

Abstract
Down syndrome (DS) is a common disorder with enormous medical and social costs, caused by trisomy for chromosome 21 (Chr21). We tested the concept that gene imbalance across an extra chromosome can be de facto corrected by manipulating a single gene, XIST. Using genome editing with zinc finger nucleases, we targeted a large, inducible XIST transgene into the Chr21 DYRK1A locus, in DS pluripotent stem cells. XIST RNA coats Chr21 and triggers stable heterochromatin modifications, chromosome-wide transcriptional silencing and DNA methylation to form a “Chr21 Barr Body.” This provides a model to study human chromosome inactivation and creates a system to investigate genomic expression changes and cellular pathologies of trisomy 21, free from genetic and epigenetic noise. Remarkably, deficits in proliferation and neural rosette formation are rapidly reversed upon silencing one Chr21. Successful trisomy silencing in vitro also surmounts the major first step towards potential development of “chromosome therapy”.
In the U.S., about 1 in 300 live births carry a trisomy, half of which are for chromosome 21 (Chr21), which causes Down syndrome (DS). DS is the leading genetic cause of intellectual disabilities and the millions of DS patients across the world also face multiple other health issues, including congenital heart defect, hematopoietic disorders, and Early-onset Alzheimer disease1,2. DS researchers have sought to define the more “DS critical” genes on Chr21, but this has proven difficult due to high genetic complexity and phenotypic variability of DS, confounded by normal variation between individuals1–3. Despite progress with DS mouse models4,5, there remains critical need for better ways to understand the underlying cell and developmental pathology of human DS, key to therapeutic design of any kind2.
The last decade has seen great advances in strategies to correct single-gene defects of rare monogenic disorders, beginning with cells in vitro and in several cases advancing to in vivo and clinical trials6. In contrast, genetic correction of the over-dose of genes across a whole extra chromosome in trisomic cells has remained outside the realm of possibility. Our effort was motivated by the idea that functional correction of living trisomic cells may be feasible by inserting a single gene that can epigenetically silence a whole chromosome. An inducible system for such “trisomy silencing” would have immediate translational relevance as a resource to investigate the cellular pathology and gene pathways impacted in DS, in a setting free from pervasive genetic or epigenetic variation that exists between individuals, sub-clones, or even isogenic cell-isolates3,7,8.
Nature has evolved a mechanism to compensate the difference in dosage of X-linked gene copies between mammalian females (XX) and males (XY). This is driven by a large (~17 kb in human) non-coding RNA, XIST, which is produced exclusively from the inactive X (Xi)9, and “paints” (accumulates across) the interphase chromosome structure10. During early development, the XIST RNA induces numerous heterochromatin modifications and architectural changes which transcriptionally silence the Xi and manifest cytologically as a condensed Barr Body (reviewed in11,12). There is evidence for some DNA sequence specificity to XIST function, since certain human genes escape X-inactivation13; however, autosomal chromatin has substantial capacity to be silenced14–16. Understanding the full potential of an autosome to be silenced, however, requires examination under conditions that avoid creating a deleterious functional monosomy. The strategy pursued here meets that requirement and creates a tractable model to study the distinct biology of human chromosome inactivation.
As outlined in Figure 1a, we set out to determine if the human X-inactivation gene, XIST, could be inserted on one Chr21, and enact a chromosome-wide change in its epigenetic state. We pursued zinc finger nuclease (ZFN)-driven targeted addition17 of an inducible XIST transgene to the gene-rich core of Chr21, in induced pluripotent stem cells (iPSCs), derived from DS patient cells. If accomplished, this milestone would provide a system to study DS cell pathology and the first step towards a potential genetic/epigenetic approach to “chromosome therapy”.
Figure 1. Genome-editing integrates XIST into Chr21 in trisomic iPSCs.
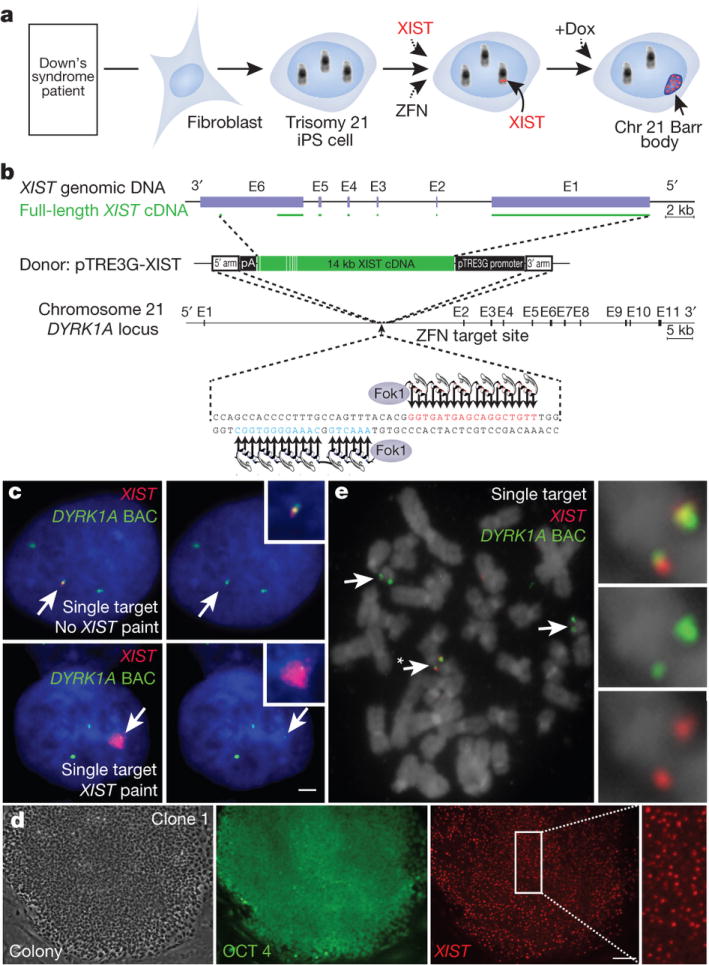
a. Concept for translating dosage compensation to trisomy 21. b. XIST construct (19kb): two homologous arms and 14kb XIST cDNA with inducible pTRE3G promoter. c. DNA/RNA FISH in interphase DS iPSCs shows XIST overlaps one of three DYRK1A genes in non-expressing cell (top, arrows), and a large XIST RNA territory with DYRK1A after 3 days in dox (bottom, arrows). Scale: 2 μm. d. OCT4 immunostaining & XIST RNA FISH in a transgenic colony: highly consistent XIST expression throughout the colony. Scale: 100 μm. e. Metaphase DNA FISH shows one targeted Chr21. XIST gene (asterisk and close-up) overlaps one of three DYRK1A genes (arrows).
Insertion of XIST into a trisomic Chr21
Given its large size, neither the XIST gene nor its cDNA has previously been integrated in a targeted fashion. Thus our first goal was to demonstrate that ZFNs could accurately insert the largest transgene to date, substantially larger than sequences commonly used for genome editing18. We first attempted this with a ~16 kb XIST transgene in a transformed cell line (HT1080), using established ZFNs to the AAVS1 locus on Chr1919. This proved highly successful (unpublished). To extend this to Chr21, we engineered ZFNs to a 36 bp sequence in intron 1 of the DYRK1A locus at Chr21q22 (as in Fig. 1b), and validated their robust activity (Supplementary Fig. 1a, b). We tested an even larger (~21 kb) construct containing near full-length XIST cDNA in HT1080 cells and demonstrated efficient, accurate addition to this gene-rich region (Supplementary Fig. 2a, b).
We next determined whether this was achievable in technically challenging iPSCs, which have unique therapeutic and developmental potential to form various cell types, and thus would be important for any future ex vivo cellular therapy efforts. We used a male DS iPSC line20 which we confirmed maintains pluripotency markers and trisomy 21. While a constitutively transcribed transgene could be used, we engineered an inducible system to maximize utility for investigating DS biology. In one step, we integrated both the doxycycline(dox)-controlled XIST transgene into Chr21 (Fig. 1b) and a transgene carrying the dox control component (rtTA) into the AAVS1 Chr19 safe harbor19 (Supplementary Fig. 3b).
We analyzed 245 colonies from the pooled transformants by interphase in situ RNA/DNA FISH (Fig. 1c) to determine if XIST was present and overlapped one of three DYRK1A alleles. Remarkably, 98.5% of XIST RNA-positive colonies carried XIST at this location on Chr21, and also contained the rtTA/selection transgene (Supplementary Table 1). Efficiency was sufficiently high that, through modifications to editing conditions, we obtained a few sub-clones with XIST integrated into two or even all three alleles of DYRK1A (see Methods and Supplementary Fig. 3c and Supplementary Table 1). Six independent sub-clones were chosen for further study based on: an XIST transgene on one of three Chr21s, pluripotent colony morphology and OCT4 staining (Fig. 1d and Supplementary Fig. 4a), and formation of embryoid bodies. FISH to metaphase chromosomes (Fig. 1e and Supplementary Fig. 3d) and Southern blotting (Supplementary Fig. 1c–e) confirmed the gene addition accuracy, with 47 chromosomes, for all six clones. High-resolution cytogenetic banding and/or array CGH on selected clones showed no significant abnormalities other than full Chr21 trisomy (Supplementary Fig. 4c–e).
XIST RNA induces a Chr21 Barr Body
In the panel of six independent genome-edited clones, we induced transgene expression and detected XIST RNA by FISH three days later. A localized XIST RNA “territory” over one Chr21 (Fig. 1c) was seen in over 85% of cells in all six clones (Fig. 1d and Supplementary Fig. 4a, b). This mirrored the unique behavior of endogenous XIST RNA which “paints” the Xi nuclear territory10.
The natural Xi forms a condensed “Barr Body” which carries repressive histone marks11. Similarly, five days after XIST induction, the edited Chr21 became markedly enriched in all heterochromatin marks examined, including H3K27Me3, UbH2A, and H4K20Me in 90%–100% of cells and, later, with macroH2A (Fig. 2a, b and Supplementary Fig. 5a). Supplementary Figure 5b illustrates that H3K27me spreads across the whole metaphase Chr21. Moreover, Chr21 DNA in many nuclei became notably condensed, further evidence that we successfully generated a heterochromatic “Chr21 Barr Body” (Fig. 2c).
Figure 2. XIST induces heterochromatin modifications and condensed Chr21 Barr Body.
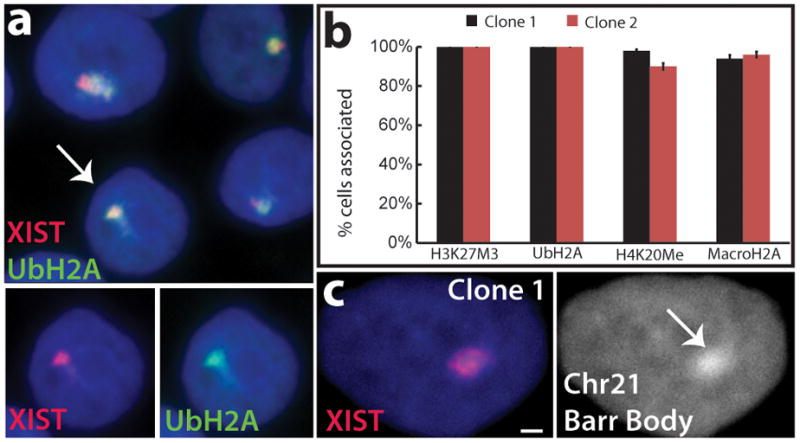
a. XIST RNA recruits heterochromatic epigenetic marks (e.g. UbH2A). Channels separated for indicated cell. b. Percentage of XIST territories with heterochromatin marks. Mean ± SE, 100 nuclei in ~5 colonies. c. XIST RNA induces “Chr21 Barr Body” visible by DAPI stain (arrow). Scale: 2 μm.
Allele-specific silencing across Chr21
To measure overall transcription across the XIST-targeted Chr21, we used an approach we developed to broadly assay nuclear hnRNA expression and to distinguish Xi from Xa16, based on in situ hybridization to CoT-1 repeat RNA. This showed that the Chr21 XIST RNA territory was depleted for hnRNA detected by CoT-1 (Supplementary Fig. 5c), similar to the inactive X chromosome16.
We next used multi-color RNA FISH to determine the presence of transcription foci at each allele for six specific Chr21 genes, an established approach we earlier showed discriminates active versus silenced genes on Xi. Without XIST expression, there are three bright transcription foci from each DYRK1A allele (Fig. 1c, top), but following XIST expression, the targeted allele becomes weaker or undetectable, indicating repression of DYRK1A (Fig. 1c, bottom).
The APP gene on Chr21 encodes amyloid beta precursor protein; mutations in APP which cause accumulation of β–amyloid lead to early onset familial Alzheimer disease (EOFAD), and APP over-expression is linked to Alzheimer disease in DS1. Initially, three bright RNA transcription foci are apparent (Fig. 3a, top). Short-term XIST expression resulted in incomplete repression of the targeted allele (Fig. 3a, middle); which after 20 days was completely silenced, as shown in two independent clones (Fig. 3a, bottom and Fig. 3b).
Figure 3. XIST induces long-range silencing in targeted iPSCs.
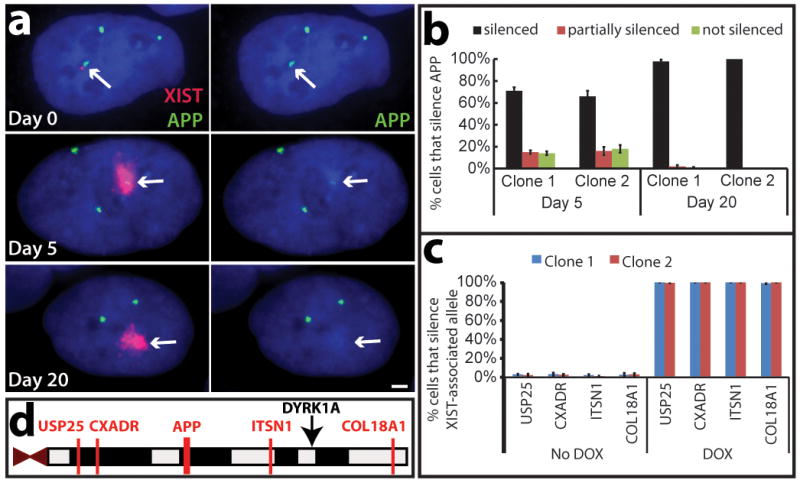
a. RNA FISH. APP RNA transcribes from three loci in uninduced cells (Day 0), and is progressively silenced following induction (targeted Chr21, arrows). Scale: 2 μm. b. Quantification of APP silencing. Mean ± SE, 100 nuclei. c. Silencing for four more Chr21-linked genes by RNA FISH. Mean ± SE from 100 nuclei. d. Long range silencing of Chr21 genes by XIST RNA. USP25 is ~21 Mb from XIST integration site (black arrow).
We examined four more loci, 3 to 21 Mb from XIST: ITSN1, USP25, CXADR, and COL18A1. Complete silencing of each allele on the edited Chr21 was seen in ~100% of cells accumulating XIST RNA (Fig. 3c, d and Supplementary Fig. 6a). Allele-specific silencing was further validated using SNP analysis. RT-PCR products for eight known polymorphic sites (in four genes) were sequenced (ADAMTS1, ETS2, TIAM1, and HSPA13) (Supplementary Fig. 6b, c). Interestingly, clones 2 and 3 showed an identical pattern of eight SNP alleles repressed, whereas clone 1 showed an alternate pattern of SNPs repressed. As summarized in Supplementary Fig. 6c, this chromosome-wide pattern allows extrapolation of the haplotype for each of the three Chr21s, and indirectly identifies for each clone which Chr21was silenced by an XIST transgene.
We also examined clones carrying XIST on two or all three copies of Chr21 and found that after 20 days in dox, most or all cells lost XIST localization or expression, and the targeted chromosomes did not silence the APP gene (Supplementary Fig. 7a, b). Thus, there is in vitro selection and epigenetic adaptation to circumvent creating a functional monosomy or nullisomy, consistent with observations that monosomic cells do not persist in mosaic patients.
Genome-wide silencing & methylation
Having demonstrated allele-specific repression for the ten genes examined above, we extended this to genome-wide expression profiling. We treated three transgenic clones and the parental line with dox for three weeks, and compared their transcriptomes to parallel cultures without XIST-transcription, all in triplicate. Only on Chr21 is there overwhelming change, in all three clones (Fig. 4a), with ~95% of significantly expressed genes becoming repressed (Supplementary Table 2).
Figure 4. Genomic expression and methylation reveal widespread silencing of Chr21.

a. Microarray: expression difference for three transgenic clones in dox versus no dox, compared to disomic line versus trisomic parental line. Total change in gene expression (N=3) per chromosome shows Chr21 “correction” near disomic levels, with only limited changes on other chromosomes. Right Y-axis scaled for percent gene expression change. b. Distribution of individual gene repression across Chr21. c. Methylation of CpG island promoters. In treated clones, 97% of Chr21 genes increased by at least 5% (2 fold greater than average), compared to none in the parental line. P: parental line; 1: Clone 1; 2: Clone 2.
Dosage compensation corrects Chr21 expression to near normal disomic levels, based on the change in total output of expressed genes per chromosome after XIST is induced. Since evidence suggests many Chr21 genes are not increased the theoretical 1.5 fold in trisomy21,22, we also directly compared trisomic to disomic cells. This provides a baseline for evaluating the degree to which Chr21 over-expression is corrected by XIST. After XIST induction, overall Chr21 expression is reduced by 20%, 15%, and 19% for clones 1, 2, and 3, respectively; this mirrors very well the 22% reduction for disomic iPSCs that lack the third Chr21 altogether (Fig. 4a). This disomic line is representative, as a similar difference (21%) was seen for another isogenic disomic sub-clone we isolated from the trisomic parental iPSCs (not shown). Individual genes repressed by XIST distribute across Chr21, as do genes over-expressed in trisomic versus disomic cells (Fig. 4b). In addition, qRT-PCR confirmed repression for individually examined genes (Supplementary Fig. 7c). Clearly, XIST induces robust dosage compensation of most Chr21 genes over-expressed in trisomy.
Trisomy 21 may impact genome-wide expression pathways, but differences attributable to trisomy 21 are confounded by genetic and epigenetic variability21. This inducible trisomy silencing system provides a new foothold into this important question. For example, even the three isogenic transgenic sub-clones show many expression differences (>1000), but upon XIST induction, ~200 genes change in all three clones (but not the dox-treated parental), most likely directly due to Chr21 over-expression. Therefore, “trisomy correction in a dish” has promise as a means to identify genome-wide pathways perturbed by trisomy 21.
In addition to transcriptional silencing, X-inactivation is stabilized by hypermethylation of promoter CpG islands23,24, which occurs late in the silencing process. Therefore, we also examined the promoter methylome in two genome-edited clones three weeks after XIST induction and found it largely unaltered, with one striking exception, genes on Chr21 (p value<2.2e-16) (Fig. 4c). Here, 97% of CpG-island-containing genes exhibited a robust increase in promoter DNA methylation, within range of that seen for Xi24 (adjusted for active/inactive chromosomes: see Methods). This change swept the entire chromosome, with the interesting exception of a handful of genes that “escape” methylation in both clones.
In summary, data from eight different approaches demonstrate impressive competence of most Chr21 genes to undergo epigenetic modification and silencing in response to an RNA that evolved to silence the X-chromosome.
Phenotypic correction in vitro
Dosage compensation of chromosome imbalance presents a new paradigm, with opportunities to advance DS research in multiple directions, including a new means to investigate human DS cellular pathologies, which are largely unknown. Inducing trisomy silencing in parallel cultures of otherwise identical cells may reveal cellular pathologies due to trisomy 21, which could be obscured by differences between cell isolates. We examined cell proliferation and neural rosette formation to look for an impact on cell phenotype.
There is some evidence of proliferative impairment in DS brains4,25; however, we observed this varied in vitro between our DS fibroblast samples, and this would be highly sensitive to culture history. A clear answer emerged from comparing identical cell cultures, grown ± dox for one week. XIST-induction in six independent transgenic sub-clones rapidly and consistently resulted in larger, more numerous and tightly packed colonies in just seven days (Fig. 5a and Supplementary Fig. 8a), with 18–34% more cells (Fig. 5b). Dox did not enhance growth of the parental DS cells or sub-clone (Fig. 5b and Supplementary Fig. 8a). Thus, a proliferative impairment linked to Chr21 over-expression can be rapidly ameliorated by dosage compensation.
Figure 5. “Trisomy correction” effects cell proliferation and neurogenesis.
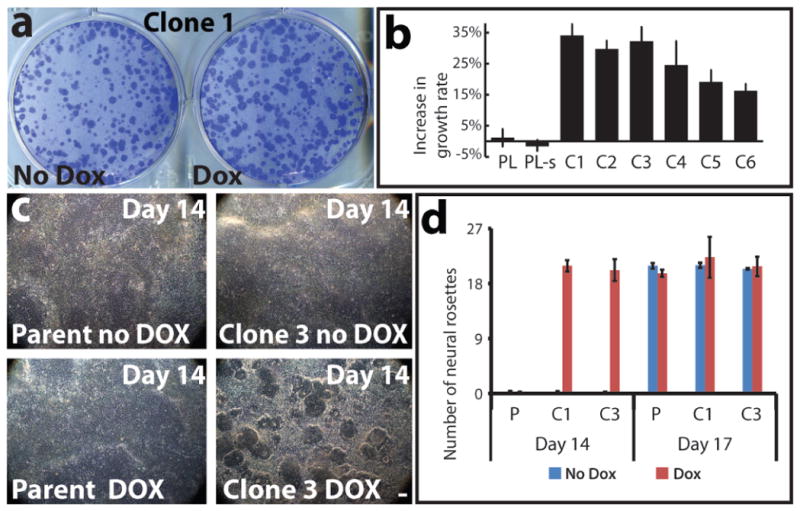
a. One week of XIST expression resulted in larger, more numerous colonies (representative sample). b. Changes in cell number for parental line (PL), parental line subclone (PL-s), and six transgenic clones (C1–C6). Mean ±SE. (n=4–6). c. Corrected cultures formed neural rosettes by day 14; trisomic (parental and non-induced) cultures took longer (17days). Scale: 100μm. e. Number of rosettes formed on day 14 and day 17. Mean ±SE, 10–12 random fields in triplicate. P: parental; C1: Clone 1; C3: Clone 3.
We next examined differentiation of targeted DS iPSCs into neural progenitor cells. In 11–12 days after neural induction of already confluent cultures, all three XIST-expressing cultures began to form neural rosettes, and in 1–2 days were replete with neural rosettes (Fig. 5c), a signature of neural progenitors (confirmed by PAX6 and SOX1 staining) (Supplementary Fig. 8b). Remarkably, even at day 14, parallel uninduced cultures remained devoid of rosettes (Fig. 5c). Uncorrected cultures required 4–5 more days in neural-induction media to fill with neural rosettes of similar size and number, as they did on day 17 (Fig. 5d and Supplementary Fig. 8d). There was no effect of dox on neurogenesis in the parental line (Fig. 5c, d and Supplementary Fig. 8d). This marked delay in neural differentiation appears primarily independent of cell proliferation (Methods). Variability in the kinetics of neural differentiation between various iPSC lines would likely obscure differences due to trisomy 2126. We circumvented this using parallel cultures and on-demand Chr21 silencing, which made clear these important phenotypic differences. This highlights the potential of this new experimental model to illuminate cellular pathologies directly attributable to Chr21 over-expression in iPSCs and their differentiated progeny.
Towards future applications
The supplement summarizes two significant points relevant to potential applications and therapeutic strategies. First, we show that heterochromatic silencing is stably maintained, even upon removal of dox and XIST expression (Supplementary Fig. 9a, b), consistent with prior studies23. Second, while not investigated extensively, we targeted XIST in non-immortalized fibroblasts from a female DS patient, which generated many cells carrying XIST (and some heterochromatin marks) on Chr21 (Supplementary Fig. 9c, d). Finally, we note that our XIST transgene lacks X-chromosome “counting” sequences, and thus is compatible with natural female X-inactivation.
DISCUSSION
We set out to bridge the basic biology of X-chromosome dosage compensation with the pathology of chromosomal dosage disorders, particularly Down syndrome. In so doing, the present work yields advances that impact three important areas, one basic and two translational.
While not our primary focus here, a significant impact of this work is that we have created a tractable, inducible system to study human chromosome silencing. Importantly, unlike random integration into a diploid cell, silencing a trisomic autosome avoids selection against full autosomal silencing, and this demonstrated remarkably robust competence of Chr21 to be silenced. Thus, XIST RNA evolved for the X-chromosome utilizes epigenome-wide mechanisms12. The ability to insert a single XIST transgene in any locus provides a more powerful tool to study XIST function, and our effort also almost triples the size of transgenes that can be thus targeted for a host of other compelling applications.
From a translational perspective, trisomy silencing has immediate impact as a new means to define the poorly understood cellular pathways deregulated in DS, and creates the opportunity to derive and study various patient-compatible cell types potentially relevant to DS therapeutics. Inducible “trisomy silencing in a dish” compares otherwise identical cultures, allowing greater discrimination of differences directly due to Chr21 over-expression distinct from genetic and epigenetic variation between transgenic sub-clones, or potentially even rare disomic sub-clones isolated from a trisomic population (27,28 and this study). XIST expression triggers not only Chr21 repression, but a defined effect on the genomic expression profile, and reverses deficits in cell proliferation and neural progenitors, which has implications for hypocellularity in the DS brain4,25. This new approach can illuminate the cohort of genes and cognate pathways most consistently impacted in DS, to inform the search for drugs that may rebalance those pathways and cell pathologies. This general strategy could be extended to study other chromosomal disorders, such as trisomy 13 and 18, often fatal in the first 1–2 years.
Finally, the more forward-looking implication of this work is to bring Down syndrome into the realm of consideration for future gene therapy research. While development of any clinical gene therapy is a multi-step process, any prospect requires that the first step, functional correction of the underlying genetic defect in living cells, is achievable. We have demonstrated that this step is no longer insurmountable for chromosomal imbalance in DS. Our hope is that for individuals and families living with DS, the proof-of-principle demonstrated here initiates multiple new avenues of translational relevance for the fifty years of advances in basic X-chromosome biology.
METHODS
Cell culture
HT1080 TetR cells (Invitrogen) and Female DS human primary fibroblast line (Coriell) (AG13902) were cultured as recommended by supplier. DS iPSC parental line (DS1-iPS4) was provided by George Q. Daley (Children’s Hospital Boston, USA) and maintained on irradiated mouse embryonic fibroblasts (iMEFs) (R & D Systems, PSC001) in hiPSC medium containing DNEM/F12 supplemented with 20% knockout Serum Replacement (Invitrogen), 1mM glutamine (Invitrogen), 100 μM non-essential amino acids (Invitrogen), 100 μM β-mercaptoethanol (Sigma) and 10 ng/ml FGF-β (Invitrogen, PHG0024). Cultures were passaged every 5–7 days with 1 mg/ml of collagenase type IV (Invitrogen).
ZFN design
ZFNs against the human AAVS1 locus (PPP1R12C) on Chr19 have been previously described19. ZFNs against the DYRK1A locus were designed using an archive of pre-validated zinc finger modules18,29, and validated for genome editing activity by transfection into K562 cells and Surveyor endonuclease-based measurement of endogenous locus disruption (“Cel1”30,31) exactly as described29. Southern blotting for targeted gene addition was performed exactly as described17,32 on SphI-digested genomic DNA probed with a fragment corresponding to positions Chr21:38825803+38826056 (hg19).
XIST and rtTA/puro plasmid construction
14 kb human XIST cDNA, a splicing isoform of full length XIST cDNA was subcloned into pTRE3G (Clontech, Cat#: 631167). Two homologous arms (left arm, 690 bp; right arm, 508 bp) of DYRK1A gene on Chr21 were amplified by PCR from primary DS fibroblasts (AG13902) (Coriell) and cloned into the pTRE3G vector (Human Chr21 DYRK1A left arm primers: forward 5′-GCCGTATACCATTAACTCTTTACTGTTC-3′, reverse 5′-TCTGTATACGTAAACTGGCAAAGGGGTGG-3′; Human Chr21 DYRK1A right arm primers: forward 5′-ATTTCGCGAACGGGTGATGAGCAGGCTGT-3′, reverse 5′-CCGTCGCGAAAACCAGAAAGTATTCTCAG-3′). The pEF1α-3G rtTA-pA cassette from pEF1α-Tet3G vector (Clontech) was subcloned into a plasmid for targeted gene addition to the PPP1R12C/AAVS1 locus19, which contains a unique HindIII site flanked by two 800 bp stretches of homology to the ZFN-specified position in the genome.
Dual-Targeted-addition of human DS iPSCs and generation of stable targeted clones
The DS iPSC line was cultured in 10 μM of Rho-associated protein kinases (ROCK) inhibitor (Calbiochem; Y27632) 24 h before electroporation. Single cells (1×107) were harvested using TryPLE select (Invitrogen), resuspended in 1 x PBS and electroporated with a total of 55 μg DNA including five plasmids (XIST, DYRK1A ZFN1, DYRK1A ZFN2, rtTA/puro, and AAVS1 ZFN) with both 3:1 and 5:1 ratios of XIST: rtTA/puro. The electroporation conditions were 220v, and 750 μF (BioRad Gene Pulser II System). Cells were subsequently plated on puromycin-resistant DR4 MEF feeders (Open Biosystems, Cat#: MES3948) in hiPSC medium supplemented with ROCK inhibitor for the first 24 h. Over 300 colonies remained after 12 days of 0.4 μg/ml puromycin selection and 245 randomly chosen individual colonies across 36 pooled wells were examined by interphase DNA/RNA FISH for the presence and expression of XIST, correct targeting and retention of trisomy (since some subclones lacked XIST or showed just two DYRK1A DNA signals). Over 100 individual clones were isolated and characterized, and those of interest, containing targeted XIST on one of three DYRK1A loci, were frozen. Six single target clones with good pluripotent morphology, OCT4 positive staining, correct targeting to one trisomic chromosome, and good XIST RNA paint were expanded for further characterization. One double and one triple target line, two non-target clones, and one disomic clone were also isolated and frozen. Targeting and correct chromosome number (47) was confirmed by interphase and metaphase FISH and genome integrity by high resolution G-band karyotype and CGH array.
Chromosome preparation
iPSCs were treated with 100 ng/ml KaryoMAX colcemid (Invitrogen) for 2–4 h at 37°C in a 5% CO2 incubator. Cells were trypsinized, treated with hypotonic solution, and fixed with methanol:acetic acid (3:1). Metaphases were spread on microscope slides, and at least 20 analyzed per clone. Karyotype analysis was done on pro-metaphase chromosomes using Standard Giemsa-trypsin G band methods.
CGH array
CGH was performed in the Cytogenetics Laboratory at UMass Medical School. Genomic Microarray analysis using UMass Genomic Microarray platform (Human Genome Build hg19) was performed with 1 ug of DNA. The array contains approximately 180,000 oligonucleotides (60 mers) that represent coding and noncoding human sequences and high density coverage for clinically relevant deletion/duplication syndromes and the telomeric and pericentromeric regions of the genome. Data was analyzed by BlueFuse Multi, v3.1 (BlueGnome, Ltd).
DNA/RNA FISH and Immunostaining
DNA and RNA FISH were carried out as previously described10,15,16,33. The XIST probe is a cloned 14 kb XIST cDNA (the same sequence as XIST transgene in Fig. 1b) in pGEM-7Zf(+) (Promega). Six Chr21 gene probes are BACs from BACPAC Resources (DYRK1A: Rp11-105O24, APP: RP11-910G8, USP25: RP11-840D8, CXADR: RP11-1150I14, ITSN1: RP11-1033C16, COL18A1: RP11-867O18). DNA probes were labeled by nick translation with either biotin-11-dUTP or digoxigenin-16-dUTP (Roche). In simultaneous DNA/RNA FISH (interphase targeting assay), cellular DNA was denatured and hybridization performed without eliminating RNA and also treated with 2 U/μl of RNasin Plus RNase inhibitor (Promega). For immunostaining with RNA FISH, cells were immunostained first with RNasin Plus and fixed in 4% paraformaldehyde before RNA FISH. Antibodies: H3K27me3 (Millipore, 07–449), UbH2A (Cell Signaling, 8240), H4K20Me (Abcam, ab9051), MacroH2A (Millipore, 07–219), OCT4 (Santa Cruz, sc-9081), PAX6 (Stemgent, 09-0075), SOX1 (R & D Systems, AF3369).
Allele-specific SNP analysis
Primers were designed to amplify 3′ UTR regions of Chr21 genes reported to contain SNPs (Supplementary Table 3). Total cDNA was used from three transgenic clones with and without XIST induction for 22 days. RT-PCR products were sequenced by GENEWIZ. Of ~10 genes examined, four were heterozygous and informative in the patient DS iPSC line used here.
Microarray analysis
Three independently targeted subclones plus the parental Chr21 trisomic (non-targeted) iPSC line were grown ± dox (2 μg/ml) for 22 d. Normal male iPSC and disomic isogenic lines were also cultured for 22 d and total RNA was extracted with a High Pure RNA extraction kit (Roche) in triplicate for each, processed with a Gene Chip 3′ IVT Express Kit (Affymetrix), and hybridized to Affymetrix human gene expression PrimeView arrays. Array normalization was performed with Affymetrix Expression Console Software with Robust Multichip Analysis (RMA)34. Probesets with the top 60% of signal values were considered present and “expressed” and were used for all further analysis. Data in Figure 4 has no other threshold applied. When designated, a gene expression change significance threshold was applied using a two-tailed T-test comparing samples ± dox in triplicate (N=3) (Supplementary Table 2. p < 0.01). For the ~200 genes found to significantly change in all three clones (in text), a T-test with p < 0.001 was applied.
Microarray Data Interpretation.
Using extraction-based methods, changes on just one of three alleles (from the XIST-bearing chromosome) will be diluted by the other two. If all three chromosomes are fully expressed, this would predict a 33% reduction in Chr21 expression levels per cell when one Chr21 is fully silenced. However, 33% would apply only if Chr21 genes are fully over-expressed to start, and prior evidence and results in this study show this is not the case for many genes. Previous microarray studies have analyzed expression levels of Chr21 in DS patient cells, although such analyses are hampered by the extensive genetic and epigenetic differences between any two individuals3. The fraction of Chr21 genes detected as over-expressed varies with the study and tissue, but generally is in the 19–36% range3,22, with individual gene increases often in the ~1.2–1.4 range (less than the theoretical 1.5). For example, one study of DS embryoid bodies showed only 6–15% of genes appeared significantly up-regulated, but this was comparing non-isogenic samples of different ES cell isolates22.
Our trisomy correction system allows direct comparison of the same cells grown in identical parallel cultures, with and without XIST-mediated chromosome silencing. Our data shows a ~20% reduction in Chr21 expression overall; importantly this level of reduction is seen either when the third chromosome is silenced in trisomic cells, or when disomic and trisomic cells are compared. This 20% reduction represents an average per cell for all three chromosomes, but corresponds to a 60% reduction in expression for just one Chr21 (the one silenced by XIST RNA, as shown here).
Apart from our goal here of trisomy dosage compensation, these results add significantly to understanding the extent of Chr21 over-expression in Down syndrome, by providing a more comprehensive analysis which shows expression of most genes is increased, but less than the theoretical 1.5 fold.
qRT-PCR
qRT-PCR was performed for eight down-regulated Chr21 genes determined by microarray on an BIO-RAD MyiQ™ Real-Time PCR Detection System in triplicate for clone 3 with/without dox treatment for 22 d. The β-actin gene was used as an internal standard for calculation of expression levels. Primers for eight Chr21 genes and β-actin were described in Supplementary Table 4.
DNA Methylation analysis
The parental line and two independent targeted lines were grown with and without dox for 22 d, in duplicate cultures. Genomic DNA was extracted using PureLink Genomic DNA Mini Kit (Invitrogen) and 750 ng bisulfite modified with the Alternative Incubation Conditions from the EZ DNA Methylation Kit (Zymo Research). 160 ng of bisulfite DNA was amplified, fragmented and hybridized to Illumina Infinium HumanMethylation450 array following standard protocol as outlined in the user guide. CpG islands were defined as high and intermediate CpG densities using the CpG density classifications based on those used by Weber et al35. The program CpGIE was used to locate HC and IC islands on the X chromosome and chromosomes 21 and 22. When multiple probes in CpG islands were associated with the same TSS, an average genic methylation value calculated. These average genic values were compared pre and post dox induction using the Mann-Whitney test. Analysis was based on CpG islands within promoters of 143 Chr21 genes (Fig. 4c).
The average methylation value was 6% on Chr21 before XIST induction, and increased to 20–21% in both subclones after induction. Since any methylation increase on the transgenic chromosome would be diluted by the presence of three Chr21s, this suggests the range of 60% methylation on the one XIST-coated chromosome, which is within the range seen for the inactive X chromosome24.
Cell proliferation analysis
Eight different iPSC lines (parental line, one non-targeted subclone, and six independent targeted subclones) were passaged onto 6-well plates at equal cell densities per well of each line and grown ± dox for 7 d. At least four replicates of each line were analyzed in two independent experiments. Rigorous measures were taken to minimize and control for any minor variations in seeding densities of iPSCs, which cannot be plated as single cell suspensions. First, the analysis was done twice for six different transgenic clones, in each case comparing triplicate plates of corrected versus not corrected (dox versus no dox). To avoid differences in plating efficiencies of dox and no dox cells, we performed the experiments over a time course that did not require passage. For each of the six transgenic clones, the parental line and one negative control (non-targeted) subclone, a single well of DS iPSCs (without dox) was used to generate a cell suspension (cells and small disaggregated clumps). Next, equal aliquots of the cell suspension were plated into each of six wells four times (not relying on one measurement but the average of four for seeding each well). After plating, dox was added to three of the six wells, and the cultures were maintained for 7 d. For images, plates were fixed, stained with 1 mg/ml crystal violet (Sigma) in 70% ethanol for 30 min and scanned to generate TIFF images. For cell counts, single cells were harvested by TryPLE select and counted using Beckman Coulter Z1 Particle Counter.
Differentiation of neural progenitors and irreversibility in cortical neurons
Differentiation: Independent XIST-transgenic iPSC clones, and the parental DS iPSC line were dissociated with Accutase (Innovative Cell Technologies) and 4×105 single cells were plated on Matrigel-coated 6-well plates in mTeSR1 medium (Stemcell technologies). Once the cell culture reached 90%–100% confluence, neural induction was initiated by changing the culture medium to neural induction medium, a 1:1 mixture of N2- and B27-containing media supplemented with 500 ng/ml Noggin (R&D Systems), 10 μM SB431542 (Tocris Bioscience), and 1 μM retinoic acid (Sigma, cat#: R2625), with/without treatment of dox for the specified times. The neural rosettes were counted and their diameter measured for at least 300 rosettes (sampled in random areas from triplicate dishes). At Day 14, the dox-induced culture had an average rosette diameter of 142 μm ± 0.55 μm in Clone 1 and 141 μm ± 3.49 μm in Clone 3. Rosettes could not be measured at the same time point in the uncorrected culture, since they had not formed. At day 17, the uncorrected culture had neural rosettes of similar number and size for both Clones 1 (140 μm ± 0.87 μm) and 3 (140 μm ± 1.09 μm). The corrected culture could not be accurately compared for Day 17 because the rosettes had become so mature and often had merged. After 17 d, neural rosettes were collected by dissociation with Dispase and replated on poly-ornithine and laminin-coated plastic dishes in N2- and B27-containing media including 20 ng/ml FGF2. After a further 2 d, FGF2 was withdrawn to promote differentiation of cortical neurons. Test of the irreversibility of silencing: Two independent clones were differentiated to cortical neurons in the presence of dox for 70 days to initiate silencing. They were then split into parallel cultures grown with and without dox for another 30 days, and XIST and APP expression analyzed by RNA FISH.
Targeted addition to primary fibroblasts
We used non-immortalized primary human female DS fibroblasts, which like all primary fibroblasts have a limited lifespan in culture (potentially more limited for DS fibroblasts). We reasoned that the robustness of ZFN-driven editing, combined with reduction to disomy for the DRYK1A gene, may make it possible to observe some edited cells before they senesce. We used a transgene carrying near full length (~14 kb) XIST cDNA under a TetO2 inducible promoter, and a selectable marker on the same construct, with ~600 bp homology arms to the DYRK1A gene (vector is ~21 kb, with a ~17kb insert) (data not shown). When introduced without the Tet-repressor construct, the TetO2 CMV promoter is constitutively active. Two ZFN containing vectors and the 21 kb XIST transgene were transfected into primary DS fibroblasts (Coriell AG13902) using Stemfect polymer (Stemgent) (10:1 ratio of XIST to ZFN, and 13 ug DNA to 1.3 ul Stemfect per well of 6-well plate). The frequency of stable integrants was such that a sparse monolayer of transgenic fibroblasts emerged, rather than a few individual colonies following selection with hygromycin (75 ug/ml). The pooled population of selected cells was analyzed by FISH and immunostaining for targeting, XIST expression and heterochromatin marks. XIST RNA was observed over the DYRK1A locus in ~74% of cells, indicating accurate transgene targeting, which was also verified by metaphase FISH (Supplementary Fig. 9c). In many cells there was notable enrichment of H3K27me, UbH2A & H4K20me heterochromatic marks (Supplementary Fig. 9d). Due to the limited lifespan of primary cells and the progressive silencing of the CMV promoter used in this construct, these cells were not more fully characterized.
Supplementary Material
Acknowledgments
We appreciate recent initiatives by administrators of NIGMS and NIH to support more high-risk, high-impact research. Research began with support from GM053234 to JBL for basic X chromosome research, and was made fully possible by GM085548 and GM096400 RC4 to JBL. CJB and AMC were supported by CIHR (MOP-13680) to CJB. We thank Terence Flotte for encouragement and advice regarding genome editing strategies, and similarly appreciate the support of Stephen Jones and Peter Newburger. We thank Trevor Collingwood for initial discussions regarding this project, and the George Daley lab (Harvard) for the DS iPSC line. Zdenka Matijasevic, Kelly Smith, Laurie Lizotte and Eric Swanson provided various assistance. Dr. MS Kobor and Lucia Lam (Kobor lab) assisted with methylation analysis. DMC is supported by an NIH fellowship 1F32CA154086 and BRC (O. Rando lab) is supported by NIH training grant 2T32HD007439 (G. Witman, PI).
Footnotes
Supplementary Information is available in the online version of the paper.
Author contributions JJ with assistance of YJ: designed and produced all constructs, edited all cell lines, designed and performed most experiments. JBL, JJ, LLH: were the main contributors to designing experiments, and interpreting results. JBL, JJ, LLH, & FDU wrote the manuscript. FDU, PDG, GJC: engineered and validated ZFNs. JRP: performed Cel1 and Southern analysis. JC: performed SNP analysis, characterized three sub-clones, and helped with proliferation experiments. JJ, YJ, with help from JC, MB, HJK & LLH: initial screening of targeted iPSC sub-clones. HJK: edited and characterized primary DS fibroblast line. AMC, CJB: DNA methylation analysis and provided XIST cDNA. JJ, FDU: prepared microarray library. DMC and BRC analyzed microarray data with help from DAS, DYG, EJR.
Microarray for 27 samples is deposited in GEO under accession number GSE47014.
Reprints and permissions information is available at www.nature.com/reprints.
Readers are welcome to comment on the online version of the paper.
Requests for ZFNs can be directed to FDU (furnov@sangamo.com).
Conflict of interest statement. JBL and LLH are the inventors on an issued patent describing the concept of epigenetic chromosome therapy by targeted addition of non-coding RNA. GJC, DAS, DYG, JRP, EJR, PDG, FDU are full-time employees of Sangamo BioSciences.
References
- 1.Megarbane A, et al. The 50th anniversary of the discovery of trisomy 21: the past, present, and future of research and treatment of Down syndrome. Genet Med. 2009;11:611–616. doi: 10.1097/GIM.0b013e3181b2e34c. [DOI] [PubMed] [Google Scholar]
- 2.Gardiner KJ. Molecular basis of pharmacotherapies for cognition in Down syndrome. Trends Pharmacol Sci. 2010;31:66–73. doi: 10.1016/j.tips.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prandini P, et al. Natural gene-expression variation in Down syndrome modulates the outcome of gene-dosage imbalance. Am J Hum Genet. 2007;81:252–263. doi: 10.1086/519248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haydar TF, Reeves RH. Trisomy 21 and early brain development. Trends Neurosci. 2012;35:81–91. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Doherty A, et al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005;309:2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee B, Davidson BL. Gene therapy grows into young adulthood: special review issue. Human molecular genetics. 2011;20:R1. doi: 10.1093/hmg/ddr188. [DOI] [PubMed] [Google Scholar]
- 7.Hall LL, et al. X-inactivation reveals epigenetic anomalies in most hESC but identifies sublines that initiate as expected. J Cell Physiol. 2008;216:445–452. doi: 10.1002/jcp.21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nazor KL, et al. Recurrent variations in DNA methylation in human pluripotent stem cells and their differentiated derivatives. Cell Stem Cell. 2012;10:620–634. doi: 10.1016/j.stem.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown CJ, et al. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–542. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- 10.Clemson CM, McNeil JA, Willard HF, Lawrence JB. XIST RNA paints the inactive X chromosome at interphase: evidence for a novel RNA involved in nuclear/chromosome structure. J Cell Biol. 1996;132:259–275. doi: 10.1083/jcb.132.3.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heard E. Delving into the diversity of facultative heterochromatin: the epigenetics of the inactive X chromosome. Curr Opin Genet Dev. 2005;15:482–489. doi: 10.1016/j.gde.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Hall LL, Lawrence JB. XIST RNA and architecture of the inactive X chromosome: implications for the repeat genome. Cold Spring Harb Symp Quant Biol. 2010;75:345–356. doi: 10.1101/sqb.2010.75.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 14.Lee JT, Strauss WM, Dausman JA, Jaenisch R. A 450 kb transgene displays properties of the mammalian X-inactivation center. Cell. 1996;86:83–94. doi: 10.1016/s0092-8674(00)80079-3. [DOI] [PubMed] [Google Scholar]
- 15.Hall LL, Clemson CM, Byron M, Wydner K, Lawrence JB. Unbalanced X; autosome translocations provide evidence for sequence specificity in the association of XIST RNA with chromatin. Hum Mol Genet. 2002;11:3157–3165. doi: 10.1093/hmg/11.25.3157. [DOI] [PubMed] [Google Scholar]
- 16.Hall LL, et al. An ectopic human XIST gene can induce chromosome inactivation in postdifferentiation human HT-1080 cells. Proc Natl Acad Sci U S A. 2002;99:8677–8682. doi: 10.1073/pnas.132468999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moehle EA, et al. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci U S A. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 19.DeKelver RC, et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Research. 2010;20:1133–1142. doi: 10.1101/gr.106773.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park IH, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ait Yahya-Graison E, et al. Classification of human chromosome 21 gene-expression variations in Down syndrome: impact on disease phenotypes. Am J Hum Genet. 2007;81:475–491. doi: 10.1086/520000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biancotti JC, et al. Human embryonic stem cells as models for aneuploid chromosomal syndromes. Stem Cells. 2010;28:1530–1540. doi: 10.1002/stem.483. [DOI] [PubMed] [Google Scholar]
- 23.Csankovszki G, Nagy A, Jaenisch R. Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol. 2001;153:773–783. doi: 10.1083/jcb.153.4.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cotton AM, et al. Chromosome-wide DNA methylation analysis predicts human tissue-specific X inactivation. Hum Genet. 2011;130:187–201. doi: 10.1007/s00439-011-1007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guidi S, Ciani E, Bonasoni P, Santini D, Bartesaghi R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathol. 2011;21:361–373. doi: 10.1111/j.1750-3639.2010.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Y, et al. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med. 2012;4:124ra129. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lavon N, et al. Derivation of euploid human embryonic stem cells from aneuploid embryos. Stem cells. 2008;26:1874–1882. doi: 10.1634/stemcells.2008-0156. [DOI] [PubMed] [Google Scholar]
- 28.Li LB, et al. Trisomy correction in down syndrome induced pluripotent stem cells. Cell Stem Cell. 2012;11:615–619. doi: 10.1016/j.stem.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doyon JB, et al. Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nat Cell Biol. 2011 doi: 10.1038/ncb2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller JC, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 31.Guschin DY, et al. A rapid and general assay for monitoring endogenous gene modification. Methods in molecular biology. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 32.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 33.Byron M, Hall LL, Lawrence JB. A multifaceted FISH approach to study endogenous RNAs and DNAs in native nuclear and cell structures. Curr Protoc Hum Gen. 2013;Chapter 4(Unit 4):15. doi: 10.1002/0471142905.hg0415s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irizarry RA, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic acids research. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weber M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.