

Abstract
Wingless (Wg)/Wnt signaling is essential for patterning invertebrate and vertebrate embryos, and inappropriate Wnt activity is associated with a variety of human cancers. Despite intensive study, Wnt pathway mechanisms are not fully understood. We have discovered a new mechanism for regulating the Wnt pathway: activity of a Rho guanine nucleotide exchange factor (GEF) encoded by pebble (pbl) in Drosophila and ECT2 in humans. This RhoGEF has an essential role in cytokinesis, but also plays an unexpected, conserved role in inhibiting Wg/Wnt activity. Loss and gain of pbl function in Drosophila embryos cause pattern defects that indicate altered Wg activity. Both Pbl and ECT2 repress Wg/Wnt target gene expression in cultured Drosophila and human cells. The GEF activity is required for Wnt regulation, whereas other protein domains important for cytokinesis are not. Unlike most negative regulators of Wnt activity, Pbl/ECT2 functions downstream of Armadillo (Arm)/beta-catenin stabilization. Our results indicate GTPase regulation at a novel point in Wg/Wnt signal transduction, and provide new insight into the categorization of ECT2 as a human proto-oncogene.
Keywords: Wingless/Wnt signaling, Pebble/ECT2, RhoGEF
INTRODUCTION
The Wnt family of secreted growth factors is highly conserved in metazoan animals, playing crucial roles in development, cell proliferation and tissue homeostasis (reviewed by MacDonald et al., 2009). Wnt signaling generates embryonic pattern in many tissue types from epidermal epithelia to the nervous system. In mature animals, Wnts maintain stem cell populations in a variety of tissues, including blood, bone, hair follicles and gut (reviewed by Clevers, 2006). Mutations that result in excess Wnt signaling are associated with a number of human cancers especially colorectal cancer (Polakis, 2007). The diverse and widespread function of Wnts highlights the importance of understanding the mechanism of Wnt signal transduction. We use Drosophila as a model system for studying Wnt signaling, because its powerful genetics allows the discovery of new pathway components.
Wnt signaling is easily assayed by examining the highly Wnt-sensitive cuticle pattern of the Drosophila embryo. At the end of embryogenesis, epidermal cells secrete a layer of cuticle with a stereotyped segmental pattern consisting of an array of hooked elements, called denticles, interspersed with naked cuticle. The wingless (wg) gene, which encodes the primary fly Wnt, is expressed in a single row of epidermal cells in each segment and generates positional information across the segment (reviewed by Bejsovec, 2006). Loss-of-function wg mutations result in a uniform lawn of denticles at the expense of naked cuticle (Nüsslein-Volhard et al., 1984; Nüsslein-Volhard et al., 1985), whereas high levels of Wg signaling lead to excess naked cuticle and loss of denticles (Noordermeer et al., 1992). Epistasis experiments from many laboratories have used these phenotypes to identify genes required for proper Wg signaling. The signaling components identified in Drosophila are conserved with vertebrates, and so this body of work has led to the following general model for Wg/Wnt signaling. In the absence of Wg/Wnt signaling, Axin, Apc, Casein kinase 1 and Zeste-white3 (Zw3; also known as Shaggy, Sgg)/glycogen synthase kinase 3β (GSK) form the destruction complex, which phosphorylates Armadillo/beta-catenin (Arm/beta-cat) and targets it for degradation. Wg/Wnt binds to a receptor complex of Frizzled (Fz) and Arrow (Arr)/LRP5/6, which inactivates the destruction complex and stabilizes Arm/beta-cat. Arm/beta-cat then enters the nucleus where it displaces a transcriptional co-repressor, Groucho (Gro), from the transcription factor Tcf (Pan in Drosophila). This converts Tcf from a transcriptional repressor to a transcriptional activator as Arm recruits Legless (Lgs)/BCL9 and Pygopus (Pygo), leading to transcription of Wg target genes (reviewed by Bejsovec, 2006; Clevers, 2006).
Additional negative regulators of the Wg pathway have been identified in genetic screens using Drosophila cuticle pattern as an assay system. Some showed pleiotropic phenotypes that had prevented them from being connected with the Wg/Wnt pathway prior to these screens. For example, the RacGTPase Activating Protein Tumbleweed (Tum) and the kinesin-like protein Pavarotti (Pav) have essential roles in cytokinesis (Somers and Saint, 2003), but also can downregulate the Wg/Wnt pathway (Jones and Bejsovec, 2005; Jones et al., 2010). In their cytokinesis role, they interact with Pebble (Pbl), a Rho guanine nucleotide exchange factor (RhoGEF), to form the centralspindlin complex. This complex is essential for positioning the actin contractile ring during cell division (Prokopenko et al., 1999; Somers and Saint, 2003). The maternal contributions of tum, pav or pbl gene product allow zygotically mutant embryonic cells to divide normally until mitotic cycle 14, after which division fails and binucleate cells accumulate in each mutant (Adams et al., 1998; Prokopenko et al., 1999; Zavortink et al., 2005). Pbl plays an additional role in regulating mesodermal spreading in Drosophila embryos, and so pbl mutant embryos also show gastrulation defects (Schumacher et al., 2004; Smallhorn et al., 2004). The human homolog of Pbl, ECT2, is likewise essential for cytokinesis and has additional roles in cell polarity and oncogenesis (Tatsumoto et al., 1999; Fields and Justilien, 2010). Epithelial cell transforming sequence 2 (ECT2) was designated a proto-oncogene based on the transforming capacity of truncated forms that lack the N-terminus or the nuclear localization signal (Miki et al., 1993; Saito et al., 2004). Full-length ECT2 does not show transforming potential in cultured cells, but it is overexpressed in a number of human cancers (Saito et al., 2004; Hirata et al., 2009; Justilien and Fields, 2009). This has led to debate about whether ECT2 overexpression contributes to or is a consequence of oncogenic transformation.
Because Tum and Pav downregulate Wg/Wnt activity, we tested whether their binding partner, Pbl, and its human homolog ECT2, also affect the Wg/Wnt pathway. Previous work in Drosophila hinted that Pbl might influence Wg-mediated patterning. Mutations in pbl were isolated in a genetic screen for mutations that disrupt cuticle pattern (Jürgens et al., 1984), but the epidermal defect was not characterized further. In addition, a deletion that removes pbl, as well as several other genes, was identified as an enhancer of arm gain-of-function phenotypes in the wing (Greaves et al., 1999). Here, we show that Pbl inhibits Wg signaling in Drosophila embryos and that both Pbl and ECT2 repress Wg/Wnt target gene expression in Drosophila and mammalian cells in culture.
RESULTS
Pbl is a negative regulator of Wg signaling
Loss-of-function pbl mutants die at the end of embryogenesis, with epidermal pattern defects that suggest a de-regulation of Wg signaling. Zygotic pbl3 mutants show pleiotropic effects on mesoderm development that disrupt gastrulation; these defects complicate the analysis of mutant cuticle pattern. However, we found that the ventral cuticle of pbl mutants consistently showed replacement of denticles with ectopic naked cuticle, compared with wild type (Fig. 1A,B). This epidermal phenotype was seen more clearly when the gastrulation defects were rescued. We used a twist-Gal4 driver to provide wild-type pbl in the mesoderm, causing less distortion to the mutant cuticle and a better view of its excess naked cuticle specification (Fig. 1C), which is diagnostic of Wg hyperactivity (Noordermeer et al., 1992). We also examined molecular markers of Wg signaling in pbl mutants. Although we did not see significant changes in expression of the Wg target genes engrailed (en) or wg itself (not shown), we did detect a very slight elevation of Arm protein levels in the normal striped pattern of Arm accumulation (Fig. 1E, compare with wild-type sibling to right, marked with GFP balancer chromosome in 1F). Because Arm is stabilized by Wg signaling, any increase in Arm levels also would be consistent with Wg hyperactivity.
Fig. 1.
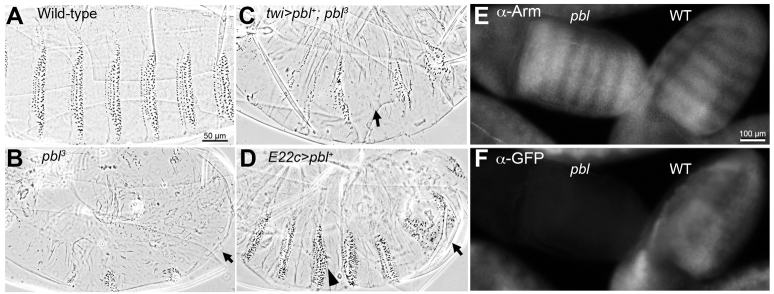
pbl mutant embryos show defects consistent with Wg pathway regulation. (A) Wild-type embryos secrete a ventral cuticle pattern with naked cuticle separating eight abdominal denticle belts. Abdominal segments 2-7 are shown here. (B) Homozygous pbl3 zygotic mutants showed severe gastrulation defects, with reduced denticle belts (arrow). (C) Rescuing mesodermal defects by driving UAS-pbl with twist(twi)-Gal4 provided a better view of the epidermal pattern: pbl3 mutants showed a reduced number of denticle belts (arrow) as well as a reduced number of denticle rows per belt compared with wild type. (D) Driving ubiquitous epidermal UAS-pbl expression with E22c-Gal4 resulted in an expansion of denticle belts, replacing naked cuticle (arrowhead), as well as in head and dorsal closure defects. In addition, 40.2% of the embryos showed denticle belt fusions due to complete loss of naked cuticle in the affected segment (arrow). (E) Arm antibody staining in pbl3 mutants showed normal striping (left), with slightly higher intensity of fluorescence in mutants than in wild-type siblings (right) stained under identical conditions (n>100). (F) GFP staining revealed presence of the twi >GFP-marked balancer. Anterior is to the left in all panels.
We next tested whether pbl gain of function would produce the opposite effect on epidermal patterning. We used the E22c-Gal4 driver to express full-length pbl ubiquitously in embryonic epidermis. This overexpression resulted in 100% embryonic lethality (n=385), with 40.2% of the embryos showing some loss of naked cuticle that normally separates the denticle belts (Fig. 1D; Table 1). As naked cuticle reflects Wg signaling activity in epidermal cells, its loss suggests antagonism of signaling. In addition, overexpressing pbl in the epidermis caused other developmental defects, such as missing head structures, failure to retract the germ band, and incomplete dorsal closure (Fig. 1D). We also found that overexpressing pbl in the epidermis slightly reduced Arm protein levels. This was most easily visualized by driving transgene expression in alternate segments with prd-Gal4, leaving even-numbered abdominal segments unaffected. Driving UAS-GFP in the prd domain had no effect on Arm stripes (Fig. 2A,B), whereas driving UAS-pbl diminishes Arm antibody staining in the prd-expressing cells (Fig. 2C,D). Thus, in embryos, both loss- and gain-of-function pbl phenotypes are consistent with a role for wild-type Pbl in negatively regulating Wg signaling.
Table 1.
Phenotypes of Pbl overexpression in embryonic epidermis

Fig. 2.

Pbl acts to repress Wg pathway activity in embryos and cultured cells. (A,B) Normal Arm stripes were not altered by prd-Gal4-driven expression of UAS-GFP. B shows merged signals: Arm in red, GFP in green. (C,D) prd-Gal4 driving UAS-pbl diminished Arm accumulation in the prd domain, marked by co-expression with UAS-GFP (D shows merge). (E) S2R+ cells activate DROPflash reporter in response to Wg-CM. This was unaffected by addition of GFP control DNA. Addition of pbl or Axin, a component of the destruction complex, repressed DROPflash activation to 37% and 9% of GFP control, respectively (*P<0.001). (F) qPCR showed that pbl repressed activation of the endogenous Wg target genes nkd and CG6234 to 28% and 68%, respectively, compared with control GFP. The Axin positive control repressed nkd and CG6234 to 4% and 30%, respectively (*P<0.001). (G) Conversely, knocking down pbl with RNAi elevated expression of both nkd and CG6234. (H) pbl RNAi resulted in accumulation of multinucleate cells (right) due to defects in cytokinesis, whereas mock RNAi-treated cells continued to divide normally (left). Rhodamine phalloidin stains actin (red), DAPI stains DNA (blue). Error bars represent s.e.m.
We also observed a negative regulatory effect on cells in culture. We used the DROPflash reporter to monitor pathway activity in Wg-responsive Drosophila S2R+ cells (Yanagawa et al., 1998). DROPflash contains six tandem TCF binding sites that drive luciferase expression when the Wg pathway is activated (Li et al., 2007). We treated S2R+ cells with Wingless-conditioned media (Wg-CM), and found that overexpressing pbl reduced DROPflash activity to 37% of control levels (P<0.001; Fig. 2E). We compared this to the effect of Axin, which is a known negative regulator of the Wg pathway. As expected, Axin expression repressed Wg-CM induction of DROPflash to 9% of control levels (P<0.001; Fig. 2E). Thus, excess Pbl downregulates Wg reporter activity in cultured fly cells, but does not do so as strongly as overexpression of a destruction complex protein.
Ectopic Pbl also inhibited the expression of endogenous genes in S2R+ cells. The Wg target genes nkd and CG6234 are transcriptionally activated by addition of Wg-CM (Fang et al., 2006). We quantified their expression levels in Wg-CM-induced cells by real-time PCR, and found that overexpressing pbl reduced nkd and CG6234 expression to 28% and 68% of control levels, respectively (P<0.001; Fig. 2F). As expected, overexpression of Axin repressed nkd and CG6234 to 4% and 30% of control levels, respectively (P<0.001; Fig. 2F). Moreover, knocking down pbl gene function, using RNA interference (RNAi), produced the opposite effect of overexpressing it. We treated cells with pbl double-stranded RNA and found that expression of both target genes was elevated above control levels (Fig. 2G), as they are when cultured cells are treated with Axin RNAi (Blauwkamp et al., 2008). Although overexpression of pbl did not disrupt cell division (not shown), knocking it down with RNAi did: treated cultures accumulated multinucleate cells whereas control-treated cultures continued to divide normally (Fig. 2H). Thus, native promoters of Wg target genes are responsive to Pbl repression in vitro, independently of its effect on cell division.
Pbl requires GEF domain activity to regulate Wg signaling
Pbl is a complex protein with multiple protein-binding motifs in addition to its GEF domain (Fig. 3A). A series of mutated or truncated Pbl cDNA clones were constructed and used to determine which domains were important for cytokinesis, mesodermal spreading and subcellular protein localization (Smallhorn et al., 2004; van Impel et al., 2009). We tested a similar series of mutated Pbl constructs in S2R+ cells to determine which domains of Pbl are important for Wg regulation, assaying both DROPflash reporter activity and expression of endogenous genes (Fig. 3C,D). The N-terminus contains two BRCT protein-protein interaction domains, which are necessary for cytokinesis, but are not essential for mesodermal spreading (Somers and Saint, 2003; Smallhorn et al., 2004; van Impel et al., 2009). Tum physically interacts with the first BRCT domain of Pbl (Somers and Saint, 2003). We found that expression of pblΔBRCT1 or pblΔBRCT1&2 repressed DROPflash significantly compared with control values (P<0.001; Fig. 3C). Indeed, these constructs consistently repressed DROPflash more strongly than did full-length Pbl. pblΔBRCT1 or pblΔBRCT1&2 also significantly repressed expression of nkd and CG6234 (P<0.001; Fig. 3D). Thus, the BRCT domains that are required for cytokinesis and for Tum binding are not necessary for Pbl repression of Wg signaling.
Fig. 3.

Pbl requires GEF activity to regulate Wg signaling. (A) Protein domains of full-length and mutated forms of Pbl. Domains indicated from N to C terminus are BRCT (BRCA1 C-terminal domain), NLS (nuclear localization sequence), PEST (rich in proline, glutamic acid, serine, and threonine), DH (Dbl homology) and PH (pleckstrin homology). Yellow line marks the position of the V531D mutation, which disrupts the GEF domain. (B) Mutating the NLS of Pbl prevented localization to the nucleus (right panel) compared with wild-type hemagglutinin (HA)-tagged Pbl (left panel). Overlap of anti-HA (red) and DAPI (blue) appears bright pink. (C) In S2R+ cells induced with Wg-CM, DROPflash reporter activity was inhibited by pblΔBRCT1, pblΔBRCT1&2, pblΔNLS, pblΔPEST, pblΔN-term, pblDH-PH or Axin compared with GFP control (*P<0.001). Disrupting the GEF domain (pblΔN-termV531D, pblDH-PHV531D and pblV531D) abolished this repression. (D) Wg-induced expression of nkd and CG6234 was repressed by pblΔBRCT1, pblΔBRCT1&2, pblΔNLS, pblΔN-term, pblDH-PH or the Axin positive control (*P<0.001). The V531D mutation abolished this repression. (E-L) Wild-type cuticle pattern (E) was altered by E22c-Gal4 driven epidermal expression of UAS-pblΔBRCT1&2 (F), UAS-pblDH-PH (G), UAS-pblΔN-term (H) and UAS-pblΔNLS (I), but not of UAS-pblV531D (J), UAS-pblDH-PHV531D (K) or UAS-pblΔN-termV531D (L). Denticle belt fusions (arrows in F,G) indicate antagonism of Wg signaling. UAS-pblΔN-term (H) and UAS-pblΔNLS (I) produced severely fragmented cuticles. Mutating the GEF domain rescued this fragmentation: E22c-driven UAS-pblΔN-termV531D (L) was indistinguishable from wild type. Error bars represent s.e.m.
The central region of Pbl contains a nuclear localization sequence (NLS) and a PEST degradation sequence (Fig. 3A). A construct that lacks this region, pblΔN-term, localized constitutively to the cytosol, disrupting cytokinesis and mesoderm invagination (van Impel et al., 2009). Because Tum and Pav require functional NLSs to act as negative regulators of Wnt signaling (Jones et al., 2010), we investigated whether nuclear localization was important for Pbl function. Mutating the NLS (pblΔNLS) or deleting the N-terminus (pblΔN-term and pblDH-PH) prevented these proteins from localizing to the nucleus (van Impel et al., 2009). These forms of Pbl, however, were still able to repress DROPflash and endogenous Wg target genes (Fig. 3C,D). Although expression of pblΔN-term in embryos led to multi-nucleated cells, we did not observe this phenotype during the time course of our cell culture experiments (data not shown). We also mutated the PEST sequence alone but did not observe any alteration in the repression by Pbl of DROPflash induction by Wg-CM (Fig. 3C).
The C-terminal region of Pbl contains the DH-PH domain, which is crucial for GEF activity (Prokopenko et al., 1999). The pblDH-PH construct consists only of this portion of the molecule, yet was sufficient to mediate Wg repression (Fig. 3C,D), although somewhat less effectively than the full-length protein. To test directly the role of the GEF activity of Pbl in Wg repression, we introduced a point mutation (V531D) in the DH domain. This missense change is known to compromise the catalytic activity of Pbl: it abolished all dominant phenotypes associated with pblΔN-term expression (van Impel et al., 2009). Constructs containing the V531D mutation (pblV531D, pblΔNtermV531D, pblDH PHV531D) were no longer able to repress DROPflash activity or Wg target gene induction by Wg-CM (Fig. 3C,D). Thus, GEF activity is essential for Pbl regulation of Wg signaling.
We next tested the function of these mutated Pbl proteins in vivo, using transgenic fly lines to overexpress each construct in the embryonic epidermis with E22c-Gal4. Overexpression of pblΔBRCT1&2 resulted in 95.7% embryonic lethality with loss of naked cuticle separating the denticle belts in 40.6% (n=352) of embryos; pblDH-PH resulted in 57.3% embryonic lethality with loss of naked cuticle separating the denticle belts in 49% (n=363) of embryos (Fig. 3F,G; Table 1). These results confirm that Tum binding is not essential for antagonism by Pbl of Wg signaling, and that the DH-PH domain alone is sufficient for this function. Expression of pblΔBRCT1&2, like full-length pbl, produced other developmental phenotypes, such as missing head structures and defects in germ band retraction and in dorsal closure (note the curvature of embryonic cuticle in Fig. 1D and Fig. 3F, due to dorsal defects). These pleiotropic phenotypes were not observed with pblDH-PH overexpression, indicating that they were not related to the role of Pbl in Wg signaling. Instead, these defects may stem from regulatory factors that interact with the C-terminus: this region is present in pblΔBRCT1&2 but absent in pblDH-PH. Lethality and loss of naked cuticle phenotypes observed with full-length pbl or pblDH-PH were eliminated by introducing the V531D GEF mutation into these constructs (Fig. 3J,K; Table 1). Thus, GEF activity is required for Pbl to antagonize Wg-mediated patterning in vivo, just as it is in vitro.
Expression of the pblΔN-term (n=271) or pblΔNLS (n=529) transgenes resulted in epidermal fragmentation that prevented analysis of cuticle pattern (Fig. 3H,I; Table 1). The forced cytosolic localization of these mutant proteins, which lack the signals for nuclear translocation, may trigger inappropriate Rho1 activation early in development. Consistent with this idea, overexpression of constitutively active Rho1V12 in the epidermis yielded a similar phenotype (data not shown). Again, the dramatic dominant phenotype depended on GEF activity: effects of pblΔN-term were completely suppressed when the V531D GEF mutation was introduced (Fig. 3L; Table 1). We did not observe any adverse effect on cell morphology when overexpressing pblΔN-term or pblΔNLS in cell culture (data not shown). Like the full-length pbl, these constructs repressed Wg response in a GEF-dependent fashion (Fig. 3C,D). Thus, the epidermal fragmentation we observed in embryonic cuticles probably results from disruption of cell-to-cell contacts, which are not essential for cells growing in culture.
Pbl functions downstream of Arm and does not act through Arm destabilization
Most negative regulators of the Wg/Wnt pathway are associated with the destruction complex, which controls Arm/beta-cat stability. We expected that Pbl functions at this level as well, because it appeared to modulate Arm accumulation in the embryo. However, when we tested its effects on cells induced by overexpression of arm or an artificially stabilized arm, we were surprised to find that Pbl can still repress pathway activation. The stabilized form, arm*, carries mutations in the Casein kinase 1α phosphorylation sites; these sites are required to prime subsequent Zw3/GSK phosphorylation and therefore arm* protein is resistant to degradation (Yanagawa et al., 2002; Blauwkamp et al., 2008). Wild-type arm overexpression induced DROPflash expression in S2R+ cells at levels similar to those induced by Wg-CM, suggesting that this is sufficient to overwhelm destruction complex activity. This reflects another difference between cultured cells and intact embryos: overexpressing wild-type arm in embryos does not disrupt pattern (Orsulic and Peifer, 1996), only the stabilized form does so. Overexpressing full-length pbl repressed the induction of DROPflash by either arm or arm* to 53% of control GFP expression (P<0.001; Fig. 4A). As expected, overexpression of Axin, a component of the destruction complex, was able to repress DROPflash induced by arm (P<0.001), but not DROPflash induced by the degradation-resistant arm* (Fig. 4A). These data indicate that pbl functions downstream not only of the Wg receptor complex, but also downstream of Arm stabilization. Furthermore, all of the mutated forms of Pbl affected arm- and arm*-induced pathway activation in similar fashion to their effect on Wg-CM induced cells. Expressing pblΔBRCT1, pblΔBRCT1&2, pblΔNLS, pblΔN-term and pblDH-PH repressed arm and arm* induction of DROPflash, but constructs bearing the GEF mutation did not show repression (Fig. 4A).
Fig. 4.

Pbl acts downstream of Arm stabilization. (A) pbl repressed induction of DROPflash by arm and arm* to 53% of control GFP. By contrast, Axin repressed arm-induced DROPflash to 26% of GFP control but was unable to repress arm*-induced DROPflash, consistent with its destruction complex role. pblΔBRCT1, pblΔBRCT1&2, pblΔNLS, pblΔN-term and pblDH-PH repressed arm and arm* induction of DROPflash (*P<0.001), whereas GEF-mutated pblΔN-termV531D, pblDH-PHV531D and pblV531D did not (P>0.05). (B) In fly embryos, arm-Gal4 driven expression of the stabilized UAS-armS10 resulted in excess naked cuticle and few denticle belts. Co-expressing UAS-GFP did not alter this cuticle phenotype. Arrowhead indicates position of the ventral midline in B-D. (C) Co-expressing UAS-pblDH-PH diminished excess naked cuticle specification and increased the proportion of embryos with partial denticle belts, where denticles were continuous across the ventral midline. (D) Co-expressing UAS-pblDH-PHV531D did not alter excess naked cuticle specification. (E) Quantification of denticle belt status in larval cuticles (n>100 for each cross). Cuticles representing the median phenotype for each genotype are shown in B-D. Error bars represent s.e.m.
To verify that Pbl functions downstream of Arm stabilization in vivo, we overexpressed the armS10 transgene in the fly embryo. This transgene carries a deletion of phosphorylation sites that results in a stabilized form of arm; when ubiquitously expressed in the embryonic epidermis, armS10 produces excess naked cuticle at the expense of denticles (Pai et al., 1997). We found that co-expressing armS10 with full-length pbl or pblΔBRCT1&2 produced severe developmental defects, which prevented us from analyzing the cuticle pattern (data not shown). However, co-expressing armS10 with pblDH-PH suppressed the specification of excess naked cuticle, compared with armS10 co-expressed with the UAS-GFP control (Fig. 4B,C,E). Again, this effect requires GEF activity because the GEF-mutated pblDH-PHV531D construct did not suppress naked cuticle when co-expressed with armS10 (Fig. 4D,E). These observations are consistent with the idea that pbl, like tum (Jones et al., 2010), functions downstream of Arm degradation both in Drosophila cultured cells and in embryos.
ECT2 is a negative regulator of Wnt signaling in human HEK293T cells
ECT2, the human homolog of Pbl, has a protein structure similar to Pbl but with a longer N-terminus and with additional NLSs in the central domain and C-terminus (reviewed by Fields and Justilien, 2010) (Fig. 5A). ECT2 was first isolated as a cDNA that exhibits transforming activity in cell lines (Miki et al., 1991). Subsequently, it was discovered that the original cDNA encoded an N-terminal deletion that eliminates the BRCT domains and the two N-terminal NLSs (Saito et al., 2004). Eliminating the NLSs alone also resulted in a form of ECT2 with transforming activity, although the phenotype was not as strong as the longer N-terminal deletion (Saito et al., 2004). Full-length ECT2 is highly expressed in several human cancers, including those of the brain, lung, bladder, esophagus and pancreas (reviewed by Fields and Justilien, 2010). Because Wnt signaling is misregulated in a variety of cancers, we investigated whether ECT2, like Pbl, negatively regulates Wg signaling.
Fig. 5.

ECT2 and Pbl modulate Wnt signaling in HEK293T cells. (A) ECT2 domains are similar to Pbl, but ECT2 has a longer N-terminus, no PEST sequence, and multiple NLS motifs. The N-terminus contains regions homologous to human XRCC1, a DNA repair enzyme, and yeast Cyclin B6, a cell cycle regulator. Ect2ΔNLS1,2,&3 mutates all three NLSs. (B) ECT2 repressed Wg-CM-induced DROPflash to 32% of control GFP in S2R+ cells. Mutating the GEF domain as described by Saito et al. (Saito et al., 2004) diminished this effect to 66% of control GFP (*P<0.001). (C) In human HEK293T cells induced with Wnt3a-CM, pbl or ECT2 reduced TOPflash reporter activity to 57% or 56%, respectively, of control GFP, compared with Axin inhibition to 3% of control GFP. Ect2ΔBRCT1, Ect2ΔBRCT1&2, Ect2ΔNLS1,2,&3, EctΔN-term and Ect2DH-PH repressed Wnt3a-CM-induced TOPflash to an extent similar to full length ECT2 (*P<0.001). GEF domain mutations diminished ECT2 repression to 77% of GFP control (**P<0.05). (D) TOPflash induced in HEK293T cells by transfection with ΔGSK beta-cat cDNA, was repressed to 55% and 53% of control levels by co-transfecting with pbl or ECT2, respectively. Ect2ΔBRCT1, Ect2ΔBRCT1&2, Ect2ΔNLS1,2,&3, Ect2ΔN-term or Ect2DH-PH showed similar repression ofΔGSK-induced TOPflash (*P<0.001). GEF mutations in Pbl abolished repression ofΔGSK-induced TOPflash whereas analogous GEF mutations in ECT2 substantially diminished repression (**P<0.05). No changes were observed in FOPflash activity. Error bars represent s.e.m.
We first expressed human ECT2 in Drosophila S2R+ cells and found that it inhibited the Wg-CM-induced activation of DROPflash by 32% compared with control (P<0.001; Fig. 5B). This effect was diminished, but not completely eliminated, when mutations predicted to disrupt GEF activity (Liu et al., 1998) were introduced (Fig. 5B). Unlike the potent V531D mutation in Pbl, mutating the corresponding conserved V residue and adjacent residues in mammalian DH-PH domain GEFs only partially reduced their GEF activity (Liu et al., 1998). We next tested the effects of both ECT2 and Pbl on Wnt signaling in mammalian cells. We monitored TOPflash reporter activity in human embryonic kidney 293T (HEK293T) cells. The TOPflash construct carries three copies of Tcf-optimal binding sites that drive luciferase expression in response to Wnt pathway activation, whereas the FOPflash negative control carries mutated copies of the Tcf sites. Expression of either Drosophila pbl or human ECT2 in HEK293T cells repressed Wnt3a-CM-induced TOPflash activity to 57% and 56%, respectively, of control (P<0.001; Fig. 5C). As expected, neither had any effect on FOPflash (Fig. 5C).
We used the TOPflash system to test each predicted ECT2 functional domain for possible contribution to Wnt regulation. We constructed mutations in ECT2 analogous to those made in Pbl, and expressed Ect2ΔBRCT1, Ect2ΔBRCT1&2, Ect2ΔNLS1,2,&3 (in which all NLSs are mutated), Ect2ΔN-term and Ect2DH-PH in HEK293T cells. All repressed Wnt3a-CM-induced TOPflash activity to a similar degree, with no effect on FOPflash (Fig. 5C). We further found that mutating the GEF domain diminished the ability of ECT2 to repress TOPflash induction (Fig. 5C), just as it did for DROPflash induction in Drosophila S2R+ cells (Fig. 5B). Thus, ECT2 probably requires GEF activity to regulate Wnt signaling, consistent with what we had observed for Pbl function in Drosophila embryos and cultured cells.
We next tested whether ECT2, like Pbl, acts downstream of beta-catenin stabilization. We induced TOPflash activity using ΔGSK-beta-cat (ΔGSK), a constitutively active form of beta-catenin carrying mutations in the GSK-3beta and Casein Kinase 1alpha phosphorylation sites. We expressed pbl, ECT2, Ect2ΔBRCT1, Ect2ΔBRCT1&2, Ect2ΔNLS1,2,&3, EctΔN-term or Ect2DH-PH in HEK293T cells and found that all repressed ΔGSK-induced TOPflash activity (Fig. 5D). The V531D mutation in Pbl, which eliminated the activity of Pbl in S2R+ cells, also eliminated the ability of Pbl to repress ΔGSK-induced TOPflash activity (Fig. 5D). Similarly, mutations to the ECT2 GEF domain partially eliminated ECT2 repression of ΔGSK induction, just as was observed with Wnt3a induction. Thus, ECT2 RhoGEF activity blocks Wnt signal transduction downstream of beta-catenin stabilization, just as Pbl does, indicating that this novel mechanism for modulating Wnt signaling is highly conserved.
Pbl may regulate Wg signaling by modulating the activity of Rho1
Because GEF activity was crucial to Pbl-mediated Wg regulation, we tested known GTPases to determine which might be responsible for the Pbl effect. Pbl is known to act as an exchange factor for Rho1 during cytokinesis (Prokopenko et al., 1999), and in vitro exchange assays using the Pbl DH domain demonstrated that Pbl also has GEF activity towards Rac1 and Rac2 (van Impel et al., 2009). However, as full-length Pbl was not used in these experiments it is possible that Pbl has activity towards other Rho family GTPases as well. Therefore, we examined Cdc42, RhoL and Mtl as well as Rho1, Rac1 and Rac2 to determine whether any of these GTPases might play a role in Wg regulation. We found that all GTPases tested significantly reduced DROPflash activity (P<0.015) when overexpressed in S2R+ cells, with Rho1, Rac1 and Rac2 showing the greatest effect (Fig. 6A). This was observed with induction either by Wg-CM (not shown) or by transfection with wild-type arm (shown). Pbl co-expression (pink columns) further repressed DROPflash activity (P<0.0015), except in the case of Rho1 (P>0.05). Thus, Rho family GTPase activation in general can interfere with Wg/Wnt signal transduction, but Rho1 appears to be the one through which Pbl acts.
Fig. 6.

Pbl acts through Rho1 to modulate Wg/Wnt signaling. (A) arm-induced DROPflash activity in S2R+ cells was monitored after addition of GFP control DNA (green bars) versus pbl DNA (pink bars). All GTPases tested showed some repression compared with GFP alone (P<0.015). Further repression was observed when pbl was added (P<0.0015) except in the case of Rho1 (P>0.05). (B) Dominant-negative GTPases did not diminish Pbl-mediated repression of activity (lavender bars) except for dominant-negative Rho1 (P<0.01). Only Rac1N17 showed significant repression of DROPflash on its own (P<0.001). (C) Rho1720, a strong allele, shows some haploinsufficiency when outcrossed away from the CyO balancer. More than 25%, the expected proportion of homozygous embryos, fail to hatch. The proportion of embryos showing the two most severe classes of pattern defects (25.4%; right-hand panels), was consistent with these classes representing Rho172O homozygotes. These two classes show an excess of naked cuticle at the expense of denticles. Arrows indicate pattern disruptions. (D) Model for how Pbl RhoGEF could influence the Wg/Wnt pathway, either directly through activation of a Wnt-specific effector or indirectly through general alteration of actin cytoskeleton properties. Error bars represent s.e.m.
We also co-expressed pbl with dominant-negative forms of Rho1, Rac1, Rac2, Cdc42, RhoL and Mtl to test loss-of-function rather than gain-of-function phenotypes. If Pbl influences Wg signaling through Rho1 activation, then one would predict that dominant-negative Rho1 should abrogate the effects of Pbl overexpression. This is exactly what we observed. None of the dominant-negative GTPases tested interfered with Pbl-mediated Wg/Wnt repression except for dominant-negative Rho1 (Fig. 6B). We next examined Rho172O mutant embryos and found that in addition to well-characterized defects in head involution and dorsal closure (Strutt et al., 1997; Bloor and Kiehart, 2002; Jacinto et al., 2002), zygotic mutants show excess naked cuticle replacing the ventral denticles (Fig. 6C). Thus. loss of Rho1 function results in patterning defects consistent with hyperactivation of the Wg/Wnt pathway, just as Pbl loss of function does. We propose that the Pbl RhoGEF acts through Rho1 to negatively regulate Wg/Wnt signaling.
DISCUSSION
We had originally investigated Pbl because of its interaction with the Wnt regulators Tum and Pav during cytokinesis. Our data show that although Pbl does regulate Wnt signaling, it does so independently of Tum and Pav. First, Pbl regulation of Wg signaling does not require the Tum-binding BRCT1 domain. Indeed, some constructs lacking this region were more effective than full-length Pbl in repressing DROPflash. Second, Tum and Pav require nuclear localization to regulate Wg signaling (Jones et al., 2010). By contrast, we found no nuclear role for Pbl or ECT2 in Wg/Wnt regulation. Third, Pbl regulation of Wg/Wnt signaling correlates with its GEF activity, whereas Tum does not require GAP activity to regulate Wg signaling (Jones and Bejsovec, 2005; Jones et al., 2010). Together, these results suggest that although Tum, Pav and Pbl act together in a complex during cytokinesis, they play distinctly different roles in their influence on the Wg/Wnt pathway. Our analysis of Pbl and ECT2 domain requirements further established distinctions between cytokinesis and signaling roles: for example, the BRCT domains of Pbl or ECT2 are essential for cytokinesis (Saito et al., 2003; van Impel et al., 2009) but are not required for Wg/Wnt inhibition (Fig. 3C,D; Fig. 5C,D).
Most negative regulators of Wnt signaling alter Arm/beta-cat stabilization; this is true of Axin, which consistently repressed the pathway more effectively than did Tum, Pav or Pbl. However, we found that Pbl/ECT2 RhoGEF can repress Wg/Wnt activity induced by stabilized forms of Arm/beta-cat (Fig. 4; Fig. 5D), which Axin cannot do (Fig. 4A). Thus Pbl/ECT2 functions downstream of the destruction complex to modulate expression of target genes. Although the mild changes in endogenous Arm stability observed in pbl loss- and gain-of-function conditions (Fig. 1E,F; Fig. 2C,D) are consistent with mutant phenotypes, they may instead be a secondary effect of RhoGEF on Arm localization. Our data suggest that Pbl/ECT2 acts on Arm/beta-cat at a point between its stabilization and its translocation into the nucleus. We can imagine several ways in which this could occur (Fig. 6D). First, Wg/Wnt repression could result from a general effect on the actin cytoskeleton. Rho family GTPases regulate formins and other effector molecules that control actin dynamics (Symons and Settleman, 2000; Bustelo et al., 2007). Both Arm/beta-cat and Apc are known to interact with the actin cytoskeleton (Weis and Nelson, 2006; Moseley et al., 2007; Okada et al., 2010) and to assume a cortical localization in polarized epithelial cells (Peifer, 1993; McCartney et al., 1999). Changes in the actin network might alter subcellular localization or activity of Arm. Alternatively, GTPase activation of an effector molecule might have a direct effect on some component of the Wnt pathway or of a parallel signaling pathway that antagonizes Wnt signaling. For instance, Diaphanous, a Rho1 effector, interacts genetically and physically with Apc2 (Webb et al., 2009). All GTPases tested had some capacity to repress Wg/Wnt signaling, raising the possibility that any change in the actin cytoskeleton can negatively influence signaling and/or that multiple effectors are involved. Alternatively, these GTPases may cross-activate Rho1, and only Rho1 activation mediates the Pbl regulation of Wnt signaling. We are currently exploring these possibilities.
The role of Rho GTPases in planar cell polarity, a non-canonical Wnt signaling pathway, has been well-studied, but growing evidence suggests that Rho GTPases modulate canonical Wnt signaling in mammalian cells. Several GEFs potentiate Wnt signaling by activating Rac1: the GEF DOCK4 promotes beta-cat degradation, whereas the GEF TiamI is recruited to Wnt-responsive promoters where it activates Rac1 and promotes transcription (Buongiorno et al., 2008; Upadhyay et al., 2008). Rac1 also appears to be activated by Wnt signaling, leading to activation of JNK2 (MAPK9) kinase and nuclear import of beta-catenin (Wu et al., 2008). By contrast, we found that Pbl antagonizes Wnt signaling through Rho1 activation. Thus, Rho GTPases can have either positive or negative effects on Wnt signaling. The challenge now is to identify the effector molecules with which these G proteins interact in their activated, GTP-bound forms to modulate Wnt pathway output.
Our finding that ECT2 negatively regulates Wnt signaling in human cells suggests that it would be protective against oncogenesis. Prior work categorizing ECT2 as a proto-oncogene was based on clones that lacked the N-terminus or nuclear localization signals. Although full-length ECT2 cannot transform cells (Saito et al., 2004), full-length ECT2 is overexpressed in many human tumors (Saito et al., 2003). It is unclear if the upregulation observed in tumors is a cause versus a consequence of oncogenesis; it may simply reflect the essential role of ECT2 in the cell cycle. No upregulation of ECT2 is observed in colorectal cancer, the cancer most strongly associated with aberrations in Wnt signaling (Fields and Justilien, 2010). The full role that ECT2 plays in human tumorigenesis is just beginning to be explored (Justilien and Fields, 2009) and our work demonstrates that any proposed role for ECT2 in cancer must consider its conserved function in inhibiting Wnt signaling.
MATERIALS AND METHODS
Drosophila stocks, embryo collection, and imaging
All stocks were obtained from the Bloomington Stock Center (Bloomington, IN, USA) except UAS-pbl, UAS-pblΔBRCT (for clarity referred to in this manuscript as UAS-pblΔBRCT1&2), UAS-pblΔN-term, UAS-pblΔN-termV531D, UAS-pblDH-PH and UAS-pblDH-PHV531D, which were a gift from Arno Muller (University of Dundee, UK). UAS-pblV531D and UAS-pblΔNLS were created by Gateway cloning (Invitrogen). To examine cuticles, eggs were allowed to develop for 24 hours at 25°C, dechorionated and mounted in Hoyer’s medium (Wieschaus and Nüsslein-Volhard, 1986). Antibody staining was performed on embryos at developmental stage 9, as described by Chao et al. (Chao et al., 2007). Images were captured with SPOT camera (Diagnostic Instruments, Sterling Heights, MI, USA) on a Zeiss Axioplan microscope, and were processed with SPOT imaging and Adobe Photoshop.
Plasmids
cDNAs for pbl, Rho1, Rac1, Rac2, Cdc42, RhoL and Mtl were obtained from the Drosophila Genomics Research Center (DGRC, Bloomington, IN, USA). ECT2 cDNA is from Open Biosystems (Fisher Scientific). Deletions and mutation of Pbl and ECT2 were made as described previously (van Impel et al., 2009; Saito et al., 2004). The following are not described in the cited papers and encode the following amino acids of Pbl or ECT2 proteins: pblΔBRCT1 consists of aa 207-853; pblΔNLS mutates aa 316, 318, 319 and 320 to alanine; pblΔPEST mutates aa 373-377 to alanine; and Ect2ΔBRCT1 only encodes aa 221-883. pblV531D, pblΔNLS, pblΔPEST, Ect2ΔGEF and all dominant-negative Rho1, Rac1, Rac2, Cdc42, RhoL and Mtl were made using site-directed mutagenesis (QuikChange II, Agilent Technologies). arm and arm* were constructed by PCR amplifying transgenic sequences from the UAS-armS2 (Orsulic and Peifer, 1996) and UAS-arm* (Chang et al., 2008) fly stocks, respectively. For Drosophila cell culture, we used the pAW vector (DGRC), which drives expression with the Drosophila Actin 5C promoter, and for HEK293T cells we used the pcDNA3.1/nV5-DEST vector (Invitrogen), which drives expression with the CMV promoter. The Renilla luciferase from phRG-TK (Promega) was moved into the pHW vector (DGRC), driven by Drosophila hsp70 promoter, to make the pHW-Renilla control.
Cell culture and luciferase assays
S2, S2-tub-wg and S2R+ Drosophila cell lines were obtained from the DGRC and grown in Schneiders Drosophila media (Gibco/Invitrogen) with 10% fetal bovine serum (FBS) and 1% Antibiotic and Antimycotic (Invitrogen). S2 and S2-tub-wg cells were used to isolate Wg-conditioned media (Wg-CM) as described by Blauwkamp et al. (Blauwkamp et al., 2008). DROPflash assays were performed in S2R+ cells transfected with Fugene HD (Promega), using 20 ng of DROPflash (a gift from K. Cadigan, University of Michigan, Ann Arbor, MI, USA), 0.03 ng of pHW Renilla transfection control, 40 ng pA-GFP, arm, or arm* and 40 ng test DNA. Wg-CM and arm induce DROPflash to similar levels, arm* induction is threefold higher. TOPflash assays were carried out in HEK293T cells transfected with Lipofectamine 2000 (Invitrogen) using 20 ng TOPflash or FOPflash (Upstate/Millipore), 0.05 ng phRG-TK Renilla transfection control plasmid (Promega), 50 ng pc-GFP or pc-ΔGSK-beta-cat and 40 ng test DNA. All TOPflash, FOPflash and DROPflash assays were performed in triplicate with at least two independent experiments for each condition. Expression was monitored within 24 hours using the Dual-Luciferase Reporter Assay System (Promega) on a Veritas luminometer (Promega), and values were divided by the Renilla luciferase transfection control. The fold induction was determined by dividing induced by uninduced values. Data were normalized to compare across experiments by setting the average pA-GFP or pc-GFP values to 1 and normalizing. P-values were calculated using Student’s t-test or an ANOVA with Bonferonni post-test to compare pA-GFP or pc-GFP to all and pA-pbl or pc-Ect2 to all. Error bars in all graphs are s.e.m.
Quantitative real time-PCR and RNA interference
RT-PCR assays were carried out using S2R+ cells transfected with 750 ng pA-IL2Rα control and 1750 ng test DNA. nkd, CG6234 and beta-tubulin primers were the same as those used by Fang et al. (Fang et al., 2006). Transfected cells were isolated from non-transfected cells using Dynabeads (Invitrogen) 5 hours after addition of conditioned media [as described by Blauwkamp et al. (Blauwkamp et al., 2008)], RNA was harvested using an RNeasy mini kit with DNase digestion (Qiagen), and cDNA was made using Superscript II kit (Invitrogen). Gene expression was analyzed using SYBR green (Bio-Rad) on a Mastercycler realplex2 (Eppendorf). Samples were run in duplicate and fold induction represents averages from over three experiments, normalized to beta-tubulin expression. To compare across experiments, data were normalized by setting average fold induction of pA-GFP to 1.
RNAi experiments were performed as outlined by the Drosophila RNAi Screening Center (http://www.flyrnai.org/DRSC-PRS.html) except that two rounds of RNAi treatment were performed. Primers used for pbl RNAi were: TAATACGACTCACTATAGGGGAGATCAAGACGATCTTTGGC (forward) and TAATACGACTCACTATAGGGGTGTTGAATCCTTTAGAACGCC (reverse). Double-stranded RNA was synthesized using the MEGAscript T7 kit (Invitrogen).
Acknowledgments
We are grateful to Arno Müller for encouragement and for providing transgenic pbl fly stocks. We thank K. Cadigan for the DROPflash reporter, E. Verheyen for UAS-arm* flies, J. Wang for technical assistance, and lab members for comments on the manuscript. We also thank FlyBase for essential information and the Bloomington Stock Center and Drosophila Genomics Resource Center for reagents.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
All three authors designed and performed the research, analyzed data and contributed to writing the manuscript.
Funding
This work was supported by a National Institutes of Health grant [R01 GM86620 to A.B.]. Deposited in PMC for release after 12 months.
References
- Adams R. R., Tavares A. A., Salzberg A., Bellen H. J., Glover D. M. (1998). pavarotti encodes a kinesin-like protein required to organize the central spindle and contractile ring for cytokinesis. Genes Dev. 12, 1483–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejsovec A. (2006). Flying at the head of the pack: Wnt biology in Drosophila. Oncogene 25, 7442–7449 [DOI] [PubMed] [Google Scholar]
- Blauwkamp T. A., Chang M. V., Cadigan K. M. (2008). Novel TCF-binding sites specify transcriptional repression by Wnt signalling. EMBO J. 27, 1436–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloor J. W., Kiehart D. P. (2002). Drosophila RhoA regulates the cytoskeleton and cell-cell adhesion in the developing epidermis. Development 129, 3173–3183 [DOI] [PubMed] [Google Scholar]
- Buongiorno P., Pethe V. V., Charames G. S., Esufali S., Bapat B. (2008). Rac1 GTPase and the Rac1 exchange factor Tiam1 associate with Wnt-responsive promoters to enhance beta-catenin/TCF-dependent transcription in colorectal cancer cells. Mol. Cancer 7, 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo X. R., Sauzeau V., Berenjeno I. M. (2007). GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays 29, 356–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M. V., Chang J. L., Gangopadhyay A., Shearer A., Cadigan K. M. (2008). Activation of wingless targets requires bipartite recognition of DNA by TCF. Curr. Biol. 18, 1877–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao A. T., Jones W. M., Bejsovec A. (2007). The HMG-box transcription factor SoxNeuro acts with Tcf to control Wg/Wnt signaling activity. Development 134, 989–997 [DOI] [PubMed] [Google Scholar]
- Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- Fang M., Li J., Blauwkamp T., Bhambhani C., Campbell N., Cadigan K. M. (2006). C-terminal-binding protein directly activates and represses Wnt transcriptional targets in Drosophila. EMBO J. 25, 2735–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields A. P., Justilien V. (2010). The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv. Enzyme Regul. 50, 190–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves S., Sanson B., White P., Vincent J. P. (1999). A screen for identifying genes interacting with armadillo, the Drosophila homolog of beta-catenin. Genetics 153, 1753–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata D., Yamabuki T., Miki D., Ito T., Tsuchiya E., Fujita M., Hosokawa M., Chayama K., Nakamura Y., Daigo Y. (2009). Involvement of epithelial cell transforming sequence-2 oncoantigen in lung and esophageal cancer progression. Clin. Cancer Res. 15, 256–266 [DOI] [PubMed] [Google Scholar]
- Jacinto A., Wood W., Woolner S., Hiley C., Turner L., Wilson C., Martinez-Arias A., Martin P. (2002). Dynamic analysis of actin cable function during Drosophila dorsal closure. Curr. Biol. 12, 1245–1250 [DOI] [PubMed] [Google Scholar]
- Jones W. M., Bejsovec A. (2005). RacGap50C negatively regulates Wingless pathway activity during Drosophila embryonic development. Genetics 169, 2075–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones W. M., Chao A. T., Zavortink M., Saint R., Bejsovec A. (2010). Cytokinesis proteins Tum and Pav have a nuclear role in Wnt regulation. J. Cell Sci. 123, 2179–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jürgens G., Wieschaus E., Nüsslein-Volhard C., Kluding H. (1984). Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster: II. Zygotic loci on the third chromosome. Rouxs Arch. Dev. Biol. 193, 283–295 [DOI] [PubMed] [Google Scholar]
- Justilien V., Fields A. P. (2009). Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene 28, 3597–3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Sutter C., Parker D. S., Blauwkamp T., Fang M., Cadigan K. M. (2007). CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 26, 2284–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Wang H., Eberstadt M., Schnuchel A., Olejniczak E. T., Meadows R. P., Schkeryantz J. M., Janowick D. A., Harlan J. E., Harris E. A., et al. (1998). NMR structure and mutagenesis of the N-terminal Dbl homology domain of the nucleotide exchange factor Trio. Cell 95, 269–277 [DOI] [PubMed] [Google Scholar]
- MacDonald B. T., Tamai K., He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney B. M., Dierick H. A., Kirkpatrick C., Moline M. M., Baas A., Peifer M., Bejsovec A. (1999). Drosophila APC2 is a cytoskeletally-associated protein that regulates Wingless signaling in the embryonic epidermis. J. Cell Biol. 146, 1303–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T., Fleming T. P., Bottaro D. P., Rubin J. S., Ron D., Aaronson S. A. (1991). Expression cDNA cloning of the KGF receptor by creation of a transforming autocrine loop. Science 251, 72–75 [DOI] [PubMed] [Google Scholar]
- Miki T., Smith C. L., Long J. E., Eva A., Fleming T. P. (1993). Oncogene ect2 is related to regulators of small GTP-binding proteins. Nature 362, 462–465 [DOI] [PubMed] [Google Scholar]
- Moseley J. B., Bartolini F., Okada K., Wen Y., Gundersen G. G., Goode B. L. (2007). Regulated binding of adenomatous polyposis coli protein to actin. J. Biol. Chem. 282, 12661–12668 [DOI] [PubMed] [Google Scholar]
- Noordermeer J., Johnston P., Rijsewijk F., Nusse R., Lawrence P. A. (1992). The consequences of ubiquitous expression of the wingless gene in the Drosophila embryo. Development 116, 711–719 [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C., Wieschaus E., Kluding H. (1984). Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster: I. Zygotic loci on the second chromosome. Rouxs Arch. Dev. Biol. 193, 267–282 [DOI] [PubMed] [Google Scholar]
- Nüsslein-Volhard C., Kluding H., Jürgens G. (1985). Genes affecting the segmental subdivision of the Drosophila embryo. Cold Spring Harb. Symp. Quant. Biol. 50, 145–154 [DOI] [PubMed] [Google Scholar]
- Okada K., Bartolini F., Deaconescu A. M., Moseley J. B., Dogic Z., Grigorieff N., Gundersen G. G., Goode B. L. (2010). Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J. Cell Biol. 189, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S., Peifer M. (1996). An in vivo structure-function study of Armadillo, the beta-catenin homologue, reveals both separate and overlapping regions of the protein required for cell adhesion and for Wingless signaling. J. Cell Biol. 134, 1283–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai L. M., Orsulic S., Bejsovec A., Peifer M. (1997). Negative regulation of Armadillo, a Wingless effector in Drosophila. Development 124, 2255–2266 [DOI] [PubMed] [Google Scholar]
- Peifer M. (1993). The product of the Drosophila segment polarity gene Armadillo is part of a multi-protein complex resembling the vertebrate adherens junction. J. Cell Sci. 105, 993–1000 [DOI] [PubMed] [Google Scholar]
- Polakis P. (2007). The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 17, 45–51 [DOI] [PubMed] [Google Scholar]
- Prokopenko S. N., Brumby A., O’Keefe L., Prior L., He Y., Saint R., Bellen H. J. (1999). A putative exchange factor for Rho1 GTPase is required for initiation of cytokinesis in Drosophila. Genes Dev. 13, 2301–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S., Tatsumoto T., Lorenzi M. V., Chedid M., Kapoor V., Sakata H., Rubin J., Miki T. (2003). Rho exchange factor ECT2 is induced by growth factors and regulates cytokinesis through the N-terminal cell cycle regulator-related domains. J. Cell. Biochem. 90, 819–836 [DOI] [PubMed] [Google Scholar]
- Saito S., Liu X. F., Kamijo K., Raziuddin R., Tatsumoto T., Okamoto I., Chen X., Lee C. C., Lorenzi M. V., Ohara N., et al. (2004). Deregulation and mislocalization of the cytokinesis regulator ECT2 activate the Rho signaling pathways leading to malignant transformation. J. Biol. Chem. 279, 7169–7179 [DOI] [PubMed] [Google Scholar]
- Schumacher S., Gryzik T., Tannebaum S., Müller H. A. (2004). The RhoGEF Pebble is required for cell shape changes during cell migration triggered by the Drosophila FGF receptor Heartless. Development 131, 2631–2640 [DOI] [PubMed] [Google Scholar]
- Smallhorn M., Murray M. J., Saint R. (2004). The epithelial-mesenchymal transition of the Drosophila mesoderm requires the Rho GTP exchange factor Pebble. Development 131, 2641–2651 [DOI] [PubMed] [Google Scholar]
- Somers W. G., Saint R. (2003). A RhoGEF and Rho family GTPase-activating protein complex links the contractile ring to cortical microtubules at the onset of cytokinesis. Dev. Cell 4, 29–39 [DOI] [PubMed] [Google Scholar]
- Strutt D. I., Weber U., Mlodzik M. (1997). The role of RhoA in tissue polarity and Frizzled signalling. Nature 387, 292–295 [DOI] [PubMed] [Google Scholar]
- Symons M., Settleman J. (2000). Rho family GTPases: more than simple switches. Trends Cell Biol. 10, 415–419 [DOI] [PubMed] [Google Scholar]
- Tatsumoto T., Xie X., Blumenthal R., Okamoto I., Miki T. (1999). Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J. Cell Biol. 147, 921–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyay G., Goessling W., North T. E., Xavier R., Zon L. I., Yajnik V. (2008). Molecular association between beta-catenin degradation complex and Rac guanine exchange factor DOCK4 is essential for Wnt/beta-catenin signaling. Oncogene 27, 5845–5855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Impel A., Schumacher S., Draga M., Herz H. M., Grosshans J., Müller H. A. (2009). Regulation of the Rac GTPase pathway by the multifunctional Rho GEF Pebble is essential for mesoderm migration in the Drosophila gastrula. Development 136, 813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb R. L., Zhou M. N., McCartney B. M. (2009). A novel role for an APC2-Diaphanous complex in regulating actin organization in Drosophila. Development 136, 1283–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis W. I., Nelson W. J. (2006). Re-solving the cadherin-catenin-actin conundrum. J. Biol. Chem. 281, 35593–35597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieschaus E., Nüsslein-Volhard C. (1986). Looking at embryos. In Drosophila, A Practical Approach (ed. Roberts D. B.). Oxford, England: IRL Press; [Google Scholar]
- Wu X., Tu X., Joeng K. S., Hilton M. J., Williams D. A., Long F. (2008). Rac1 activation controls nuclear localization of beta-catenin during canonical Wnt signaling. Cell 133, 340–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa S., Lee J. S., Ishimoto A. (1998). Identification and characterization of a novel line of Drosophila Schneider S2 cells that respond to wingless signaling. J. Biol. Chem. 273, 32353–32359 [DOI] [PubMed] [Google Scholar]
- Yanagawa S., Matsuda Y., Lee J. S., Matsubayashi H., Sese S., Kadowaki T., Ishimoto A. (2002). Casein kinase I phosphorylates the Armadillo protein and induces its degradation in Drosophila. EMBO J. 21, 1733–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavortink M., Contreras N., Addy T., Bejsovec A., Saint R. (2005). Tum/RacGAP50C provides a critical link between anaphase microtubules and the assembly of the contractile ring in Drosophila melanogaster. J. Cell Sci. 118, 5381–5392 [DOI] [PubMed] [Google Scholar]