

Abstract
Chronic injury often triggers maladaptive wound healing responses leading to the development of tissue fibrosis and subsequent organ malfunction. Inflammation is a key component of the wound healing process and promotes the development of organ fibrosis. Here, we review the contribution of Toll-like receptors (TLRs) to wound healing with a particular focus on their role in liver, lung, kidney, skin and myocardial fibrosis. We discuss the role of TLRs on distinct cell populations that participate in the repair process following tissue injury, and the contribution of exogenous and endogenous TLR ligands to the wound healing response. Systemic review of the literature shows that TLRs promote tissue repair and fibrosis in many settings, albeit with profound differences between organs. In particular, TLRs exert a pronounced effect on fibrosis in organs with higher exposure to bacterial TLR ligands, such as the liver. Targeting TLR signaling at the ligand or receptor level may represent a novel strategy for the prevention of maladaptive wound healing and fibrosis in chronically injured organs.
1. Introduction
The capacity to detect tissue injury and to initiate adequate repair mechanisms is indispensable for the survival of all higher species. A common aspect of all types of injury – caused by infectious, physical, chemical or immune processes – is a compositional change of the cellular environment leading to the presence of novel molecular patterns. These patterns are recognized by a group of receptors termed pattern recognition receptors (PRRs), and trigger specific responses that promote the restoration of tissue function, including inflammation and wound healing.
Pathogen recognition is critical to survive in an essentially hostile environment that is full of potentially infective microorganisms. Detection systems for molecular patterns characteristic for pathogens (pathogen-associated molecular patterns = PAMPs) developed early in evolution, and are present in most species including plants and invertebrates [1, 2]. Toll-like receptors (TLRs) are a group of highly conserved pattern recognition receptors that signal the presence of various PAMPs to cellular constituents of the innate and adaptive immune system. Upon binding to distinct biochemical components of protozoa, bacteria and viruses, TLRs trigger immune responses via NF-κB -dependent and interferon regulatory factor (IRF)-dependent mechanisms. In addition to their function as pathogen recognition receptors, TLRs may also be activated by endogenous ligands termed damage-associated molecular patterns (DAMPs), that are either inaccessible to the immune system under physiologic conditions or undergo changes in response to injury, leading to recognition by PRRs. After tissue injury, these patterns are unmasked or released from damaged cells, and subsequently trigger inflammation via TLRs and other PRRs. Accordingly, TLRs can be considered as master safeguards of the structural integrity of tissue: activated through molecular indicators of infection or injury, they exert a key role in initiating countermeasures that repair the wound and protect the host from further damage.
Inflammation and wound healing are tightly linked processes as demonstrated by the need for inflammatory signals to recruit both fibroblasts and macrophages for tissue repair and removal of debris. However, sustained inflammation may lead to maladaptive processes like loss of functional parenchyma, fibrosis and carcinogenesis in a variety of organs [3–5] (see Fig. 1). TLR signaling in particular has been identified as a trigger of inflammation leading to dysfunctional wound healing in several chronic diseases [5]. Here, we will review the role of TLRs in the regulation of wound repair and fibrogenesis in the setting of chronic injury, with a particular focus on five main organs, which are commonly affected by chronic inflammation and maladaptive wound healing responses: the liver, kidney, lung, skin and heart. Insight into the complex interactions between TLRs and their ligands may reveal novel targets for the prevention or treatment of these maladaptive responses to chronic injury.
Figure 1.
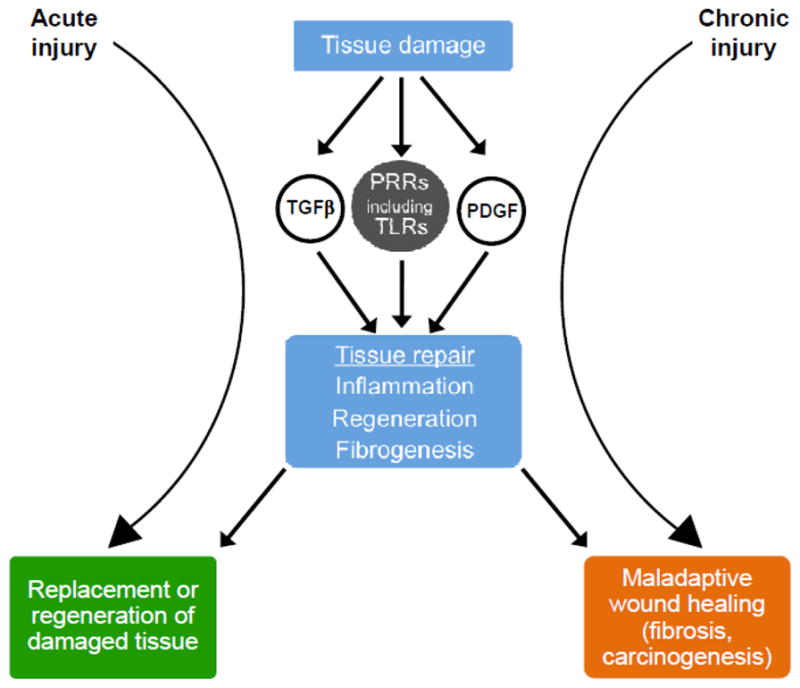
Central role for pattern recognition receptors (PRRs) in wound healing responses. After tissue damage, molecular indicators of injury or stress activate pattern recognition receptors to induce measures that contribute to the restoration of tissue integrity. Chronic injury may trigger maladaptive wound healing through the same effector systems, and ultimately lead to fibrosis and cancer.
2. Wound healing and the danger hypothesis
Despite being sterile, tissue injury often leads to profound inflammation [6]. Inflammatory, fibrogenic and regenerative responses are part of a complex and intertwined injury response system that serves to contain damage and to restore and maintain tissue function through recruitment of several types of specialized cells: platelets and fibroblasts provide mechanical stability to the wound in the short and long term, phagocytic leukocytes combat potential pathogens and clear the site of injury from dead cells and debris, and progenitor or stem cells replace functional epithelium. Inflammation may be initiated via acellular biochemical reaction cascades such as the complement system and coagulation cascade, and via the activation of resident immune cells. Both lead to the recruitment of circulating white blood cells into the injured tissue and the production of different cytokines, which further amplify inflammation and cell recruitment, or regulate important aspects of tissue repair such as regeneration and fibrogenesis. The inflammatory phase of wound healing resolves within several days and transitions into a regenerative phase, characterized by extracellular matrix (ECM) production through activated and proliferating fibroblasts, as well as angiogenesis, re-epithelialization and wound contraction. Ultimately, the provisional matrix remodels into the “pre-injury state”, or matures into scar tissue.
Numerous mediators that are essential for inflammation, wound healing and fibrogenic responses are well-established including inflammatory cytokines, TGFβ, PDGF and several chemokines [7]. However, it is not well understood through which mechanisms cells sense the presence of tissue injury and initiate these responses in the first place. One hypothesis is that the activation of the coagulation cascade following damage of vascular endothelial cells is a major stimulus for wound healing and inflammation, as demonstrated by proinflammatory and profibrogenic effects of cleaved clotting factors on tissue-resident and circulating cells [8–11], as well as the release of fibrogenic mediators such as serotonin, PDGF and TGFβ from activated platelets or endothelial cells [7, 12–14]. A second hypothesis is that the encounter with unknown molecular patterns released from damaged or injured cells (DAMPs) triggers inflammatory, fibrogenic and regenerative responses in specific cell types [15–17]. Finally, physical changes in the extracellular environment such as an increase in pressure and tissue stiffness may also trigger or amplify fibrosis via activation of fibrogenic cell populations through mechanoreceptors [18–20]. These hypotheses are not mutually exclusive, and it is likely that the coagulation cascade, DAMPs and physical forces co-operatively regulate wound healing responses.
DAMPs are a growing group of signature molecules comprising biochemical entities as diverse as nucleic acids, extracellular matrix fragments, cytoskeleton components, small molecules like uric acid and ATP, as well as large proteins such as heat shock proteins (HSPs), S100 proteins or high mobility group box protein 1 (HMGB1) [21–26]. Under physiologic conditions, these molecules do not trigger inflammation due to seclusion in intracellular compartments or physicochemical properties that render them inert to PRRs. After tissue injury, however, DAMPs are either released into the extracellular space or undergo chemical changes, leading to recognition by PRRs expressed by immune cells and other cell types. Based on these insights, it has been suggested that the immune system has not only evolved to detect patterns that signal foreignness, but to globally detect patterns that signal danger [27]. In the setting of chronic injury, DAMPs and their receptors may contribute to maladaptive wound healing by triggering chronic inflammation and activation of fibrogenic cell populations [5, 28, 29].
3. Toll-like receptors and signaling
Toll, the founding member of the TLR family, was described more than 20 years ago as a regulator of dorsoventral polarization processes in the embryogenesis of Drosophila melanogaster [30]. Subsequently, a vital role of Toll in the induction of an immunological defense against fungal and bacterial infections was uncovered, and homologous receptors in vertebrates were identified [31–33]. In contrast to Drosophila, vertebrate species exhibit a diversification of TLRs into up to 12 different transmembrane proteins, allowing the detection of a wide range of ligands. In combination and in cooperation with other pathogen recognition receptors, TLRs precisely exert the function of pathogen recognition, which was postulated by Charles Janeway more than 20 years ago [34]. As conserved germline-encoded pattern recognition receptors, TLRs empower cells of the innate immune system to reliably differentiate “self” from “non-self” patterns through detection of conserved microbial PAMPs. The recognized patterns are unique to microbes and essential products of their metabolism, so that mutations cannot readily render these organisms undetectable by TLRs. Finally, the interactions between PAMPs with TLRs are perceived as signatures of infection and consequently induce an immunological response.
TLR ligands
TLRs efficiently recognize distinct components of various pathogens through direct or adaptor-mediated binding (see Table 1): TLR1, TLR2 and TLR6 recognize cell wall components of gram-positive bacteria such as peptidoglycan, N-acyl lipoproteins or lipoteichoic acid. TLR3 and TLR7 are activated by double- or single-stranded RNA, indicating viral infection, and TLR9 senses unmethylated CpG dinucleotides, a characteristic feature of bacterial and viral pathogens. TLR4 recognizes distinct components of lipopolysaccharide (LPS), a cell wall component of all gram-negative bacteria that consists of a lipophilic region (“lipid A”), a covalently linked hydrophilic oligosaccharide and a polysaccharide chain termed O-polysaccharide [35, 36]. Lipid A constitutes the biologically active compound of LPS, whereas the presence (“smooth” LPS) or absence (“rough” LPS) of O-polysaccharide varies depending on the microbial source of LPS and determines the need for additional adaptor molecules for TLR4 activation [36, 37].
Table 1.
Toll-like receptors and their respective ligands.
| TLR | PAMP ligands | DAMP ligands | references |
|---|---|---|---|
| TLR1 | triacyl lipoproteins | [223] | |
| TLR2 | peptidoglycan, lipoproteins, zymosan, lipoteichoic acid, virus envelope proteins (CMV, HSV1) | heat shock proteins, hyaluronan, HMGB1 | [37, 43, 176, 224–228] |
| TLR3 | double-stranded RNA (dsRNA), poly-(I:C) | [229] | |
| TLR4 | lipopolysaccharide, taxol, mannan, RSV fusion protein | heat shock proteins, HMGB1, heparin sulfate, hyaluronic acid, glycoinositolphospholipids, fibrinogen | [31, 37, 43, 230–234] |
| TLR5 | flagellin | [235] | |
| TLR6 | diacyl lipopeptides, lipoteichoic acid | [236] | |
| TLR7 | single-stranded RNA (ssRNA), imidazoquinoline | [237–239] | |
| TLR8 | single-stranded RNA (ssRNA) | [237] | |
| TLR9 | non-methylated CpG | chromatin-IgG complexes | [240,241] |
| TLR10 | unknown | ||
| TLR11 | uropathogenic bacteria | [242] | |
| TLR12 | unknown |
Importantly, most TLRs may be activated by diverse ligands, which often do not share any obvious structural similarities. It is therefore believed that TLRs may have a wide ligand spectrum that possibly includes patterns not derived from pathogens [38, 39]. This concept is exemplified by hyaluronic acid, a major component of the extracellular matrix in virtually all organs. In its physiological, unfragmented form (Mw>1,000 kDa), hyaluronan is biologically inert or may even exert anti-inflammatory and anti-angiogenic effects. Following modulation by hyaluronidases and reactive oxygen species (ROS) during tissue injury, the emerging small molecular weight fragments (Mw~100–250kDa) stimulate inflammatory and wound healing responses through TLR4 and TLR2 on immune cells [40–44]. HMGB1 is another postulated endogenous TLR ligand that contributes to inflammation in various models of injury via signaling through TLR2, TLR4 and RAGE on inflammatory cells [25, 45]. This abundantly expressed nucleoprotein can be secreted by activated immune cells or passively released from necrotic cells, but is retained within cells undergoing apoptosis, which may account for the virtual lack of inflammation in response to programmed cell death [45, 46].
The detection of DAMPs empowers the immune system to sense tissue damage or danger even in the absence of infection, mostly mediated through interactions with TLR2 or TLR4, thus sharing receptors with their PAMP counterparts [41, 47]. Others, such as ATP and its receptor P2X7, cooperate with other inflammatory stimuli to induce activation of the inflammasome (see also the chapter on the role of the inflammasome in fibrosis by W. Mehal in this issue). It needs to be noted, however, that the bona fide DAMP properties of several candidate molecules remain yet to be confirmed due to limitations that are inherent to the methodology employed in this field of research: recombinant molecules used in experimental studies are commonly derived from bacterial systems, and contamination with bacterial components may account for some of the observed proinflammatory effects. HMGB1 was originally described as a direct TLR2/4 activator, yet an intrinsic proinflammatory activity was not confirmed in more recent studies [48]. Instead, HMGB1 appears to amplify the inflammatory response to different TLR agonists, as demonstrated by the increased production of tumor necrosis factor (TNF) in monocytes after stimulation with HMGB1 and LPS in comparison to LPS alone [49, 50], as well as impaired TLR3, TLR7 and TLR9 activation in vivo in the absence of HMGB1 [51]. Furthermore, the biological properties of the HMGB1 molecule may depend on the redox status of critical cysteine residues, which is largely determined by the wound microenvironment and the mode of HMGB1 release [52]. Future studies employing clean genetic inactivation methods will help circumvent some of these limitations and provide clearer insights into the contribution of individual DAMP candidates to disease processes.
Beyond ligand specificity, TLR diversification extends to the subcellular localization of the respective receptor molecules: TLR 1, 2, 4, 5, 6 and 10 are cell surface receptors, whereas TLR 3, 7, 8 and 9 are localized in the endosome. The differential distribution contributes to optimal recognition of their respective ligands, as many of the patterns that activate endosomal TLRs – mostly pathogenic DNA and RNA - enter through this cellular compartment. In addition, the restriction of TLRs to specific cellular compartments may reduce the risk for exposure to endogenous molecules under physiologic conditions. Finally, the temporal and spatial patterns of TLR expression emerge as a highly efficient “response network”, which orchestrates the function of the involved inflammatory and resident cell populations into a coordinated immune response following tissue injury [53, 54].
TLR signaling
Although individual TLRs recognize distinct ligands, the mechanisms of TLR activation and signal transduction are highly conserved (see Fig. 2). Ligand binding occurs via leucin-rich repeats (LRRs) of extracellular TLR domains and triggers signal transduction pathways through interaction of Toll/interleukin-1 receptor (TIR) domains on the intracellular side with conserved adaptor molecules (reviewed in [55]). TLR4 signals as a homodimer, but requires the presence of multiple other proteins, including MD-2 and CD14 on macrophages and RP105 on B cells, to activate downstream signaling in response to LPS. In contrast to TLR4, TLR2 forms heterodimers with either TLR1 or TLR6 to recognize components of gram-positive bacteria. TLRs signal through two upstream signaling molecules, myeloid differentiation factor 88 (MyD88) or Toll-Interleukin-1R domain-containing adaptor inducing interferon-b (Trif), explaining the similarity of downstream effects after activation of different TLRs. MyD88 is the adaptor for all TLRs except for TLR3 which exclusively signals through Trif. TLR4 signals through both MyD88 and Trif. MyD88 and Trif in turn trigger multiple proinflammatory signaling pathways including NF-κB, JNK/AP1, ERK and p38, and also induce a potent activation of the interferon pathway through Interferon-regulatory factors (IRFs) [56–58].
Figure 2.

Toll-like receptor signaling as exemplified by TLR4. Toll-like receptors bind to endogenous and exogenous ligands that indicate tissue injury or infection. Upon stimulation, TLRs activate adaptor molecules (MyD88, Trif), which trigger conserved signaling pathways culminating in a “danger response”, consisting of the induction of inflammatory and anti-viral genes. Other TLR signaling pathways can be reviewed in [55, 57].
TLR activation also triggers regulatory feedback loops, which mostly result in a subsequent hyporesponsiveness towards exposure to their respective ligands, called “TLR tolerance”. This negative regulation of TLR activity is mediated by a plethora of molecular mechanisms including interference with ligand binding, TLR downregulation and degradation, inhibition of downstream signaling, alterations of target structures through chromatin remodeling, and histone modification (reviewed in [59] and [60]). Due to the use of common signaling cascades, the activation of regulatory pathways through one TLR can lead to decreased responsiveness of other TLRs, termed “crosstolerance” [61].
4. TLR responsive cell types in wound healing
Cells of the innate immune system constitute the “first line of defense” in response to tissue injury. This group of cells consists of tissue resident sentinel cells - mast cells, phagocytes and dendritic cells – as well as circulating cells: neutrophils, basophils, eosinophils, natural killer (NK) cells, and γδ T cells. In contrast to the effector cells of the adaptive immune system, which use highly specific, randomly generated antigen receptors to recognize target epitopes, cells of the innate immune system carry germ-line encoded and evolutionarily conserved receptors (such as the TLRs) for pattern recognition. Other non-myeloid effector cells of the wound healing response, including epithelial and endothelial cells, vascular smooth muscle cells and fibroblasts also express TLRs, and modulation of TLR signaling in these cell populations affects tissue regeneration [62–64]. In this section, we focus on TLR-expressing cell types that play a role in wound healing processes.
TLRs and fibroblasts
Depending on the organ and the type of injury, wound fibroblasts originate from tissue-resident, circulating or bone marrow-derived precursor cells [65–67] and are the main producers of the extracellular matrix, which replaces the injured or disrupted tissue and constitutes the scaffold for tissue regeneration. The identification of TLRs on fibroblasts in a variety of organs has spurred significant interest into the role of this cell population in inflammation in addition to their well-documented fibrogenic effects. Synovial fibroblasts isolated from patients with rheumatoid arthritis exhibit a robust expression of TLRs, particularly TLR3 and TLR4, and respond to their respective ligands with an increased production of inflammatory cytokines [68]. Similar findings were described for human intestinal fibroblasts: they express baseline levels of TLR1–9, which are upregulated in response to LPS or lipoteichoic acid and trigger downstream signal cascades that culminate in enhanced IL-8 secretion [69]. Likewise, pulmonary fibroblasts are responsive to LPS and unmethylated CpG DNA via the respective TLRs [70–73]. In the liver, myofibroblasts are mainly derived from hepatic stellate cells (HSC), which are normally quiescent and serve as storage cells for the majority of the body’s vitamin A content [74]. Already under baseline conditions these cells express TLR4, suggesting a sentinel function for increased levels of LPS. After liver injury, HSCs become activated - they lose their vitamin A-containing droplets, express alpha-smooth muscle actin and produce large amounts of collagen and other extracellular matrix proteins. HSC stimulation with LPS induces the production of chemokines (MCP-1, MIP-1α, MIP-1β, RANTES, IP-10) and downregulates Bambi, a repressor of TGFβ signaling [75]. In a similar fashion, HMGB1 activates hepatic stellate cells to enhance their inflammatory phenotype and to stimulate fibrogenic gene expression [76]. Unstimulated HSCs are hyporesponsive to TLR2 ligands, yet pretreatment with TNFα or IL-1β increases expression of TLR2 and responsiveness to its ligands, underscoring the role of activated HSCs in the regulation of inflammation after tissue injury [77]. Finally, HSCs may respond to host-derived denatured DNA from apoptotic hepatocytes as well as non-methylated CpG DNA, a prototypical TLR9-agonistic PAMP resulting in differentiation processes into fibroblasts via TLR9 activation [78, 79]. Another study did not find a role for CpG DNA in activating HSCs [80] suggesting that TLR9 agonists from apoptotic cells or TLR9 activation via macrophage-released factors may be the predominant pathway through which TLR9 promotes fibrosis.
With regards to these findings, it needs to be acknowledged that our understanding of fibroblast function and behavior is largely derived from in vitro experiments, which do not necessarily reflect the situation in vivo in an accurate manner. This is exemplified by the observation that in vivo-stimulation of HSCs leads to highly correlated gene expression patterns across different injury models, whereas stimulation of isolated HSCs only partially reproduces this pattern [81]. Future studies using conditional genetic deletion of TLRs will allow a more precise assessment of TLR function in fibroblasts in vivo and circumvent limitations that are inherent to in vitro approaches.
TLRs and macrophages
Mononuclear phagocytes arise from at least two distinct precursor cell populations. The yolk sac is the origin of hematopoietic cells that migrate into developing organs and form progenitors that continuously renew tissue resident phagocyte populations [82]. Bone-marrow-derived hematopoietic stem cells give rise to dendritic cells as well as circulating monocytes in later stages [83]. After tissue damage, these circulating monocytes are rapidly recruited from the bloodstream into the site of injury, where they acquire macrophage phenotypic traits and cooperate with activated tissue-resident phagocytes in the regulation of inflammation, the destruction of invading pathogens as well as the processing and presentation of antigens [84–87]. Through phagocytosis of necrotic tissue, degradation of ECM components via matrix-metalloproteinases (MMPs) and production of anti-inflammatory and pro-fibrogenic mediators (TGFβ, IL10), macrophages also contribute to the restoration of tissue homeostasis following the initial inflammatory phase of wound healing [88, 89]. This crucial role in wound healing is highlighted by profound effects of depletion or inactivation strategies: in the liver, home of the largest macrophage population in the body, genetic or pharmacologic depletion of macrophages ameliorates liver fibrosis in several models of chronic liver damage, including carbon tetrachloride hepatotoxicity, bile duct ligation and non-alcoholic steatohepatitis [75, 88, 90–92]. In the kidney, experimental fibrosis after aminonucleoside nephrosis, renal ablation and unilateral ureteral obstruction is reduced in the absence of macrophages [93–96], and in the skin, macrophage-depleted mice exhibit delayed re-epithelialization, diminished collagen deposition, impaired angiogenesis and decreased cellular proliferation compared to wild-type control animals [97]. Interestingly, the timing of macrophage depletion after injury critically affects distinct aspects of the wound healing response in the skin and liver: macrophage depletion at early stages of the repair response results in decreased fibrosis in response to chronic liver damage, and reduces granulation tissue formation, re-epithelialization and scarring in the skin [88, 98]. In striking contrast, macrophage depletion at late stages of repair increases liver fibrosis through an MMP13-dependent mechanism [88, 89]. The wide spectrum of macrophage functions reflects the diversity and plasticity of this cell population and has led to the conceptual distinction between M1 and M2 subtypes with pro- and anti-inflammatory properties, respectively. This distinction, however, still falls short to reflect the complexity and plasticity of these cell populations, and remains a continuously evolving topic in macrophage biology [99, 100].
Many functions of monocytes and macrophages have been linked to TLR activation. TLR stimulation of bone marrow hematopoietic stem and progenitor cells induces differentiation into macrophages and thus supports the rapid generation of immune cells in response to injury or infection [101]. Circulating monocytes are highly responsive to TLR agonists and activate a wide range of genes after TLR stimulation, including cytokines, chemokines and growth factors, leading to the recruitment and activation of other cell types after injury [102, 103]. TLR signaling enhances the phagocytic and bactericidal activity of macrophages by directing their energy metabolism towards autophagy and ROS production [104, 105]. Cooperative effects between immune cell populations at the site of injury are highlighted by the dependence of neutrophils on macrophages to fully exert their TLR-induced effector functions. Macrophages and dendritic cells sustain neutrophil function by releasing “survival signals” that prevent neutrophil apoptosis after LPS challenge; an effect that has also been observed in the interplay with eosinophils and other cells of the immune system [106–108]. Conversely, at later stages of wound healing, macrophages actively suppress further neutrophil recruitment into the wound and thus help to terminate the inflammatory response [109].
In contrast to the well-documented effects of global macrophage deletion or inactivation, the specific contribution of TLRs on macrophages to wound repair and fibrosis is less clear. Profibrogenic effects of bone-marrow derived macrophages in chronic liver injury are independent of TLR4 signaling [75], but depend on TLR9-dependent production of IL-1β by Kupffer cells [80]. In other organs, the contribution of macrophage TLR activation to fibrosis remains elusive as most experimental studies were carried out in mice with global TLR deletions. As macrophages exert major functions in the regulation of inflammation and injury in the liver [110–112], the lung [113–116] and kidney [117–120], it is likely that TLR activation on macrophages has a more pronounced role in these responses. Their precise role in tissue fibrosis remains to be defined, and it is possible that differences in fibrogenic responses between wildtype and TLR-deficient mice may be indirectly caused by alterations of inflammation or injury.
TLRs and neutrophils
Neutrophils constitute ~70% of all circulating leukocytes and are usually the first cell type to be recruited into the wound in response to microbial or host-derived stimuli. Except for intracellular TLR3, human neutrophils express all other TLRs, highlighting their importance in the induction of an immune response [121]. TLR stimulation on neutrophils contributes to a prolonged lifespan of this otherwise short-lived cell population [122]. It also modulates cell surface receptor expression and initiates degranulation processes as well as ROS production and phagocytosis - all of which add to their potent microbicidal and proinflammatory effects [123]. In addition to these functions, activated neutrophils release a variety of soluble mediators that regulate the evolving inflammatory response in a context-dependent manner. Both PAMPs and DAMPs are potent neutrophil activators, however with differences in neutrophil “fine tuning”. In one study, stimulation of human neutrophils with LPS or HMGB1 showed an overlapping upregulation of 95 out of 470 genes; 140 genes were upregulated exclusively by HMGB1, and 235 by LPS [124]. While this partially reflects the “receptor promiscuity” of HMGB1, which extends to other pattern recognition receptors such as TLR2 and RAGE, it also demonstrates how the fine composition of the wound microenvironment influences neutrophil activity. Other DAMPs that stimulate neutrophil activation include mitochondrial DNA fragments (through TLR9 and other receptors), S100 proteins (through TLR4), as well as ATP and formyl peptides, which trigger neutrophil recruitment and activation in infectious and sterile models of tissue injury via activation of cell surface receptors [125–129].
In contrast to the well-established pro-inflammatory role of neutrophils in various organs in response to injury [130–134], their contribution to the resolution of inflammation and wound healing is less clear. Cutaneous wounds show accelerated closure in neutrophil-depleted mice, and heal without scarring in the absence of macrophages and neutrophils [135, 136]. Similarly, in the lung, genetic or pharmacologic inhibition of neutrophil elastase attenuates bleomycin-induced pulmonary fibrosis [137, 138]. Most of these effects, however, may be secondary to alterations of the inflammatory response rather than evidence of a direct involvement of neutrophils in fibrosis. Hepatic neutrophils contribute to injury and fibrogenic wound healing in chronic liver disease [139, 140], however, liver fibrosis occurs independently of the presence or activation of neutrophils after bile-duct ligation or chronic α-naphthylisothiocyanate-induced liver injury [141, 142]. Following activation, neutrophils undergo apoptosis induced by endogenous or exogenous triggers and are efficiently removed from the wound by macrophages and dendritic cells, in part mediated by TLR-dependent mechanisms [143, 144].
TLRs and epithelial cells
TLR expression on “classical” immune cells is paramount for the wound healing response, reflecting that the appearance of TLR ligands indicates a breach in tissue integrity, which necessitates containment and repair. In contrast, the epithelial linings of the body’s outer and inner surfaces are constantly exposed to PAMPs under physiologic conditions, particularly in the skin, digestive tract, and – albeit to a far lesser degree – the lung [145]. These epithelia maintain the delicate balance between shielding the organism from potentially lethal luminal pathogens, while at the same time providing a surface that tolerates microorganisms and favors the exchange of nutrients and gases. Moreover, epithelial organs need to maintain their ability to detect injury and infection and to control the inflammatory response after injury has occurred - the profound consequences of excess epithelial inflammation are illustrated by devastating diseases such as Crohn’s disease, bronchial asthma or atopic dermatitis.
Multitudes of reciprocal mechanisms regulate the dynamic interplay between the commensal microbiota and the host [146], and TLRs are important mediators in this interaction with functions that by far exceed a mere sentinel role for invading bacteria. Mice deficient for the TLR adaptor molecule MyD88 exhibit severe morbidity and mortality upon induction of intestinal injury and inflammation through dextran sodium sulfate (DSS) [147]. While at first counterintuitive regarding the proinflammatory effects of MyD88 activation, TLR-mediated recognition of commensals was shown to induce the production of tissue protective factors in epithelial cells, and mice devoid of a luminal microbiota are more prone to intestinal injury than control animals [147]. Similar findings have been made in other organs such as the liver [148] and the lung [41], where TLR4 deficiency or absence of microbiota increases susceptibility to experimental injury, possibly indicating a critical role for baseline TLR activity in cellular homeostasis. In a similar fashion, MyD88−/− mice show a markedly delayed wound healing response after experimental skin injury with impairments in wound contraction and angiogenesis [149]. These findings were more recently corroborated by the notion that the release of TLR ligands from commensal bacteria actively suppresses TLR2-induced epithelial inflammation in the skin in a TLR3-dependent manner [150]. While this finding supports the idea of a critical role for the microbiota in epithelial homeostasis, it may also represent a mechanism by which commensals evade immune responses via TLR crosstolerance. In the lung, MyD88 deficiency attenuates experimentally induced inflammation and injury, but impairs epithelial regeneration and ultimately increases mortality [41, 121, 151]. Despite the beneficial role of the microbiota in epithelial homeostasis, the host needs to prevent and be able to detect bacterial translocation across the epithelium into underlying tissues or the bloodstream. This protection is in part provided by immune cells in the subepithelial layers of the epidermis and mucous membranes, which constitutively express TLRs and are exposed to their respective ligands right after the epithelium has been crossed [146, 152, 153]. Moreover, TLR expression on the basolateral cell surface of epithelial cells enables the recognition of bacterial components and other indicators of an epithelial breach, and the subsequent induction of inflammation even before deeper tissue layers have been penetrated [154, 155].
5. TLRs and organ fibrosis
Toll-like receptors and liver fibrosis
Chronic hepatic inflammation leads to liver fibrosis and is predominantly caused by chronic viral hepatitis, alcohol abuse and nonalcoholic steatohepatitis (NASH) in industrialized countries. Progressive hepatic fibrosis can lead to cirrhosis, hepatocellular carcinoma and liver failure, and liver transplantation remains the only curative option for a subset of patients with advanced disease [156]. Due to its anatomical location, the liver is constantly exposed to components of gut-derived bacteria entering through the portal vein, and tolerance towards low levels of LPS and other patterns is crucial to avoid constant hepatic inflammation [157]. This is reflected by the comparatively low expression levels of most TLRs and signaling adaptor molecules MD-2 and MyD88 in the whole liver under physiologic conditions [74]. In fact, hepatocytes exhibit only weak upregulation of inflammatory genes after exposure to LPS, their main function may therefore be its uptake and biliary secretion rather than the induction of inflammation [158, 159]. The ability to respond to tissue damage or pathologic concentrations of TLR ligands, however, is maintained through baseline TLR expression on Kupffer cells and hepatic stellate cells, which serve as cellular sentinels of liver homeostasis. Prolonged or repeated injury to the liver stimulates a maladaptive interplay of hepatocytes, hepatic stellate cells (HSCs) and Kupffer cells, ultimately resulting in the excessive deposition of collagen and other extracellular matrix proteins in the liver [156, 160].
Increased bacterial translocation and elevated levels of circulating LPS are characteristic for patients with acute and chronic liver diseases [161, 162]. These increases are caused by ultrastructural alterations of the intestinal mucosa, changes in the composition of the luminal microbiota, and bacterial overgrowth [163–166]. A causal involvement of bacteria or bacterial PAMPs in the pathogenesis of liver fibrosis was first demonstrated in experimental animal studies in the 1950s and 1960s, and has been extended to a broad range of models and diseases since then. In rats, a reduction of the intestinal microbiota through administration of non-absorbable antibiotics prevents experimental liver fibrosis and cirrhosis [167, 168], an effect that can be reverted by adding LPS to the drinking water [168]. In mice, hepatic fibrogenesis after BDL is strongly reduced after treatment with a non-absorbable antibiotic cocktail [75, 169]. Likewise, intestinal decontamination protects from liver fibrosis in response to a choline-deficient diet (a model for NASH) as well as MDR2 deficiency (a genetic model of cholestatic liver disease) [167, 170]. Conversely, increased portal venous concentrations of bacterial PAMPs including LPS elicited by chemically-induced colitis strongly enhance experimental hepatic inflammation and fibrogenesis in mice [171].
More recently, the underlying molecular mechanisms,involved receptors and signaling pathways were uncovered: TLR4, the receptor for LPS, is a modulator of hepatic fibrosis, as demonstrated by the protective effect of TLR4 deficiency across various models of liver injury, including bile duct ligation, chronic CCl4 treatment, experimental non-alcoholic steatohepatitis and alcoholic steatohepatitis [75, 92, 172, 173]. Similar findings were demonstrated in mice deficient for the TLR4 binding molecules CD14 [174], the LPS co-receptor MD-2 [173] as well as the adaptor molecules MyD88 and Trif [75, 175], thus confirming a critical role of TLR4 signaling in inflammatory responses in the liver. The cellular mediators of this TLR4-dependent profibrogenic response are non-bone marrow-derived cells, with HSCs being the primary effectors that display increased fibrogenic responses after LPS treatment in vitro, and high LPS-responsiveness in vitro and in vivo [75, 176]. TLR4 on liver endothelial cells appears to regulate fibrosis-associated neovessel formation [63] - the biological significance of this finding, however, remains to be confirmed in vivo using conditional ablation of TLR4 in endothelial cells. Intriguingly, a single nucleotide polymorphism (SNP) in the human TLR4 gene, which leads to decreased LPS responsiveness, has been associated with a reduced risk of fibrosis progression in chronic hepatitis C virus infection [177, 178]. Finally, TLR4 activation through components of the intestinal microbiota links inflammation and carcinogenesis in the chronically injured liver, suggesting that increased TLR4 signaling promotes progression of chronic liver disease at various stages [148].
Apart from LPS, TLR4 may also be activated by endogenous ligands (i.e., heat shock proteins, HMGB1) as demonstrated in a variety of studies employing different models of acute and chronic liver injury [76, 179–184]. The contribution of individual TLR4 activators, however, remains a matter of debate, and it is conceivable that both exogenous and endogenous ligands co-operatively modulate the inflammatory and wound healing response following liver damage. In addition to TLR4, several studies have demonstrated a critical involvement of other TLRs in liver disease [126, 182, 185]. In the setting of chronic hepatic injury, TLR9 is highly expressed on activated stellate cells, and TLR9-deficient mice are protected from hepatic fibrosis after BDL and in the CDAA diet model of NASH [79, 80]. In this context, it was suggested that TLR9 expressed on hepatic stellate cells may have a role in the detection of DNA from apoptotic hepatocytes, and the subsequent HSC activation to induce fibrosis [79]. TLR2 deficiency is associated with decreased liver injury and mortality in two models of acute liver damage [186]. Following the administration of a high-fat diet, liver damage is exacerbated in the absence of TLR2 [187, 188]. By contrast, no difference in liver injury and only a trend toward decreased liver fibrosis was reported between TLR2 −/− and control mice following BDL [75]. A recent study reported that liver injury is reduced in TLR2 −/− following BDL or chronic administration of the hepatotoxin carbon tetrachloride. Interestingly, TLR2-mediated protection from injury is conferred by TLR2-expressing macrophages of the intestinal lamina propria that promote increased intestinal permeability after BDL. Consequently, TLR2−/− exhibit less bacterial translocation and systemic endotoxin levels following BDL, which may indirectly account for the attenuated fibrotic response via reduced TLR4 signaling [189]. Together, these studies suggest a contribution of TLR2 to liver fibrosis, albeit to a smaller extent than TLR4 and possibly depending on the composition of the gut microbiome.
While the role of TLRs in various experimental models of hepatic fibrogenesis has been outlined, their contribution to liver fibrosis caused by hepatotropic viruses (HBV, HCV) remains widely unknown. This gap of knowledge regarding some of the most prevalent causes of human liver disease is largely explained by a lack of suitable small animal models, but may be overcome with the development of “humanized” mice [190, 191], which allow the simulation and investigation of HCV or HBV infection in animal models.
The above studies on the gut microbiome and TLRs in chronic liver disease not only illustrate the very close relationship between the liver and the gut reaching far beyond embryonic development, but also point towards novel strategies to ameliorate fibrogenic responses in the liver through modulation of the intestinal microbiota or TLR4 signaling (reviewed in [192]). For example, probiotics exert a beneficial influence on bacterial translocation, circulating endotoxin levels, liver fibrosis and bacterial infections in preclinical studies and patients with liver cirrhosis [169, 193–195]. Conclusive evidence for a beneficial effect of the modulation of the intestinal flora on fibrosis progression or HCC development in humans remains lacking, but may eventually lead to new treatment strategies in the setting of chronic liver injury.
Toll-like receptors and lung fibrosis
Pulmonary fibrosis is a devastating disease in which the replacement of functional lung parenchyma with fibrous tissue progressively impairs oxygenation and gas exchange, ultimately leading to death from respiratory failure. Causes of pulmonary fibrosis include autoimmune disorders, viral infections, radiotherapy, chemotherapy, graft-versus host disease or aerosolized environmental toxins, yet in most cases the etiology of the disease remains unknown (“idiopathic pulmonary fibrosis”, IPF) [181].
Contrasting the traditional belief that the lungs are sterile organs, recent studies have identified numerous pulmonary commensals that constitute a “lung microbiome”, which shares distinct similarities with microbiomes of other organs [196]. Despite a comparatively low microbial density in the lung, it is conceivable that these microorganisms actively partake in pulmonary physiology and in the regulation of inflammation and wound healing after lung injury [196]. In contrast to the liver, the contribution of TLRs to pulmonary fibrosis and wound healing are not well defined. Deficiency of MyD88, the common adaptor for all TLRs except TLR3, protects mice from inflammation and fibrosis after treatment with bleomycin (BLM) a glycopeptide antibiotic that causes pulmonary inflammation and fibrosis [151]. Interestingly, the effect of MyD88 on lung fibrosis is not dependent on TLR2 or TLR4, as TLR2/4 double knockout mice are not protected from BLM-induced pulmonary fibrosis [41]. Instead, IL-1R1 triggers MyD88-dependent promotion of lung fibrosis as shown by reduced fibrosis in IL-1R1-deficient mice that resembles the reduced fibrosis observed in MyD88-deficient mice (see also the chapter on the role of the inflammasome in fibrosis by W. Mehal in this issue). In fact, two studies demonstrated that TLR4 deficiency or pharmacologic TLR4 inhibition exacerbates lung injury and fibrosis induced by bleomycin and silica, respectively - pointing towards a protective role of TLR4 in these models of lung injury [197, 198]. In radiation-induced pulmonary injury, TLR-mediated signals also protect the lung from fibrosis development as seen by increased lung fibrosis of TLR2/TLR4-double deficient mice [199]. In a similar model, it was demonstrated that MyD88-deficiency decreases survival after irradiation, and that surviving MyD88-deficient mice develop increased pulmonary fibrosis in late stages, apparently as a consequence of persistent inflammatory infiltrates in the lung [200]. It thus seems that MyD88 deficiency alters the pulmonary wound healing response and fibrogenesis in a highly context-dependent manner, for reasons that remain to be clarified. In contrast to TLR2 and TLR4, TLR9 appears to promote lung fibrosis. TLR9 is present at higher concentrations in surgical lung biopsies from rapidly progressing IPF patients when compared to biopsies from slowly progressing patients. Moreover, intranasal CpG promotes fibrosis in chimeric mice transplanted with human IPF fibroblasts [72].
The main effects of TLR2 and TLR4 signaling in the injured lung are mediated by bone marrow-derived cell populations and epithelial cells – and not by fibroblasts. Inflammatory cells from TLR2/TLR4 double-knockout mice fail to upregulate mediators that are crucial for the induction and resolution of inflammation after challenge with small molecular weight hyaluronan, and increased epithelial apoptosis in these animals is held responsible for the increased mortality after bleomycin-induced injury [41]. This may be the reason why the majority of studies found either no reduction in pulmonary fibrosis or even increased fibrosis when inhibiting TLR2 and TLR4 signaling in contrast to the fibrosis-reducing effects of TLR inhibition in organs such as the liver, kidney and skin. Also, it appears that TLR ligands in the injured lung are not derived from bacteria but predominantly endogenous ligands, which cooperate with other endogenous activators of the inflammasome, such as uric acid, to attain a strong inflammatory response [201]. A role for DAMPs in lung injury has also been documented for cigarette smoking, where acute exposure to cigarette smoke increases pulmonary HSP70 levels and leads to a TLR4/MyD88 and IL-1R1/MyD88-dependent recruitment of neutrophils into the lung [202]. The contributions of TLRs to pulmonary injury and fibrosis, in particularly the role of TLR9, deserve further attention. It is conceivable that TLR2 and TLR4 stimulation prevent fibrosis, whereas TLR9 stimulation promotes fibrogenic wound healing in the lung. Knowledge about the specific contribution of individual TLRs will not only allow better understanding of the underlying pathophysiology but may allow designing targeted therapies, especially in diseases with unknown etiologies such as IPF.
Toll-like receptors and kidney fibrosis
Renal fibrosis, characterized by glomerulosclerosis and tubulointerstitial fibrosis, is the common terminal end stage of various types of chronic kidney disease. The development of kidney fibrosis and the parallel destruction of functional kidney parenchyma lead to end-stage renal failure, a devastating condition that requires dialysis or kidney replacement. Unlike the liver, lung or skin, the kidney is rarely exposed to bacterial PAMPs. However, levels of endogenous TLR ligands increase in the injured kidney [203–207]. Moreover, Tamm-Horsfall protein, a heavily glycosylated protein that is exclusively expressed in the ascending limb of the Henle’s loop and secreted into the urine, may also act as a TLR4 agonist [208]. There is strong evidence that TLR signaling regulates the inflammatory response to acute kidney injury as seen by reduced renal inflammation and tubular damage in mice deficient for TLR2, TLR4 or MyD88 after post-ischemic damage [204, 209–212]. In chronic renal injury, TLRs also regulate inflammation and fibrosis, but the contribution to fibrosis appears to moderate and largely mediated by TLR4 and not other TLRs. Similar to other organs, TLR4 protects the kidney from injury as seen by increased tubular damage after unilateral ureteral obstruction [213]. Despite increased injury after UUO, TLR4-deficient mice developed considerably less fibrosis [213]. Interestingly, TLR4-deficient mice had decreased matrix metalloproteinase activity and did not show a reduction of myofibroblast accumulation. In vitro, TLR4-deficient primary tubular epithelial cells and myofibroblasts produced significantly less type I collagen mRNA after TGFβ stimulation than WT cells. These data were confirmed in a second study, in which TLR4 deficient mice exhibited a significant reduction in obstruction-induced α-SMA expression, fibroblast accumulation, and renal fibrosis [214]. TLR2 is markedly upregulated on tubular and tubulointerstitial cells in patients with chronic renal injury. In mice with obstructive nephropathy, renal injury was associated with a marked upregulation and change in distribution of TLR2 as well as upregulation of TLR2 ligands Gp96, biglycan, and HMGB1. However, functional data on the role of TLR2 in renal fibrosis is conflicting, with some studies reporting no influence on fibrosis [215], one study showing an effect on myofibroblast numbers but not long-term fibrosis [216], and a third study showing decreased fibrosis [217]. Together, these studies suggest at best a moderate promotion of kidney fibrosis by TLR2. Despite increased expression of TLR9 and TLR9 ligands, TLR9 does not contribute to renal fibrosis development after UUO [218]. In a second study, TLR2 regulated neutrophil influx and chemokine production as well as the number of myofibroblasts, but did not affect long-term fibrosis or injury development. Deficiency of the common adaptor molecule MyD88 significantly reduced fibrosis after UUO in one study [217], but only showed a trend towards decreased fibrosis in another study [218]. In summary, TLRs appear to be critical regulators of acute inflammatory responses in the kidney, which is in contrast to their only moderate contribution to chronic injury and fibrosis. Additional experimental studies are needed to improve our understanding of TLRs in the healthy and injured kidney in order to make this knowledge applicable to therapeutic interventions.
Regulation of wound healing and fibrosis in the skin
The skin is an important barrier that shields and protects the organism from many environmental noxae and pathogens. It is also the site of a distinct and varied microbiome [145]. Following injury, the barrier becomes more permeable to bacteria and their products, resulting in potential exposure to PAMPs or even bacterial superinfection. Several lines of evidence support a crucial role of TLR-induced signals in re-epithelization and closure of skin wounds: (i) Topical application of TLR3 agonist poly(I:C) promotes re-epithelialization, granulation, and neovascularization required for wound closure in mice [219]. Notably, topical application of poly-(I:C) accelerates wound closure in patients with laser plastic surgery. Conversely, mice that lack TLR3 display delayed wound healing parameters such as re-epithelialization, granulation formation, and neovascularization [219]. (ii) Nucleic acids, released by damaged cells in skin wounds, stimulate TLR7 and TLR9 on infiltrating plasmacytoid dendritic cells, leading to transient production of type I interferons (IFN) [153]. Deficiency of MyD88 and TLR7, or pharmacologic inhibition of TLR7 or TLR9 inhibits type I IFN production. The presence of dendritic cells and production of type I IFN are required for re-epithelialization, as demonstrated by experiments in dendritic cell-depleted and IFN-α/β receptor-deficient mice, respectively [153]. As demonstrated in another study, TLR9 knockout mice exhibit a general delay in wound healing when compared with wild-type mice. Moreover, administration of the TLR9 agonist CpG ODN promotes the influx of macrophages to the wound site and increases the production of vascular endothelial growth factor, expediting neovascularization of the wound bed of mice [220], and accelerates wound closure in non-human primates [221]. (iii) Excisional skin wounds in MyD88−/− mice heal at a markedly slower rate than wounds in wild-type MyD88+/+ mice, showing delayed contraction, decreased and delayed granulation tissue formation, and reduced density of newly formed blood vessels [149].
While the role for TLRs, in particular TLR3 and TLR9 in wound healing of the skin is well documented, it is not clear whether TLR4 may contribute to hypertrophic scars (HTS) after skin injury. Expression of TLR4 and its downstream adaptor myeloid differentiation factor 88 (MyD88) in HTS fibroblasts are significantly increased compared with normal fibroblasts [222]. Moreover, TLR2, TLR3 and TLR4 ligands strongly induce IFN- and TGFβ-responsive genes in fibroblasts from normal skin and fibroblasts from patients with systemic sclerosis [223]. However, no studies have assessed skin fibrosis development in mice deficient in TLRs or TLR signaling components. It is conceivable that stimulation of TLR plays an important role in promoting normal wound healing, but that excessive TLR signaling might contribute to maladaptive or hypertrophic wound healing and fibrosis.
Regulation of cardiac remodeling and fibrosis by Toll-like receptors
Cardiac hypertrophy commonly found in response to chronic hypertension is accompanied by an abnormal accumulation of ECM components. Cardiac fibrosis increases myocardial stiffness and may thereby contribute to ventricular dysfunction. TLR2 promotes wound healing and fibrosis and may thus contribute to some complications after injury induced by myocardial infarction. While infarct size and degree of inflammatory cell infiltration were similar in wild-type and TLR2-deficient mice, myocardial fibrosis in the non-infarct areas was significantly reduced [224]. Moreover, left ventricular dimensions at end diastole were smaller, and fractional shortening as well as survival were higher in TLR2-deficient mice in comparison to wild-type animals. However, another study suggested that TLR2 deficiency results in left ventricular dilation due to reduced collagen and decorin deposition in the infarct scar [225]. Interpretation of the latter study needs to be performed carefully as wild-type and TLR2-deficient mice did not appear in an identical genetic background. TLR4 deficiency improves left ventricular function and improves remodeling by decreasing atrial natriuretic factor, total collagen and cardiac hypertrophy after myocardial infarction when compared with WT-MI mice. These changes resulted in significantly improved survival of TLR4-deficient mice compared with WT-MI mice [226, 227]. MyD88-deficient mice were protected from cardiac fibrosis induced by complete Freund's adjuvant (CFA) or MyHC-alpha/CFA-mediated myocarditis [228]. In this study, fibrosis depended on MyD88 expression in bone marrow-dependent fibroblasts. Similar results were found in a study employing aortic banding as a model of cardiac hypertrophy, where adenovirally delivered dominant-negative MyD88 significantly reduced cardiac hypertrophy, cardiomyocyte apoptosis and cardiac fibrosis, and improved cardiac function [229]. Together, these studies suggest that TLR2 and TLR4, via their common adaptor MyD88, negatively affect remodeling, cardiac function and fibrosis development after cardiac injury.
Summary and outlook
The discovery of TLRs as key activators of innate immunity has fundamentally changed our understanding of how organisms protect themselves from infection and survive in the omnipresence of potentially life-threatening microbes. A growing body of evidence on the involvement of TLRs in tissue injury greatly extends this understanding beyond innate immunity, and provides important insights into the body’s responses to danger. TLRs act as gatekeepers for several highly efficient response systems that regulate tissue homeostasis and protect the host after acute injury, but may also trigger maladaptive responses if the damaging stimulus cannot be eliminated.
From a medical point of view, this knowledge is of exceptional interest as it may be used to design new strategies for therapeutic interventions. Yet, despite significant advances in this field of research, several very important questions remain unanswered and need to be resolved before these insights can be translated into clinical practice. Conflicting roles of TLRs in different organs and different modes of tissue injury make it virtually impossible to outline distinct greater functions for individual TLRs in wound healing responses. One explanation for the observed variability may be the differential contribution of endogenous and exogenous ligands to TLR activation, which likely depends on the anatomical localization and the related exposure to microbes, as well as the type of injury and the physicochemical properties of the emerging wound. Secondly, we are at best beginning to understand the complex reciprocal interactions between the host and his commensal microbiota, which are in part regulated by TLRs and profoundly influence the host’s metabolism and overall welfare. Manipulations of this delicate interplay may have adverse long-term side effects that could exceed the benefits. Inevitably, both alterations of the resident microbiota as well as interference with immune regulatory mechanisms carry the risk of infectious complications. Finally, based on their role in sterile tissue injury, it is also conceivable that TLRs may exhibit roles in tissue homeostasis and physiology, which are not known at this point but crucial to the integrity of tissues even in the absence of damage or infection.
A scientifically sound and responsible approach to these problems requires a continuous effort to analyze TLR biology, including the identification of bona fide TLR ligands and relevant target cell populations with the help of clean genetic models, such as conditional knock-in and knock-out approaches. Together with an extended understanding of the precise role of TLRs in homeostasis and wound healing, we will undoubtedly be able to identify new therapeutic targets for the treatment of acute and chronic diseases and their maladaptive consequences.
Highlights.
TLRs detect pathogen-associated molecular patterns.
In tissue injury, TLRs may also detect endogenous molecular “danger” patterns.
TLRs are not only expressed on immune cells but also on fibroblasts and epithelium.
TLRs promote maladaptive wound healing responses such as fibrosis and carcinogenesis.
The contribution of TLRs to wound healing is highly organ- and context-dependent.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kimbrell DA, Beutler B. The Evolution and Genetics of Innate Immunity. Nat Rev Genet. 2001;2:256–267. doi: 10.1038/35066006. [DOI] [PubMed] [Google Scholar]
- 2.Hoffmann JA, Reichhart JM. Drosophila Innate Immunity: An Evolutionary Perspective. Nat Immunol. 2002;3:121–126. doi: 10.1038/ni0202-121. [DOI] [PubMed] [Google Scholar]
- 3.Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luedde T, Schwabe RF. Nf-Kappab in the Liver--Linking Injury, Fibrosis and Hepatocellular Carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kluwe J, Mencin A, Schwabe RF. Toll-Like Receptors, Wound Healing, and Carcinogenesis. J Mol Med (Berl) 2009;87:125–138. doi: 10.1007/s00109-008-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Medzhitov R. Origin and Physiological Roles of Inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 7.Wynn TA, Ramalingam TR. Mechanisms of Fibrosis: Therapeutic Translation for Fibrotic Disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunningham MA, Romas P, Hutchinson P, Holdsworth SR, Tipping PG. Tissue Factor and Factor Viia Receptor/Ligand Interactions Induce Proinflammatory Effects in Macrophages. Blood. 1999;94:3413–3420. [PubMed] [Google Scholar]
- 9.Johnson K, Choi Y, DeGroot E, Samuels I, Creasey A, Aarden L. Potential Mechanisms for a Proinflammatory Vascular Cytokine Response to Coagulation Activation. J Immunol. 1998;160:5130–5135. [PubMed] [Google Scholar]
- 10.Demetz G, Seitz I, Stein A, Steppich B, Groha P, Brandl R, Schomig A, Ott I. Tissue Factor-Factor Viia Complex Induces Cytokine Expression in Coronary Artery Smooth Muscle Cells. Atherosclerosis. 2010;212:466–471. doi: 10.1016/j.atherosclerosis.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 11.Scotton CJ, Krupiczojc MA, Konigshoff M, Mercer PF, Lee YC, Kaminski N, Morser J, Post JM, Maher TM, Nicholson AG, Moffatt JD, Laurent GJ, Derian CK, Eickelberg O, Chambers RC. Increased Local Expression of Coagulation Factor X Contributes to the Fibrotic Response in Human and Murine Lung Injury. J Clin Invest. 2009;119:2550–2563. doi: 10.1172/JCI33288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinhubl SR. Platelets as Mediators of Inflammation. Hematol Oncol Clin North Am. 2007;21:115–121. doi: 10.1016/j.hoc.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 13.Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, Jungel A, Beyer C, Kronke G, Zwerina J, Reiter R, Alenina N, Maroteaux L, Gay S, Schett G, Distler O, Distler JH. Platelet-Derived Serotonin Links Vascular Disease and Tissue Fibrosis. J Exp Med. 2011;208:961–972. doi: 10.1084/jem.20101629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, et al. Transforming Growth Factor Type Beta: Rapid Induction of Fibrosis and Angiogenesis in Vivo and Stimulation of Collagen Formation in Vitro. Proc Natl Acad Sci U S A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kono H, Rock KL. How Dying Cells Alert the Immune System to Danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bianchi ME. Damps, Pamps and Alarmins: All We Need to Know About Danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 17.Chen GY, Nunez G. Sterile Inflammation: Sensing and Reacting to Damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Discher DE, Janmey P, Wang YL. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 19.Wells RG. The Role of Matrix Stiffness in Regulating Cell Behavior. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 20.Olsen AL, Bloomer SA, Chan EP, Gaca MD, Georges PC, Sackey B, Uemura M, Janmey PA, Wells RG. Hepatic Stellate Cells Require a Stiff Environment for Myofibroblastic Differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110–118. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, Fletcher G, Durkin C, Postigo A, Skehel M, Batista F, Thompson B, Way M, Reis e Sousa C, Schulz O. F-Actin Is an Evolutionarily Conserved Damage-Associated Molecular Pattern Recognized by Dngr-1, a Receptor for Dead Cells. Immunity. 2012;36:635–645. doi: 10.1016/j.immuni.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Jiang D, Liang J, Noble PW. Hyaluronan in Tissue Injury and Repair. Annu Rev Cell Dev Biol. 2007;23:435–461. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y, Evans JE, Rock KL. Molecular Identification of a Danger Signal That Alerts the Immune System to Dying Cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. Rage Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 25.Lotze MT, Tracey KJ. High-Mobility Group Box 1 Protein (Hmgb1): Nuclear Weapon in the Immune Arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 26.Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin Activates the Inflammasome in Response to Toxins and Atp. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 27.Matzinger P. The Danger Model: A Renewed Sense of Self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 28.Lotfi R, Eisenbacher J, Solgi G, Fuchs K, Yildiz T, Nienhaus C, Rojewski MT, Schrezenmeier H. Human Mesenchymal Stem Cells Respond to Native but Not Oxidized Damage Associated Molecular Pattern Molecules from Necrotic (Tumor) Material. Eur J Immunol. 2011;41:2021–2028. doi: 10.1002/eji.201041324. [DOI] [PubMed] [Google Scholar]
- 29.Meran S, Thomas D, Stephens P, Martin J, Bowen T, Phillips A, Steadman R. Involvement of Hyaluronan in Regulation of Fibroblast Phenotype. J Biol Chem. 2007;282:25687–25697. doi: 10.1074/jbc.M700773200. [DOI] [PubMed] [Google Scholar]
- 30.Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of Dorsal-Ventral Polarity in the Drosophila Embryo: The Induction of Polarity by the Toll Gene Product. Cell. 1985;42:791–798. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- 31.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The Dorsoventral Regulatory Gene Cassette Spatzle/Toll/Cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 32.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A Human Homologue of the Drosophila Toll Protein Signals Activation of Adaptive Immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 33.Rutschmann S, Kilinc A, Ferrandon D. Cutting Edge: The Toll Pathway Is Required for Resistance to Gram-Positive Bacterial Infections in Drosophila. J Immunol. 2002;168:1542–1546. doi: 10.4049/jimmunol.168.4.1542. [DOI] [PubMed] [Google Scholar]
- 34.Janeway CA., Jr Approaching the Asymptote? Evolution and Revolution in Immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective Lps Signaling in C3h/Hej and C57bl/10sccr Mice: Mutations in Tlr4 Gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 36.Alexander C, Rietschel ET. Bacterial Lipopolysaccharides and Innate Immunity. J Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 37.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B. Cd14 Is Required for Myd88-Independent Lps Signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 38.Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. Class Switch Recombination and Somatic Hypermutation in Early Mouse B Cells Are Mediated by B Cell and Toll-Like Receptors. Immunity. 2007;27:64–75. doi: 10.1016/j.immuni.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beutler B. Neo-Ligands for Innate Immune Receptors and the Etiology of Sterile Inflammatory Disease. Immunol Rev. 2007;220:113–128. doi: 10.1111/j.1600-065X.2007.00577.x. [DOI] [PubMed] [Google Scholar]
- 40.Jiang D, Liang J, Noble PW. Hyaluronan as an Immune Regulator in Human Diseases. Physiol Rev. 2011;91:221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of Lung Injury and Repair by Toll-Like Receptors and Hyaluronan. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 42.McKee CM, Penno MB, Cowman M, Burdick MD, Strieter RM, Bao C, Noble PW. Hyaluronan (Ha) Fragments Induce Chemokine Gene Expression in Alveolar Macrophages. The Role of Ha Size and Cd44. J Clin Invest. 1996;98:2403–2413. doi: 10.1172/JCI119054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan Fragments Act as an Endogenous Danger Signal by Engaging Tlr2. J Immunol. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 44.Noble PW, Jiang D. Matrix Regulation of Lung Injury, Inflammation, and Repair: The Role of Innate Immunity. Proc Am Thorac Soc. 2006;3:401–404. doi: 10.1513/pats.200604-097AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sims GP, Rowe DC, Rietdijk ST, Herbst R, Coyle AJ. Hmgb1 and Rage in Inflammation and Cancer. Annu Rev Immunol. 2010;28:367–388. doi: 10.1146/annurev.immunol.021908.132603. [DOI] [PubMed] [Google Scholar]
- 46.Scaffidi P, Misteli T, Bianchi ME. Release of Chromatin Protein Hmgb1 by Necrotic Cells Triggers Inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 47.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. Hmgb1 Signals through Toll-Like Receptor (Tlr) 4 and Tlr2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 48.Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H. Pivotal Advance: Analysis of Proinflammatory Activity of Highly Purified Eukaryotic Recombinant Hmgb1 (Amphoterin) J Leukoc Biol. 2007;81:49–58. doi: 10.1189/jlb.0306200. [DOI] [PubMed] [Google Scholar]
- 49.Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS. High Mobility Group Box 1 Protein Binding to Lipopolysaccharide Facilitates Transfer of Lipopolysaccharide to Cd14 and Enhances Lipopolysaccharide-Mediated Tnf-Alpha Production in Human Monocytes. J Immunol. 2008;180:5067–5074. doi: 10.4049/jimmunol.180.7.5067. [DOI] [PubMed] [Google Scholar]
- 50.Hreggvidsdottir HS, Lundberg AM, Aveberger AC, Klevenvall L, Andersson U, Harris HE. High Mobility Group Box Protein 1 (Hmgb1)-Partner Molecule Complexes Enhance Cytokine Production by Signaling through the Partner Molecule Receptor. Mol Med. 2012;18:224–230. doi: 10.2119/molmed.2011.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T. Hmgb Proteins Function as Universal Sentinels for Nucleic-Acid-Mediated Innate Immune Responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 52.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME. Mutually Exclusive Redox Forms of Hmgb1 Promote Cell Recruitment or Proinflammatory Cytokine Release. J Exp Med. 2012 Aug 27;209(9):1519–28. doi: 10.1084/jem.20120189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan J, Frey RS, Malik AB. Tlr4 Signaling Induces Tlr2 Expression in Endothelial Cells Via Neutrophil Nadph Oxidase. J Clin Invest. 2003;112:1234–1243. doi: 10.1172/JCI18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Latz E, Golenbock DT. Receptor “Cross Talk” in Innate Immunity. J Clin Invest. 2003;112:1136–1137. doi: 10.1172/JCI20040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawai T, Akira S. Tlr Signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 56.Akira S, Takeda K. Toll-Like Receptor Signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 57.Barton GM, Medzhitov R. Toll-Like Receptor Signaling Pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 58.Honda K, Taniguchi T. Irfs: Master Regulators of Signalling by Toll-Like Receptors and Cytosolic Pattern-Recognition Receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 59.O'Neill LA. When Signaling Pathways Collide: Positive and Negative Regulation of Toll-Like Receptor Signal Transduction. Immunity. 2008;29:12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 60.Medzhitov R, Horng T. Transcriptional Control of the Inflammatory Response. Nat Rev Immunol. 2009;9:692–703. doi: 10.1038/nri2634. [DOI] [PubMed] [Google Scholar]
- 61.Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, Hellman J. Myd88-Dependent and Myd88-Independent Pathways in Synergy, Priming, and Tolerance between Tlr Agonists. J Immunol. 2007;178:1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- 62.Grote K, Schutt H, Schieffer B. Toll-Like Receptors in Angiogenesis. Scientific World Journal. 2011;11:981–991. doi: 10.1100/tsw.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jagavelu K, Routray C, Shergill U, O'Hara SP, Faubion W, Shah VH. Endothelial Cell Toll-Like Receptor 4 Regulates Fibrosis-Associated Angiogenesis in the Liver. Hepatology. 2010;52:590–601. doi: 10.1002/hep.23739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang X, Coriolan D, Murthy V, Schultz K, Golenbock DT, Beasley D. Proinflammatory Phenotype of Vascular Smooth Muscle Cells: Role of Efficient Toll-Like Receptor 4 Signaling. Am J Physiol Heart Circ Physiol. 2005;289:H1069–1076. doi: 10.1152/ajpheart.00143.2005. [DOI] [PubMed] [Google Scholar]
- 65.Wynn TA. Common and Unique Mechanisms Regulate Fibrosis in Various Fibroproliferative Diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage Tracing and Genetic Ablation of Adam12(+) Perivascular Cells Identify a Major Source of Profibrotic Cells During Acute Tissue Injury. Nature Medicine. 2012;18:1262–1270. doi: 10.1038/nm.2848. [DOI] [PubMed] [Google Scholar]
- 67.Duffield JS. The Elusive Source of Myofibroblasts: Problem Solved? Nat Med. 2012;18:1178–1180. doi: 10.1038/nm.2867. [DOI] [PubMed] [Google Scholar]
- 68.Pierer M, Rethage J, Seibl R, Lauener R, Brentano F, Wagner U, Hantzschel H, Michel BA, Gay RE, Gay S, Kyburz D. Chemokine Secretion of Rheumatoid Arthritis Synovial Fibroblasts Stimulated by Toll-Like Receptor 2 Ligands. J Immunol. 2004;172:1256–1265. doi: 10.4049/jimmunol.172.2.1256. [DOI] [PubMed] [Google Scholar]
- 69.Otte JM, Rosenberg IM, Podolsky DK. Intestinal Myofibroblasts in Innate Immune Responses of the Intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 70.He Z, Zhu Y, Jiang H. Inhibiting Toll-Like Receptor 4 Signaling Ameliorates Pulmonary Fibrosis During Acute Lung Injury Induced by Lipopolysaccharide: An Experimental Study. Respir Res. 2009;10:126. doi: 10.1186/1465-9921-10-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He Z, Zhu Y, Jiang H. Toll-Like Receptor 4 Mediates Lipopolysaccharide-Induced Collagen Secretion by Phosphoinositide3-Kinase-Akt Pathway in Fibroblasts During Acute Lung Injury. J Recept Signal Transduct Res. 2009;29:119–125. doi: 10.1080/10799890902845690. [DOI] [PubMed] [Google Scholar]
- 72.Trujillo G, Meneghin A, Flaherty KR, Sholl LM, Myers JL, Kazerooni EA, Gross BH, Oak SR, Coelho AL, Evanoff H, Day E, Toews GB, Joshi AD, Schaller MA, Waters B, Jarai G, Westwick J, Kunkel SL, Martinez FJ, Hogaboam CM. Tlr9 Differentiates Rapidly from Slowly Progressing Forms of Idiopathic Pulmonary Fibrosis. Sci Transl Med. 2010;2:57ra82. doi: 10.1126/scitranslmed.3001510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meneghin A, Choi ES, Evanoff HL, Kunkel SL, Martinez FJ, Flaherty KR, Toews GB, Hogaboam CM. Tlr9 Is Expressed in Idiopathic Interstitial Pneumonia and Its Activation Promotes in Vitro Myofibroblast Differentiation. Histochem Cell Biol. 2008;130:979–992. doi: 10.1007/s00418-008-0466-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Friedman SL. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. Tlr4 Enhances Tgf-Beta Signaling and Hepatic Fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 76.Zhang Z, Lin C, Peng L, Ouyang Y, Cao Y, Wang J, Friedman SL, Guo J. High Mobility Group Box 1 Activates Toll Like Receptor 4 Signaling in Hepatic Stellate Cells. Life Sci. 2012 Sep 4;91(5-6):207–12. doi: 10.1016/j.lfs.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 77.Paik YH, Lee KS, Lee HJ, Yang KM, Lee SJ, Lee DK, Han KH, Chon CY, Lee SI, Moon YM, Brenner DA. Hepatic Stellate Cells Primed with Cytokines Upregulate Inflammation in Response to Peptidoglycan or Lipoteichoic Acid. Lab Invest. 2006;86:676–686. doi: 10.1038/labinvest.3700422. [DOI] [PubMed] [Google Scholar]
- 78.Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, Mehal WZ. Apoptotic Hepatocyte DNA Inhibits Hepatic Stellate Cell Chemotaxis Via Toll-Like Receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 79.Gabele E, Muhlbauer M, Dorn C, Weiss TS, Froh M, Schnabl B, Wiest R, Scholmerich J, Obermeier F, Hellerbrand C. Role of Tlr9 in Hepatic Stellate Cells and Experimental Liver Fibrosis. Biochem Biophys Res Commun. 2008;376:271–276. doi: 10.1016/j.bbrc.2008.08.096. [DOI] [PubMed] [Google Scholar]
- 80.Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E. Toll-Like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1beta in Mice. Gastroenterology. 2010;139:323–334. e327. doi: 10.1053/j.gastro.2010.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, Schwabe RF. Gene Expression Profiles During Hepatic Stellate Cell Activation in Culture and in Vivo. Gastroenterology. 2007;132:1937–1946. doi: 10.1053/j.gastro.2007.02.033. [DOI] [PubMed] [Google Scholar]
- 82.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 83.Cumano A, Godin I. Ontogeny of the Hematopoietic System. Annu Rev Immunol. 2007;25:745–785. doi: 10.1146/annurev.immunol.25.022106.141538. [DOI] [PubMed] [Google Scholar]
- 84.Auffray C, Sieweke MH, Geissmann F. Blood Monocytes: Development, Heterogeneity, and Relationship with Dendritic Cells. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 85.Shi C, Pamer EG. Monocyte Recruitment During Infection and Inflammation. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-Mediated Defense against Microbial Pathogens. Annu Rev Immunol. 2008;26:421–452. doi: 10.1146/annurev.immunol.26.021607.090326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of Monocytes, Macrophages, and Dendritic Cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective Depletion of Macrophages Reveals Distinct, Opposing Roles During Liver Injury and Repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, Iredale JP. Scar-Associated Macrophages Are a Major Source of Hepatic Matrix Metalloproteinase-13 and Facilitate the Resolution of Murine Hepatic Fibrosis. J Immunol. 2007;178:5288–5295. doi: 10.4049/jimmunol.178.8.5288. [DOI] [PubMed] [Google Scholar]
- 90.Rivera CA, Bradford BU, Hunt KJ, Adachi Y, Schrum LW, Koop DR, Burchardt ER, Rippe RA, Thurman RG. Attenuation of Ccl(4)-Induced Hepatic Fibrosis by Gdcl(3) Treatment or Dietary Glycine. Am J Physiol Gastrointest Liver Physiol. 2001;281:G200–207. doi: 10.1152/ajpgi.2001.281.1.G200. [DOI] [PubMed] [Google Scholar]
- 91.Toyoda Y, Shida T, Wakita N, Matoba Y, Ozaki N. Mitral Valve Replacement in a Patient with an Extensively Calcified Mitral Anulus: Report of a Case. Surg Today. 1998;28:427–429. doi: 10.1007/s005950050156. [DOI] [PubMed] [Google Scholar]
- 92.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-Like Receptor-4 Signaling and Kupffer Cells Play Pivotal Roles in the Pathogenesis of Non-Alcoholic Steatohepatitis. J Hepatol. 2007;47:571–579. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Diamond JR, Pesek-Diamond I. Sublethal X-Irradiation During Acute Puromycin Nephrosis Prevents Late Renal Injury: Role of Macrophages. Am J Physiol. 1991;260:F779–786. doi: 10.1152/ajprenal.1991.260.6.F779. [DOI] [PubMed] [Google Scholar]
- 94.van Goor H, van der Horst ML, Fidler V, Grond J. Glomerular Macrophage Modulation Affects Mesangial Expansion in the Rat after Renal Ablation. Lab Invest. 1992;66:564–571. [PubMed] [Google Scholar]
- 95.Sung SA, Jo SK, Cho WY, Won NH, Kim HK. Reduction of Renal Fibrosis as a Result of Liposome Encapsulated Clodronate Induced Macrophage Depletion after Unilateral Ureteral Obstruction in Rats. Nephron Exp Nephrol. 2007;105:e1–9. doi: 10.1159/000096859. [DOI] [PubMed] [Google Scholar]
- 96.Machida Y, Kitamoto K, Izumi Y, Shiota M, Uchida J, Kira Y, Nakatani T, Miura K. Renal Fibrosis in Murine Obstructive Nephropathy Is Attenuated by Depletion of Monocyte Lineage, Not Dendritic Cells. J Pharmacol Sci. 2010;114:464–473. doi: 10.1254/jphs.10246fp. [DOI] [PubMed] [Google Scholar]
- 97.Mirza R, DiPietro LA, Koh TJ. Selective and Specific Macrophage Ablation Is Detrimental to Wound Healing in Mice. Am J Pathol. 2009;175:2454–2462. doi: 10.2353/ajpath.2009.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lucas T, Waisman A, Ranjan R, Roes J, Krieg T, Muller W, Roers A, Eming SA. Differential Roles of Macrophages in Diverse Phases of Skin Repair. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 99.Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Murray PJ, Wynn TA. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Megias J, Yanez A, Moriano S, O'Connor JE, Gozalbo D, Gil ML. Direct Toll-Like Receptor-Mediated Stimulation of Hematopoietic Stem and Progenitor Cells Occurs in Vivo and Promotes Differentiation toward Macrophages. Stem Cells. 2012;30:1486–1495. doi: 10.1002/stem.1110. [DOI] [PubMed] [Google Scholar]
- 102.Salkowski CA, Detore G, Franks A, Falk MC, Vogel SN. Pulmonary and Hepatic Gene Expression Following Cecal Ligation and Puncture: Monophosphoryl Lipid a Prophylaxis Attenuates Sepsis-Induced Cytokine and Chemokine Expression and Neutrophil Infiltration. Infect Immun. 1998;66:3569–3578. doi: 10.1128/iai.66.8.3569-3578.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang ZM, Liu C, Dziarski R. Chemokines Are the Main Proinflammatory Mediators in Human Monocytes Activated by Staphylococcus Aureus, Peptidoglycan, and Endotoxin. J Biol Chem. 2000;275:20260–20267. doi: 10.1074/jbc.M909168199. [DOI] [PubMed] [Google Scholar]
- 104.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. Tlr Signalling Augments Macrophage Bactericidal Activity through Mitochondrial Ros. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR. Toll-Like Receptor Signalling in Macrophages Links the Autophagy Pathway to Phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 106.Prince LR, Allen L, Jones EC, Hellewell PG, Dower SK, Whyte MK, Sabroe I. The Role of Interleukin-1beta in Direct and Toll-Like Receptor 4-Mediated Neutrophil Activation and Survival. Am J Pathol. 2004;165:1819–1826. doi: 10.1016/s0002-9440(10)63437-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Walmsley SR, Cowburn AS, Sobolewski A, Murray J, Farahi N, Sabroe I, Chilvers ER. Characterization of the Survival Effect of Tumour Necrosis Factor-Alpha in Human Neutrophils. Biochem Soc Trans. 2004;32:456–460. doi: 10.1042/BST0320456. [DOI] [PubMed] [Google Scholar]
- 108.Meerschaert J, Busse WW, Bertics PJ, Mosher DF. Cd14(+) Cells Are Necessary for Increased Survival of Eosinophils in Response to Lipopolysaccharide. Am J Respir Cell Mol Biol. 2000;23:780–787. doi: 10.1165/ajrcmb.23.6.4171. [DOI] [PubMed] [Google Scholar]
- 109.Soehnlein O, Lindbom L. Phagocyte Partnership During the Onset and Resolution of Inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 110.Laskin DL, Gardner CR, Price VF, Jollow DJ. Modulation of Macrophage Functioning Abrogates the Acute Hepatotoxicity of Acetaminophen. Hepatology. 1995;21:1045–1050. [PubMed] [Google Scholar]
- 111.Koop DR, Klopfenstein B, Iimuro Y, Thurman RG. Gadolinium Chloride Blocks Alcohol-Dependent Liver Toxicity in Rats Treated Chronically with Intragastric Alcohol Despite the Induction of Cyp2e1. Mol Pharmacol. 1997;51:944–950. doi: 10.1124/mol.51.6.944. [DOI] [PubMed] [Google Scholar]
- 112.Iimuro Y, Yamamoto M, Kohno H, Itakura J, Fujii H, Matsumoto Y. Blockade of Liver Macrophages by Gadolinium Chloride Reduces Lethality in Endotoxemic Rats--Analysis of Mechanisms of Lethality in Endotoxemia. J Leukoc Biol. 1994;55:723–728. doi: 10.1002/jlb.55.6.723. [DOI] [PubMed] [Google Scholar]
- 113.Zhao M, Fernandez LG, Doctor A, Sharma AK, Zarbock A, Tribble CG, Kron IL, Laubach VE. Alveolar Macrophage Activation Is a Key Initiation Signal for Acute Lung Ischemia-Reperfusion Injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1018–1026. doi: 10.1152/ajplung.00086.2006. [DOI] [PubMed] [Google Scholar]
- 114.Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. Alveolar Macrophages Contribute to Alveolar Barrier Dysfunction in Ventilator-Induced Lung Injury. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1191–1198. doi: 10.1152/ajplung.00055.2006. [DOI] [PubMed] [Google Scholar]
- 115.Prakash A, Mesa KR, Wilhelmsen K, Xu F, Dodd OJ, Hellman J. Alveolar Macrophages and Toll-Like Receptor 4 Mediate Ventilated Lung Ischemia Reperfusion Injury in Mice. Anesthesiology. 2012 Oct;117(4):822–35. doi: 10.1097/ALN.0b013e31826a4ae3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pendino KJ, Meidhof TM, Heck DE, Laskin JD, Laskin DL. Inhibition of Macrophages with Gadolinium Chloride Abrogates Ozone-Induced Pulmonary Injury and Inflammatory Mediator Production. Am J Respir Cell Mol Biol. 1995;13:125–132. doi: 10.1165/ajrcmb.13.2.7542894. [DOI] [PubMed] [Google Scholar]
- 117.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal Ischemia-Reperfusion Injury and Adenosine 2a Receptor-Mediated Tissue Protection: Role of Macrophages. Am J Physiol Renal Physiol. 2005;288:F722–731. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 118.Lu L, Faubel S, He Z, Andres Hernando A, Jani A, Kedl R, Edelstein CL. Depletion of Macrophages and Dendritic Cells in Ischemic Acute Kidney Injury. Am J Nephrol. 2012;35:181–190. doi: 10.1159/000335582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carlos CP, Mendes GE, Miquelin AR, Luz MA, da Silva CG, van Rooijen N, Coimbra TM, Burdmann EA. Macrophage Depletion Attenuates Chronic Cyclosporine a Nephrotoxicity. Transplantation. 2010;89:1362–1370. doi: 10.1097/tp.0b013e3181da0587. [DOI] [PubMed] [Google Scholar]
- 120.Duffield JS, Tipping PG, Kipari T, Cailhier JF, Clay S, Lang R, Bonventre JV, Hughes J. Conditional Ablation of Macrophages Halts Progression of Crescentic Glomerulonephritis. Am J Pathol. 2005;167:1207–1219. doi: 10.1016/S0002-9440(10)61209-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hayashi F, Means TK, Luster AD. Toll-Like Receptors Stimulate Human Neutrophil Function. Blood. 2003;102:2660–2669. doi: 10.1182/blood-2003-04-1078. [DOI] [PubMed] [Google Scholar]
- 122.Dick EP, Prince LR, Prestwich EC, Renshaw SA, Whyte MK, Sabroe I. Pathways Regulating Lipopolysaccharide-Induced Neutrophil Survival Revealed by Lentiviral Transduction of Primary Human Neutrophils. Immunology. 2009;127:249–255. doi: 10.1111/j.1365-2567.2008.02949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of Granulocyte Survival and Programmed Cell Death by Cytokines and Bacterial Products. Blood. 1992;80:2012–2020. [PubMed] [Google Scholar]
- 124.Park JS, Arcaroli J, Yum HK, Yang H, Wang H, Yang KY, Choe KH, Strassheim D, Pitts TM, Tracey KJ, Abraham E. Activation of Gene Expression in Human Neutrophils by High Mobility Group Box 1 Protein. Am J Physiol Cell Physiol. 2003;284:C870–879. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 125.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating Mitochondrial Damps Cause Inflammatory Responses to Injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Freire Lima BH, Oliveira Lopes GA, Russo RC, Avila TV, Melgaco JG, Oliveira AG, Pinto MA, Lima CX, de Paula AM, Cara DC, Leite MF, Teixeira MM, Menezes GB. Chemokines and Mitochondrial Products Activate Neutrophils to Amplify Organ Injury During Mouse Acute Liver Failure. Hepatology. 2012 Nov;56(5):1971–82. doi: 10.1002/hep.25801. [DOI] [PubMed] [Google Scholar]
- 127.Zhang Q, Itagaki K, Hauser CJ. Mitochondrial DNA Is Released by Shock and Activates Neutrophils Via P38 Map Kinase. Shock. 2010;34:55–59. doi: 10.1097/SHK.0b013e3181cd8c08. [DOI] [PubMed] [Google Scholar]
- 128.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory Activities of S100: Proteins S100a8, S100a9, and S100a8/A9 Induce Neutrophil Chemotaxis and Adhesion. J Immunol. 2003;170:3233–3242. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- 129.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular Danger Signals Guide Neutrophils to Sites of Sterile Inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 130.Gujral JS, Farhood A, Bajt ML, Jaeschke H. Neutrophils Aggravate Acute Liver Injury During Obstructive Cholestasis in Bile Duct-Ligated Mice. Hepatology. 2003;38:355–363. doi: 10.1053/jhep.2003.50341. [DOI] [PubMed] [Google Scholar]
- 131.Jaeschke H, Farhood A, Smith CW. Neutrophils Contribute to Ischemia/Reperfusion Injury in Rat Liver in Vivo. FASEB J. 1990;4:3355–3359. [PubMed] [Google Scholar]
- 132.Grommes J, Soehnlein O. Contribution of Neutrophils to Acute Lung Injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hansen PR. Role of Neutrophils in Myocardial Ischemia and Reperfusion. Circulation. 1995;91:1872–1885. doi: 10.1161/01.cir.91.6.1872. [DOI] [PubMed] [Google Scholar]
- 134.Kelly KJ, Williams WW, Jr, Colvin RB, Meehan SM, Springer TA, Gutierrez-Ramos JC, Bonventre JV. Intercellular Adhesion Molecule-1-Deficient Mice Are Protected against Ischemic Renal Injury. J Clin Invest. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dovi JV, He LK, DiPietro LA. Accelerated Wound Closure in Neutrophil-Depleted Mice. J Leukoc Biol. 2003;73:448–455. doi: 10.1189/jlb.0802406. [DOI] [PubMed] [Google Scholar]
- 136.Martin P, D'Souza D, Martin J, Grose R, Cooper L, Maki R, McKercher SR. Wound Healing in the Pu.1 Null Mouse--Tissue Repair Is Not Dependent on Inflammatory Cells. Curr Biol. 2003;13:1122–1128. doi: 10.1016/s0960-9822(03)00396-8. [DOI] [PubMed] [Google Scholar]
- 137.Chua F, Dunsmore SE, Clingen PH, Mutsaers SE, Shapiro SD, Segal AW, Roes J, Laurent GJ. Mice Lacking Neutrophil Elastase Are Resistant to Bleomycin-Induced Pulmonary Fibrosis. Am J Pathol. 2007;170:65–74. doi: 10.2353/ajpath.2007.060352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Takemasa A, Ishii Y, Fukuda T. A Neutrophil Elastase Inhibitor Prevents Bleomycin-Induced Pulmonary Fibrosis in Mice. Eur Respir J. 2012 Dec;40(6):1475–82. doi: 10.1183/09031936.00127011. [DOI] [PubMed] [Google Scholar]
- 139.Zaldivar MM, Pauels K, von Hundelshausen P, Berres ML, Schmitz P, Bornemann J, Kowalska MA, Gassler N, Streetz KL, Weiskirchen R, Trautwein C, Weber C, Wasmuth HE. Cxc Chemokine Ligand 4 (Cxcl4) Is a Platelet-Derived Mediator of Experimental Liver Fibrosis. Hepatology. 2010;51:1345–1353. doi: 10.1002/hep.23435. [DOI] [PubMed] [Google Scholar]
- 140.Louis H, Van Laethem JL, Wu W, Quertinmont E, Degraef C, Van den Berg K, Demols A, Goldman M, Le Moine O, Geerts A, Deviere J. Interleukin-10 Controls Neutrophilic Infiltration, Hepatocyte Proliferation, and Liver Fibrosis Induced by Carbon Tetrachloride in Mice. Hepatology. 1998;28:1607–1615. doi: 10.1002/hep.510280621. [DOI] [PubMed] [Google Scholar]
- 141.Saito JM, Bostick MK, Campe CB, Xu J, Maher JJ. Infiltrating Neutrophils in Bile Duct-Ligated Livers Do Not Promote Hepatic Fibrosis. Hepatol Res. 2003;25:180–191. doi: 10.1016/s1386-6346(02)00247-4. [DOI] [PubMed] [Google Scholar]
- 142.Xu J, Lee G, Wang H, Vierling JM, Maher JJ. Limited Role for Cxc Chemokines in the Pathogenesis of Alpha-Naphthylisothiocyanate-Induced Liver Injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G734–741. doi: 10.1152/ajpgi.00300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Savill JS, Wyllie AH, Henson JE, Walport MJ, Henson PM, Haslett C. Macrophage Phagocytosis of Aging Neutrophils in Inflammation. Programmed Cell Death in the Neutrophil Leads to Its Recognition by Macrophages. J Clin Invest. 1989;83:865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rovere P, Peri G, Fazzini F, Bottazzi B, Doni A, Bondanza A, Zimmermann VS, Garlanda C, Fascio U, Sabbadini MG, Rugarli C, Mantovani A, Manfredi AA. The Long Pentraxin Ptx3 Binds to Apoptotic Cells and Regulates Their Clearance by Antigen-Presenting Dendritic Cells. Blood. 2000;96:4300–4306. [PubMed] [Google Scholar]
- 145.C Human Microbiome Project, Structure, Function and Diversity of the Healthy Human Microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hooper LV, Littman DR, Macpherson AJ. Interactions between the Microbiota and the Immune System. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of Commensal Microflora by Toll-Like Receptors Is Required for Intestinal Homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 148.Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, Lefkowitch JH, Bower M, Friedman R, Sartor RB, Rabadan R, Schwabe RF. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and Tlr4. Cancer Cell. 2012;21:504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Macedo L, Pinhal-Enfield G, Alshits V, Elson G, Cronstein BN, Leibovich SJ. Wound Healing Is Impaired in Myd88-Deficient Mice: A Role for Myd88 in the Regulation of Wound Healing by Adenosine A2a Receptors. Am J Pathol. 2007;171:1774–1788. doi: 10.2353/ajpath.2007.061048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, Hooper LV, Schmidt RR, von Aulock S, Radek KA, Huang CM, Ryan AF, Gallo RL. Commensal Bacteria Regulate Toll-Like Receptor 3-Dependent Inflammation after Skin Injury. Nat Med. 2009;15:1377–1382. doi: 10.1038/nm.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, Ryffel B, Couillin I. Il-1r1/Myd88 Signaling and the Inflammasome Are Essential in Pulmonary Inflammation and Fibrosis in Mice. J Clin Invest. 2007;117:3786–3799. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kinnebrew MA, Pamer EG. Innate Immune Signaling in Defense against Intestinal Microbes. Immunol Rev. 2012;245:113–131. doi: 10.1111/j.1600-065X.2011.01081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gregorio J, Meller S, Conrad C, Di Nardo A, Homey B, Lauerma A, Arai N, Gallo RL, Digiovanni J, Gilliet M. Plasmacytoid Dendritic Cells Sense Skin Injury and Promote Wound Healing through Type I Interferons. J Exp Med. 2010;207:2921–2930. doi: 10.1084/jem.20101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting Edge: Bacterial Flagellin Activates Basolaterally Expressed Tlr5 to Induce Epithelial Proinflammatory Gene Expression. J Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 155.Abreu MT. Toll-Like Receptor Signalling in the Intestinal Epithelium: How Bacterial Recognition Shapes Intestinal Function. Nat Rev Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 156.Bataller R, Brenner DA. Liver Fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schwabe RF, Seki E, Brenner DA. Toll-Like Receptor Signaling in the Liver. Gastroenterology. 2006;130:1886–1900. doi: 10.1053/j.gastro.2006.01.038. [DOI] [PubMed] [Google Scholar]
- 158.Matsumura T, Degawa T, Takii T, Hayashi H, Okamoto T, Inoue J, Onozaki K. Traf6-Nf-Kappab Pathway Is Essential for Interleukin-1-Induced Tlr2 Expression and Its Functional Response to Tlr2 Ligand in Murine Hepatocytes. Immunology. 2003;109:127–136. doi: 10.1046/j.1365-2567.2003.01627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mimura Y, Sakisaka S, Harada M, Sata M, Tanikawa K. Role of Hepatocytes in Direct Clearance of Lipopolysaccharide in Rats. Gastroenterology. 1995;109:1969–1976. doi: 10.1016/0016-5085(95)90765-3. [DOI] [PubMed] [Google Scholar]
- 160.Hernandez-Gea V, Friedman SL. Pathogenesis of Liver Fibrosis. Annu Rev Pathol. 2011;6:425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 161.Triger DR, Boyer TD, Levin J. Portal and Systemic Bacteraemia and Endotoxaemia in Liver Disease. Gut. 1978;19:935–939. doi: 10.1136/gut.19.10.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nolan JP. The Role of Intestinal Endotoxin in Liver Injury: A Long and Evolving History. Hepatology. 2010;52:1829–1835. doi: 10.1002/hep.23917. [DOI] [PubMed] [Google Scholar]
- 163.Wiest R, Garcia-Tsao G. Bacterial Translocation (Bt) in Cirrhosis. Hepatology. 2005;41:422–433. doi: 10.1002/hep.20632. [DOI] [PubMed] [Google Scholar]
- 164.Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, Hsu WC, Huang CC, Wang SS, Lo KJ. Endotoxemia in Patients with Chronic Liver Diseases: Relationship to Severity of Liver Diseases, Presence of Esophageal Varices, and Hyperdynamic Circulation. J Hepatol. 1995;22:165–172. doi: 10.1016/0168-8278(95)80424-2. [DOI] [PubMed] [Google Scholar]
- 165.Fouts DE, Torralba M, Nelson KE, Brenner DA, Schnabl B. Bacterial Translocation and Changes in the Intestinal Microbiome in Mouse Models of Liver Disease. J Hepatol. 2012;56:1283–1292. doi: 10.1016/j.jhep.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Seki E, Schnabl B. Role of Innate Immunity and the Microbiota in Liver Fibrosis: Crosstalk between the Liver and Gut. J Physiol. 2012;590:447–458. doi: 10.1113/jphysiol.2011.219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rutenburg AM, Sonnenblick E, Koven I, Aprahamian HA, Reiner L, Fine J. The Role of Intestinal Bacteria in the Development of Dietary Cirrhosis in Rats. J Exp Med. 1957;106:1–14. doi: 10.1084/jem.106.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Broitman SA, Gottlieb LS, Zamcheck N. Influence of Neomycin and Ingested Endotoxin in the Pathogenesis of Choline Deficiency Cirrhosis in the Adult Rat. J Exp Med. 1964;119:633–642. doi: 10.1084/jem.119.4.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhu Q, Zou L, Jagavelu K, Simonetto DA, Huebert RC, Jiang ZD, DuPont HL, Shah VH. Intestinal Decontamination Inhibits Tlr4 Dependent Fibronectin-Mediated CrossTalk between Stellate Cells and Endothelial Cells in Liver Fibrosis in Mice. J Hepatol. 2012;56:893–899. doi: 10.1016/j.jhep.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Poelstra K, Popov Y, Sverdlov DY, Sharma A, Schuppan D. Gut-Derived Bacterial Products Drive Progression of Liver Fibrosis in Mdr2−/− Mice: Involvement of Lps and Tlr4. Hepatology. 2009;50:819A. [Google Scholar]
- 171.Gabele E, Dostert K, Hofmann C, Wiest R, Scholmerich J, Hellerbrand C, Obermeier F. Dss Induced Colitis Increases Portal Lps Levels and Enhances Hepatic Inflammation and Fibrogenesis in Experimental Nash. J Hepatol. 2011;55:1391–1399. doi: 10.1016/j.jhep.2011.02.035. [DOI] [PubMed] [Google Scholar]
- 172.Yoneda M, Izawa K, Hirooka A, Hayashida K, Wakitani S. Indicators of Superior Glenoid Labral Detachment on Magnetic Resonance Imaging and Computed Tomography Arthrography. J Shoulder Elbow Surg. 1998;7:2–12. doi: 10.1016/s1058-2746(98)90177-x. [DOI] [PubMed] [Google Scholar]
- 173.Ni H, Nitta M, Komatsu H, Kojima S, Suzuki S, Harada S, Tsuboi K, Banno S, Wakita A, Yazaki M, Ren L, Kato T, Ueda R. Detection of Bcr/Abl Fusion Transcripts by Semiquantitative Multiplex Rt-Pcr Combined with a Colormetric Assay in Ph Positive Leukemia. Cancer Lett. 1998;124:173–180. doi: 10.1016/s0304-3835(97)00472-2. [DOI] [PubMed] [Google Scholar]
- 174.Isayama F, Hines IN, Kremer M, Milton RJ, Byrd CL, Perry AW, McKim SE, Parsons C, Rippe RA, Wheeler MD. Lps Signaling Enhances Hepatic Fibrogenesis Caused by Experimental Cholestasis in Mice. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1318–1328. doi: 10.1152/ajpgi.00405.2005. [DOI] [PubMed] [Google Scholar]
- 175.Wakita H, Matsushita K, Nishimura K, Tokura Y, Furukawa F, Takigawa M. Sphingosylphosphorylcholine Stimulates Proliferation and Upregulates Cell Surface-Associated Plasminogen Activator Activity in Cultured Human Keratinocytes. J Invest Dermatol. 1998;110:253–258. doi: 10.1046/j.1523-1747.1998.00120.x. [DOI] [PubMed] [Google Scholar]
- 176.Paik YH, Schwabe RF, Bataller R, Russo MP, Jobin C, Brenner DA. Toll-Like Receptor 4 Mediates Inflammatory Signaling by Bacterial Lipopolysaccharide in Human Hepatic Stellate Cells. Hepatology. 2003;37:1043–1055. doi: 10.1053/jhep.2003.50182. [DOI] [PubMed] [Google Scholar]
- 177.Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, Layden TJ, Wright TL, White T, Cheung RC. A 7 Gene Signature Identifies the Risk of Developing Cirrhosis in Patients with Chronic Hepatitis C. Hepatology. 2007;46:297–306. doi: 10.1002/hep.21695. [DOI] [PubMed] [Google Scholar]
- 178.Li Y, Chang M, Abar O, Garcia V, Rowland C, Catanese J, Ross D, Broder S, Shiffman M, Cheung R, Wright T, Friedman SL, Sninsky J. Multiple Variants in Toll-Like Receptor 4 Gene Modulate Risk of Liver Fibrosis in Caucasians with Chronic Hepatitis C Infection. J Hepatol. 2009;51:750–757. doi: 10.1016/j.jhep.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Martin-Murphy BV, Holt MP, Ju C. The Role of Damage Associated Molecular Pattern Molecules in Acetaminophen-Induced Liver Injury in Mice. Toxicol Lett. 2010;192:387–394. doi: 10.1016/j.toxlet.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The Nuclear Factor Hmgb1 Mediates Hepatic Injury after Murine Liver Ischemia-Reperfusion. J Exp Med. 2005;201:1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wynn TA. Integrating Mechanisms of Pulmonary Fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Imaeda AB, Watanabe A, Sohail MA, Mahmood S, Mohamadnejad M, Sutterwala FS, Flavell RA, Mehal WZ. Acetaminophen-Induced Hepatotoxicity in Mice Is Dependent on Tlr9 and the Nalp3 Inflammasome. J Clin Invest. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel Signal Transduction Pathway Utilized by Extracellular Hsp70: Role of Toll-Like Receptor (Tlr) 2 and Tlr4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 184.Song JY, Li L, Ahn JB, Park JG, Jo JS, Park DH, Jang HK, Jang JJ, Lee MJ. Acute Liver Toxicity by Carbon Tetrachloride in Hsp70 Knock out Mice. Exp Toxicol Pathol. 2007;59:29–34. doi: 10.1016/j.etp.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 185.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-Like Receptor 9 Inhibition Confers Protection from Liver Ischemia-Reperfusion Injury. Hepatology. 2010;51:621–632. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xu J, Zhang X, Monestier M, Esmon NL, Esmon CT. Extracellular Histones Are Mediators of Death through Tlr2 and Tlr4 in Mouse Fatal Liver Injury. J Immunol. 2011;187:2626–2631. doi: 10.4049/jimmunol.1003930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Szabo G, Velayudham A, Romics L, Jr, Mandrekar P. Modulation of Non-Alcoholic Steatohepatitis by Pattern Recognition Receptors in Mice: The Role of Toll-Like Receptors 2 and 4. Alcohol Clin Exp Res. 2005;29:140S–145S. doi: 10.1097/01.alc.0000189287.83544.33. [DOI] [PubMed] [Google Scholar]
- 188.Rivera CA, Gaskin L, Allman M, Pang J, Brady K, Adegboyega P, Pruitt K. Toll-Like Receptor-2 Deficiency Enhances Non-Alcoholic Steatohepatitis. BMC Gastroenterol. 2010;10:52. doi: 10.1186/1471-230X-10-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hartmann P, Haimerl M, Mazagova M, Brenner DA, Schnabl B. Toll-Like Receptor 2-Mediated Intestinal Injury and Enteric Tumor Necrosis Factor Receptor I Contribute to Liver Fibrosis in Mice. Gastroenterology. 2012 Nov;143(5):1330–1340.e1. doi: 10.1053/j.gastro.2012.07.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bility MT, Zhang L, Washburn ML, Curtis TA, Kovalev GI, Su L. Generation of a Humanized Mouse Model with Both Human Immune System and Liver Cells to Model Hepatitis C Virus Infection and Liver Immunopathogenesis. Nat Protoc. 2012;7:1608–1617. doi: 10.1038/nprot.2012.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, Strom S, Kay MA, Finegold M, Grompe M. Robust Expansion of Human Hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− Mice. Nat Biotechnol. 2007;25:903–910. doi: 10.1038/nbt1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pradere JP, Troeger JS, Dapito DH, Mencin AA, Schwabe RF. Toll-Like Receptor 4 and Hepatic Fibrogenesis. Semin Liver Dis. 2010;30:232–244. doi: 10.1055/s-0030-1255353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kalambokis GN, Tsianos EV. Rifaximin Reduces Endotoxemia and Improves Liver Function and Disease Severity in Patients with Decompensated Cirrhosis. Hepatology. 2012;55:655–656. doi: 10.1002/hep.24751. [DOI] [PubMed] [Google Scholar]
- 194.Kalambokis GN, Mouzaki A, Rodi M, Tsianos EV. Rifaximin for the Prevention of Spontaneous Bacterial Peritonitis. World J Gastroenterol. 2012;18:1700–1702. doi: 10.3748/wjg.v18.i14.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Velayudham A, Dolganiuc A, Ellis M, Petrasek J, Kodys K, Mandrekar P, Szabo G. Vsl#3 Probiotic Treatment Attenuates Fibrosis without Changes in Steatohepatitis in a Diet-Induced Nonalcoholic Steatohepatitis Model in Mice. Hepatology. 2009;49:989–997. doi: 10.1002/hep.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Beck JM, Young VB, Huffnagle GB. The Microbiome of the Lung. Transl Res. 2012 Oct;160(4):258–66. doi: 10.1016/j.trsl.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zhao H, Leu SW, Shi L, Dedaj R, Zhao G, Garg HG, Shen L, Lien E, Fitzgerald KA, Shiedlin A, Shen H, Quinn DA, Hales CA. Tlr4 Is a Negative Regulator in Noninfectious Lung Inflammation. J Immunol. 2010;184:5308–5314. doi: 10.4049/jimmunol.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yang HZ, Wang JP, Mi S, Liu HZ, Cui B, Yan HM, Yan J, Li Z, Liu H, Hua F, Lu W, Hu ZW. Tlr4 Activity Is Required in the Resolution of Pulmonary Inflammation and Fibrosis after Acute and Chronic Lung Injury. Am J Pathol. 2012;180:275–292. doi: 10.1016/j.ajpath.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 199.Paun A, Fox J, Balloy V, Chignard M, Qureshi ST, Haston CK. Combined Tlr2 and Tlr4 Deficiency Increases Radiation-Induced Pulmonary Fibrosis in Mice. Int J Radiat Oncol Biol Phys. 2010;77:1198–1205. doi: 10.1016/j.ijrobp.2009.12.065. [DOI] [PubMed] [Google Scholar]
- 200.Brickey WJ, Neuringer IP, Walton W, Hua X, Wang EY, Jha S, Sempowski GD, Yang X, Kirby SL, Tilley SL, Ting JP. Myd88 Provides a Protective Role in Long-Term Radiation-Induced Lung Injury. Int J Radiat Biol. 2012;88:335–347. doi: 10.3109/09553002.2012.652723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B, Couillin I. Uric Acid Is a Danger Signal Activating Nalp3 Inflammasome in Lung Injury Inflammation and Fibrosis. Am J Respir Crit Care Med. 2009;179:903–913. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- 202.Doz E, Noulin N, Boichot E, Guenon I, Fick L, Le Bert M, Lagente V, Ryffel B, Schnyder B, Quesniaux VF, Couillin I. Cigarette Smoke-Induced Pulmonary Inflammation Is Tlr4/Myd88 and Il-1r1/Myd88 Signaling Dependent. J Immunol. 2008;180:1169–1178. doi: 10.4049/jimmunol.180.2.1169. [DOI] [PubMed] [Google Scholar]
- 203.Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, Chadban SJ. Hmgb1 Contributes to Kidney Ischemia Reperfusion Injury. J Am Soc Nephrol. 2010;21:1878–1890. doi: 10.1681/ASN.2009101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. Tlr4 Activation Mediates Kidney Ischemia/Reperfusion Injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Oyama Y, Hashiguchi T, Taniguchi N, Tancharoen S, Uchimura T, Biswas KK, Kawahara K, Nitanda T, Umekita Y, Lotz M, Maruyama I. High-Mobility Group Box-1 Protein Promotes Granulomatous Nephritis in Adenine-Induced Nephropathy. Lab Invest. 2010;90:853–866. doi: 10.1038/labinvest.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sato F, Maruyama S, Hayashi H, Sakamoto I, Yamada S, Uchimura T, Morita Y, Ito Y, Yuzawa Y, Maruyama I, Matsuo S. High Mobility Group Box Chromosomal Protein 1 in Patients with Renal Diseases. Nephron Clin Pract. 2008;108:c194–201. doi: 10.1159/000118942. [DOI] [PubMed] [Google Scholar]
- 207.Rosin DL, Okusa MD. Dangers Within: Damp Responses to Damage and Cell Death in Kidney Disease. J Am Soc Nephrol. 2011;22:416–425. doi: 10.1681/ASN.2010040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Saemann MD, Weichhart T, Zeyda M, Staffler G, Schunn M, Stuhlmeier KM, Sobanov Y, Stulnig TM, Akira S, von Gabain A, von Ahsen U, Horl WH, Zlabinger GJ. Tamm-Horsfall Glycoprotein Links Innate Immune Cell Activation with Adaptive Immunity Via a Toll-Like Receptor-4-Dependent Mechanism. J Clin Invest. 2005;115:468–475. doi: 10.1172/JCI22720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJ, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-Associated Tlr2 Mediates Ischemia/Reperfusion Injury in the Kidney. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shigeoka AA, Holscher TD, King AJ, Hall FW, Kiosses WB, Tobias PS, Mackman N, McKay DB. Tlr2 Is Constitutively Expressed within the Kidney and Participates in Ischemic Renal Injury through Both Myd88-Dependent and -Independent Pathways. J Immunol. 2007;178:6252–6258. doi: 10.4049/jimmunol.178.10.6252. [DOI] [PubMed] [Google Scholar]
- 211.Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of Toll-Like Receptor 4 in Endotoxin-Induced Acute Renal Failure. J Immunol. 2004;172:2629–2635. doi: 10.4049/jimmunol.172.4.2629. [DOI] [PubMed] [Google Scholar]
- 212.Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. Tlr4 Signaling Mediates Inflammation and Tissue Injury in Nephrotoxicity. J Am Soc Nephrol. 2008;19:923–932. doi: 10.1681/ASN.2007090982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Pulskens WP, Rampanelli E, Teske GJ, Butter LM, Claessen N, Luirink IK, van der Poll T, Florquin S, Leemans JC. Tlr4 Promotes Fibrosis but Attenuates Tubular Damage in Progressive Renal Injury. J Am Soc Nephrol. 2010;21:1299–1308. doi: 10.1681/ASN.2009070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Campbell MT, Hile KL, Zhang H, Asanuma H, Vanderbrink BA, Rink RR, Meldrum KK. Toll-Like Receptor 4: A Novel Signaling Pathway During Renal Fibrogenesis. J Surg Res. 2011;168:e61–69. doi: 10.1016/j.jss.2009.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Chowdhury P, Sacks SH, Sheerin NS. Endogenous Ligands for Tlr2 and Tlr4 Are Not Involved in Renal Injury Following Ureteric Obstruction. Nephron Exp Nephrol. 2010;115:e122–130. doi: 10.1159/000313493. [DOI] [PubMed] [Google Scholar]
- 216.Leemans JC, Butter LM, Pulskens WP, Teske GJ, Claessen N, van der Poll T, Florquin S. The Role of Toll-Like Receptor 2 in Inflammation and Fibrosis During Progressive Renal Injury. PLoS One. 2009;4:e5704. doi: 10.1371/journal.pone.0005704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Braga TT, Correa-Costa M, Guise YF, Castoldi A, De Oliveira CD, Hyane MI, Cenedeze MA, Teixeira SA, Muscara MN, Perez KR, Cuccovia IM, Pacheco-Silva A, Goncalves GM, Camara NO. Myd88 Signaling Pathway Is Involved in Renal Fibrosis by Favoring a Th2 Immune Response and Activating Alternative M2 Macrophages. Mol Med. 2012 Oct 24;18(1):1231–9. doi: 10.2119/molmed.2012.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Skuginna V, Lech M, Allam R, Ryu M, Clauss S, Susanti HE, Rommele C, Garlanda C, Mantovani A, Anders HJ. Toll-Like Receptor Signaling and Sigirr in Renal Fibrosis Upon Unilateral Ureteral Obstruction. PLoS One. 2011;6:e19204. doi: 10.1371/journal.pone.0019204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lin Q, Wang L, Lin Y, Liu X, Ren X, Wen S, Du X, Lu T, Su SY, Yang X, Huang W, Zhou S, Wen F, Su SB. Toll-Like Receptor 3 Ligand Polyinosinic:Polycytidylic Acid Promotes Wound Healing in Human and Murine Skin. J Invest Dermatol. 2012;132:2085–2092. doi: 10.1038/jid.2012.120. [DOI] [PubMed] [Google Scholar]
- 220.Sato T, Yamamoto M, Shimosato T, Klinman DM. Accelerated Wound Healing Mediated by Activation of Toll-Like Receptor 9. Wound Repair Regen. 2010;18:586–593. doi: 10.1111/j.1524-475X.2010.00632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Yamamoto M, Sato T, Beren J, Verthelyi D, Klinman DM. The Acceleration of Wound Healing in Primates by the Local Administration of Immunostimulatory Cpg Oligonucleotides. Biomaterials. 2011;32:4238–4242. doi: 10.1016/j.biomaterials.2011.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang J, Hori K, Ding J, Huang Y, Kwan P, Ladak A, Tredget EE. Toll-Like Receptors Expressed by Dermal Fibroblasts Contribute to Hypertrophic Scarring. J Cell Physiol. 2011;226:1265–1273. doi: 10.1002/jcp.22454. [DOI] [PubMed] [Google Scholar]
- 223.Farina GA, York MR, Di Marzio M, Collins CA, Meller S, Homey B, Rifkin IR, Marshak-Rothstein A, Radstake TR, Lafyatis R. Poly(I:C) Drives Type I Ifn- and Tgfbeta-Mediated Inflammation and Dermal Fibrosis Simulating Altered Gene Expression in Systemic Sclerosis. J Invest Dermatol. 2010;130:2583–2593. doi: 10.1038/jid.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Shishido T, Nozaki N, Yamaguchi S, Shibata Y, Nitobe J, Miyamoto T, Takahashi H, Arimoto T, Maeda K, Yamakawa M, Takeuchi O, Akira S, Takeishi Y, Kubota I. Toll-Like Receptor-2 Modulates Ventricular Remodeling after Myocardial Infarction. Circulation. 2003;108:2905–2910. doi: 10.1161/01.CIR.0000101921.93016.1C. [DOI] [PubMed] [Google Scholar]
- 225.Mersmann J, Habeck K, Latsch K, Zimmermann R, Jacoby C, Fischer JW, Hartmann C, Schrader J, Kirschning CJ, Zacharowski K. Left Ventricular Dilation in Toll-Like Receptor 2 Deficient Mice after Myocardial Ischemia/Reperfusion through Defective Scar Formation. Basic Res Cardiol. 2011;106:89–98. doi: 10.1007/s00395-010-0127-y. [DOI] [PubMed] [Google Scholar]
- 226.Riad A, Jager S, Sobirey M, Escher F, Yaulema-Riss A, Westermann D, Karatas A, Heimesaat MM, Bereswill S, Dragun D, Pauschinger M, Schultheiss HP, Tschope C. Toll-Like Receptor-4 Modulates Survival by Induction of Left Ventricular Remodeling after Myocardial Infarction in Mice. J Immunol. 2008;180:6954–6961. doi: 10.4049/jimmunol.180.10.6954. [DOI] [PubMed] [Google Scholar]
- 227.Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ, Doevendans PA, van Echteld CJ, Joles JA, Quax PH, Piek JJ, Pasterkamp G, de Kleijn DP. Toll-Like Receptor 4 Mediates Maladaptive Left Ventricular Remodeling and Impairs Cardiac Function after Myocardial Infarction. Circ Res. 2008;102:257–264. doi: 10.1161/CIRCRESAHA.107.158220. [DOI] [PubMed] [Google Scholar]
- 228.Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, Pedrazzini T, Berger CT, Dirnhofer S, Matter CM, Penninger JM, Luscher TF, Eriksson U. Myeloid Differentiation Factor-88/Interleukin-1 Signaling Controls Cardiac Fibrosis and Heart Failure Progression in Inflammatory Dilated Cardiomyopathy. Circ Res. 2009;105:912–920. doi: 10.1161/CIRCRESAHA.109.199802. [DOI] [PubMed] [Google Scholar]
- 229.Ha T, Hua F, Li Y, Ma J, Gao X, Kelley J, Zhao A, Haddad GE, Williams DL, Browder IW, Kao RL, Li C. Blockade of Myd88 Attenuates Cardiac Hypertrophy and Decreases Cardiac Myocyte Apoptosis in Pressure Overload-Induced Cardiac Hypertrophy in Vivo. Am J Physiol Heart Circ Physiol. 2006;290:H985–994. doi: 10.1152/ajpheart.00720.2005. [DOI] [PubMed] [Google Scholar]