virus polymerase with promyelocytic leukemia nuclear bodies mediated by the S100 family protein p11. Biochem Biophys Res Commun. 2003;305:1049–1056. doi: 10.1016/s0006-291x(03)00881-7. [DOI] [PubMed] [Google Scholar]
- 23.Kann M, Bischof A, Gerlich WH. In vitro model for the nuclear transport of the hepadnavirus genome. J Virol. 1997;71:1310–1316. doi: 10.1128/jvi.71.2.1310-1316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao E, Gong Y, Chen N, Tavis JE. The majority of duck hepatitis B virus reverse transcriptase in cells is nonencapsidated and is bound to a cytoplasmic structure. J Virol. 2000;74:8648–8657. doi: 10.1128/jvi.74.18.8648-8657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42:3858–3863. [PubMed] [Google Scholar]
- 26.Sells MA, Chen ML, Acs G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci USA. 1987;84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ladner SK, Otto MJ, Barker CS, Zaifert K, Wang GH, Guo JT, Seeger C, King RW. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: a novel system for screening potential inhibitors of HBV replication. Antimicrob Agents Chemother. 1997;41:1715–1720. doi: 10.1128/aac.41.8.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hafner A, Brandenburg B, Hildt E. Reconstitution of gene expression from a regulatory-protein-deficient hepatitis B virus genome by cell-permeable HBx protein. EMBO Rep. 2003;4:767–773. doi: 10.1038/sj.embor.embor903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ploen D, Hafirassou ML, Himmelsbach K, Sauter D, Biniossek ML, Weiss TS, Baumert TF, Schuster C, Hildt E. TIP47 plays a crucial role in the life cycle of hepatitis C virus. J Hepatol. 2013;58:1081–1088. doi: 10.1016/j.jhep.2013.01.022. [DOI] [PubMed] [Google Scholar]
- 30.Lupberger J, Mund A, Kock J, Hildt E. Cultivation of HepG2.2.15 on Cytodex-3: higher yield of hepatitis B virus and less subviral particles compared to conventional culture methods. J Hepatol. 2006;45:547–552. doi: 10.1016/j.jhep.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Köck J, Nassal M, MacNelly S, Baumert TF, Blum HE, von Weizsäcker F. Efficient infection of primary tupaia hepatocytes with purified human and woolly monkey hepatitis B virus. J Virol. 2001;75:5084–5089. doi: 10.1128/JVI.75.11.5084-5089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Funk A, Mhamdi M, Lin L, Will H, Sirma H. Itinerary of hepatitis B viruses: delineation of restriction points critical for infectious entry. J Virol. 2004;78:8289–8300. doi: 10.1128/JVI.78.15.8289-8300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Funk A, Mhamdi M, Hohenberg H, Will H, Sirma H. pH-independent entry and sequential endosomal sorting are major determinants of hepadnaviral infection in primary hepatocytes. Hepatology. 2006;44:685–693. doi: 10.1002/hep.21297. [DOI] [PubMed] [Google Scholar]
- 34.Kozak M. An analysis of 5’-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisher CL, Pei GK. Modification of a PCR-based site-directed mutagenesis method. Biotechniques. 1997;23:570–571, 574. doi: 10.2144/97234bm01. [DOI] [PubMed] [Google Scholar]
- 36.Bleifuss E, Kammertoens T, Hutloff A, Quarcoo D, Dorner M, Straub P, Uckert W, Hildt E. The translocation motif of hepatitis B virus improves protein vaccination. Cell Mol Life Sci. 2006;63:627–635. doi: 10.1007/s00018-005-5548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oess S, Hildt E. Novel cell permeable motif derived from the PreS2-domain of hepatitis-B virus surface antigens. Gene Ther. 2000;7:750–758. doi: 10.1038/sj.gt.3301154. [DOI] [PubMed] [Google Scholar]
- 38.Stöckl L, Berting A, Malkowski B, Foerste R, Hofschneider PH, Hildt E. Integrity of c-Raf-1/MEK signal transduction cascade is essential for hepatitis B virus gene expression. Oncogene. 2003;22:2604–2610. doi: 10.1038/sj.onc.1206320. [DOI] [PubMed] [Google Scholar]
- 39.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current protocols of molecular biology. New York: John Wiley & Sons; 2004. [Google Scholar]
- 40.Koschel M, Oed D, Gerelsaikhan T, Thomssen R, Bruss V. Hepatitis B virus core gene mutations which block nucleocapsid envelopment. J Virol. 2000;74:1–7. doi: 10.1128/jvi.74.1.1-7.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaedler S, Krause J, Himmelsbach K, Carvajal-Yepes M, Lieder F, Klingel K, Nassal M, Weiss TS, Werner S, Hildt E. Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J Biol Chem. 2010;285:41074–41086. doi: 10.1074/jbc.M110.145862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvajal-Yepes M, Himmelsbach K, Schaedler S, Ploen D, Krause J, Ludwig L, Weiss T, Klingel K, Hildt E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J Biol Chem. 2011;286:8941–8951. doi: 10.1074/jbc.M110.186684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nassal M. Hepatitis B viruses: reverse transcription a different way. Virus Res. 2008;134:235–249. doi: 10.1016/j.virusres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 44.Prange R. Host factors involved in hepatitis B virus maturation, assembly, and egress. Med Microbiol Immunol. 2012;201:449–461. doi: 10.1007/s00430-012-0267-9. [DOI] [PubMed] [Google Scholar]
- 45.Wittkop L, Schwarz A, Cassany A, Grün-Bernhard S, Delaleau M, Rabe B, Cazenave C, Gerlich W, Glebe D, Kann M. Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell Microbiol. 2010;12:962–975. doi: 10.1111/j.1462-5822.2010.01444.x. [DOI] [PubMed] [Google Scholar]
- 46.Kantelhardt VC, Schwarz A, Wend U, Schüttler CG, Willems WR, Trimoulet P, Fleury H, Gerlich WH, Kann M. Re-evaluation of anti-HBc non-reactive serum samples from patients with persistent hepatitis B infection by immune precipitation with labelled HBV core antigen. J Clin Virol. 2009;46:124–128. doi: 10.1016/j.jcv.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 47.Kann M, Schmitz A, Rabe B. Intracellular transport of hepatitis B virus. World J Gastroenterol. 2007;13:39–47. doi: 10.3748/wjg.v13.i1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo H, Mao R, Block TM, Guo JT. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J Virol. 2010;84:387–396. doi: 10.1128/JVI.01921-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stahl M, Beck J, Nassal M. Chaperones activate hepadnavirus reverse transcriptase by transiently exposing a C-proximal region in the terminal protein domain that contributes to epsilon RNA binding. J Virol. 2007;81:13354–13364. doi: 10.1128/JVI.01196-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 52.Jans DA, Hübner S. Regulation of protein transport to the nucleus: central role of phosphorylation. Physiol Rev. 1996;76:651–685. doi: 10.1152/physrev.1996.76.3.651. [DOI] [PubMed] [Google Scholar]
- 53.Jans DA, Jans P. Negative charge at the casein kinase II site flanking the nuclear localization signal of the SV40 large T-antigen is mechanistically important for enhanced nuclear import. Oncogene. 1994;9:2961–2968. [PubMed] [Google Scholar]
- 54.Harreman MT, Kline TM, Milford HG, Harben MB, Hodel AE, Corbett AH. Regulation of nuclear import by phosphorylation adjacent to nuclear localization signals. J Biol Chem. 2004;279:20613–20621. doi: 10.1074/jbc.M401720200. [DOI] [PubMed] [Google Scholar]
- 55.Howard EA, Zupan JR, Citovsky V, Zambryski PC. The VirD2 protein of A. tumefaciens contains a C-terminal bipartite nuclear localization signal: implications for nuclear uptake of DNA in plant cells. Cell. 1992;68:109–118. doi: 10.1016/0092-8674(92)90210-4. [DOI] [PubMed] [Google Scholar]
- 56.Robbins J, Dilworth SM, Laskey RA, Dingwall C. Two interdependent basic domains in nucleoplasmin nuclear targeting sequence: identification of a class of bipartite nuclear targeting sequence. Cell. 1991;64:615–623. doi: 10.1016/0092-8674(91)90245-t. [DOI] [PubMed] [Google Scholar]