

Abstract
Background
Mechanical ventilation (MV) may cause ventilator-induced lung injury (VILI) and may thereby contribute to fatal multiple organ failure. We tested the hypothesis that injurious MV of lipopolysaccharide (LPS) pre-injured lungs induces myocardial inflammation and further dysfunction ex vivo, through calcium (Ca2+)-dependent mechanism.
Materials and methods
N = 35 male anesthetized and paralyzed male Wistar rats were randomized to intratracheal instillation of 2 mg/kg LPS or nothing and subsequent MV with lung-protective settings (low tidal volume (Vt) of 6 mL/kg and 5 cmH2O positive end-expiratory pressure (PEEP)) or injurious ventilation (high Vt of 19 mL/kg and 1 cmH2O PEEP) for 4 hours. Myocardial function ex vivo was evaluated in a Langendorff setup and Ca2+ exposure. Key mediators were determined in lung and heart at the mRNA level.
Results
Instillation of LPS and high Vt MV impaired gas exchange and, particularly when combined, increased pulmonary wet/dry ratio; heat shock protein (HSP)70 mRNA expression also increased by the interaction between LPS and high Vt MV. For the heart, C-X-C motif ligand (CXCL)1 and Toll-like receptor (TLR)2 mRNA expression increased, and ventricular (LV) systolic pressure, LV developed pressure, LV +dP/dtmax and contractile responses to increasing Ca2+ exposure ex vivo decreased by LPS. High Vt ventilation aggravated the effects of LPS on myocardial inflammation and dysfunction but not on Ca2+ responses.
Conclusions
Injurious MV by high Vt aggravates the effects of intratracheal instillation of LPS on myocardial dysfunction, possibly through enhancing myocardial inflammation via pulmonary release of HSP70 stimulating cardiac TLR2, not involving Ca2+ handling and sensitivity.
Keywords: Biotrauma, Ventilator-induced lung injury, Myocardial depression, Endotoxin, Toll like receptor (TLR)2, Chemokine (C-X-C motif) ligand (CXCL)1, Heat shock protein (HSP)70
Introduction
Injurious mechanical ventilation (MV) can induce ventilator-induced lung injury (VILI) and may thereby contribute to multiple organ failure, potentially by spillover from lung-borne inflammatory mediators into the circulation which may act on distant organs such as the kidneys [1-3]. Direct or primary pulmonary injury, like pneumonia or aspiration, is associated with more severe VILI than indirect pulmonary injury, such as sepsis, in the presence of identical ventilatory settings [4].
In contrast to the kidney, the effect of VILI on the heart is less clear. Only a few studies have addressed the effect of MV on myocardial function, independent from a fall in venous return and cardiac performance following a rise in intrathoracic pressure [5]. It was, for instance, suggested that plasma from dogs ventilated with 15 cm H2O positive end expiratory pressure (PEEP) contains a negative inotropic mediator, which may adversely affect myocardial function [6]. Next, Nin et al. showed that high tidal volume (Vt) ventilation induces VILI and upregulates myocardial cyclooxygenase (COX)-1 and COX-2 mRNA expression [7]. These enzymes are known to be upregulated during inflammatory conditions. Also, Brander et al. showed that high Vt ventilation during lung injury and associated VILI increased myocardial interleukin (IL) 8 expression, which was decreased with so-called lung-protective strategies [8]. We have shown that injurious MV (ie high tidal volume (Vt) ventilation) for 4 hours augmented myocardial inflammation in a cecal ligation and puncture model through an enhanced deposition of advanced glycation end products resulting in increased myocardial inflammation [9]. These latter studies suggest that pulmonary overdistention and VILI may induce myocardial inflammation but the functional consequences were not examined.
Sepsis may hamper both pulmonary function [10], thereby necessitating MV, and myocardial function [11-14]. Sepsis may be associated with myocardial inflammation and a decrease in myocardial calcium (Ca2+) handling and sensitivity, thereby potentially contributing to impaired myocardial function [12,13]. Lung injury often results in (severe) hypoxia; as a consequence the function of the cardiomyocyte L-type Ca2+ current may be suppressed [15-17]. All of this suggests that VILI may cause myocardial dysfunction via a Ca2+ dependent pathway.
In the current study we used a two hit model of lipopolysaccharide (LPS)-induced pneumonia and subsequent high Vt ventilation to induce VILI and studied myocardial function ex vivo. Non-injurious low Vt ventilation served as control. We hypothesized that spillover of inflammatory mediators during VILI induces myocardial inflammation and decreases myocardial function through a Ca2+-dependent pathway.
Materials and methods
Animal experiments
All experiments applied with the Guide for Care and Use for laboratory animals of the National Institute of Health and were approved by the Institutional Animal Care and Use Committee of the VU University Amsterdam. Thirty-five male Wistar rats weighing 330 ± 20 g were anesthetized with 12.5 mg/kg midazolam (Pharmachemie BV, Haarlem, the Netherlands) and 85 mg/kg ketamine (Alfasan, Woerden, the Netherlands) i.p. and 10 mg/kg ketamine i.m. Anesthesia was maintained with 1.2 mg/kg/h midazolam and 20 mg/kg/h ketamine i.v. Paralysis was maintained with pancuronium 0.6 mg/kg/h i.v. to allow for high Vt ventilation. Animals were placed in supine position on a heating pad maintaining body temperature at 37°C. A tracheostomy was performed and a 14 Gauge canula was inserted into the trachea. During preparation, animals were ventilated with a Vt of 6 ml/kg and 5 cm H2O positive end-expiratory pressure (PEEP) (Avea, CareFusion, Houten, the Netherlands). Catheters were inserted into the left carotid artery and left jugular vein for arterial blood sampling and continuous measurement of the mean arterial pressure (MAP) and central venous pressure (CVP) in mmHg. Heart rate (HR) in beats per minute (BPM) was derived from MAP measurements. The femoral artery was catheterized with a thermistor from a pulmonary artery catheter to measure cardiac output. Cardiac output was obtained every 30 minutes by averaging two successive thermodilution determinations (CO Computer, 9520A, Edwards laboratory, Santa Ana, Ca, USA), for which 200 μl of cold saline was injected via the right jugular vein catheter as described previously [18]. Blood samples (150 μl) were taken every hour for gas analysis and these were replaced by equal volumes of normal saline. Blood gas analysis was performed using a pH blood-gas analyzer (ABL 50; Radiometer, Copenhagen, Denmark). Partial pressure of arterial oxygen pressure (PaO2, mmHg)/fraction of inspired oxygen (FIO2) ratios were calculated.
Experimental protocol
After preparation, hemodynamics were allowed to stabilize for 10 min after which base line values were established (t=−10). At t=−5 animals were randomized to non-LPS treated or LPS treated groups. The latter received saline-dissolved LPS (2 mg/kg, LPS L2880, LPS from E. Coli 055:B5, Sigma-Aldrich) intratracheally using a miniature nebulizer (Penn-Century, Wyndmoor, PA, USA). Five minutes after LPS administration, the protocol was started and animals were randomly assigned to one of two ventilation strategies; ventilation with either low Vt (6 ml/kg, 5 cm H2O PEEP) which is regarded as a lung protective strategy [19], or high Vt (19 ml/kg, 1 cm H2O PEEP) which is regarded as an injurious strategy [20,21]. Ventilatory settings were chosen such that mean airway pressure was similar to avoid differences among groups in cardiac preloading [5,14]. Thus four groups were studied; non-LPS treated (n=8) and LPS-treated animals (n=8) ventilated with low Vt and non-LPS treated (n=11, including N = 3 animals not included in the Langendorff setup because of technical issues) and LPS-treated animals (n=8) ventilated with high Vt. The FIO2 was set at 0.4 in all groups but was increased when oxygenation was impaired. Ventilation rate was set and adjusted if necessary throughout the experiment to maintain normocapnia. Four hours after the start of the protocol, hearts were rapidly dissected and myocardial function was measured ex vivo. If PaO2/FIO2 fell below 150 with a FIO2 of 100% and an increase in mean airway pressure of 2 cm H2O compared to t=0 was present, the experiment was terminated prematurely and myocardial function ex vivo was measured.
Myocardial function ex vivo
Myocardial function ex vivo was measured in a Langendorff set-up (n=8 per group), to study myocardial function independent of loading condition as previously described [22]. Briefly, the aorta of the isolated heart was canulated and the heart was perfused with a modified Krebs-Henseleit solution at a constant coronary perfusion pressure of 80 mmHg at 37°C. The modified Krebs-Henseleit solution contained (in mM) 118.5 NaCl, 4.7 KCl, 25 NaHCO3, 1.2 MgCl2, 1.2 KH2PO4 and 11 glucose and was equilibrated with 95% O2 and 5% CO2 at a pH of 7.4. Solutions with different with CaCl2.2(H2O) concentrations were made, resulting in final Ca2+ concentrations of 0.5; 1; 2 and 4 mM. Both right and left atria were removed and hearts were paced at 5 Hz with electrodes. Afferent coronary flow was measured with a flow meter (Transonic Systems Europe B.V., Maastricht, the Netherlands). A custom-made balloon was inserted in the left ventricle to measure isovolumic pressures with a catheter tip manometer system and the heart was allowed to stabilize for 10 minutes. Ventricular volume at maximal pressure development (Vmax) was determined and balloons were adjusted to 85% of Vmax. Hearts were allowed to stabilize for 2 min. After stabilization, myocardial function was measured by left ventricular (LV) systolic and diastolic pressure, maximal rates of pressure development (+dP/dtmax) and pressure decline (−dP/dtmin). As measurements were performed at a fixed heart rate and preload, +dP/dtmax and -dP/dtmin can be regarded as indices of contractility and relaxation, respectively [11]. Developed pressure was calculated as systolic pressure minus diastolic pressure. Force-pCa (i.e. –10log [Ca2+]) relations were fit to the Hill equation and subsequently the pCa50, the pCa at which 50% of the developed force was reached, was calculated [23]. A decrease in the pCa50 indicates a decrease in Ca2+ sensitivity. After this protocol, hearts were removed from the isolated Langendorff-perfused heart set-up, cut transversely in three sections, frozen in liquid nitrogen and stored at −80°C. The apical section of the heart was used to calculate wet to dry weight ratio.
Wet to dry weight ratios
Immediately after the animals were sacrificed, middle right lung lob was taken and weighed, dried at 37°C and weighed again. After freezing, apical section of the heart was weighed, freeze-dried and weighed again.
mRNA expression
To study myocardial inflammatory response we randomly choose animals from each group in which we studied myocardial mRNA expression of the key pro-inflammatory mediators TNFα, interleukin (IL)6, IL1β, chemokine (C-X-C motif) ligand (CXCL)1 and TLR2. Myocardial tissue from the center transverse section and pulmonary tissue from the right lung were pulverized with a mortar and RNA was extracted using Trizol. Pulmonary RNA was purified using the RNA clean up kit (Qiagen, Venlo, the Netherlands). DNA was removed by DNAse I amplification grade (Invitrogen, Breda, The Netherlands) A total of 2 μg RNA was used to synthesize copy DNA (cDNA) using a Cloned AMV First Strand cDNA Synthesis Kit (Invitrogen, Breda, The Netherlands) using oligo-dT priming. Quantitive RT-PCR analysis was performed using SYBR Green in an ABI 7500 sequence detection system (Applied Biosystems, Foster City, USA). Briefly, 8 μl mix was prepared using 25 ng cDNA, forward and reverse primers and Mesa Green qPCR Mastermix Plus for SYBR assay (Eurogentec, Maastricht, The Netherlands). The used protocol was 2 min 50°C, 10 min 95°C, 40 cycles (0:15 min 95°C, 1:00 min 58°C) and a dissociation curve. Primer sequences are shown in Table 1. Cycle threshold values (Ct), the number of cycles required for the fluorescent signal to cross the threshold, were measured. Relative expression levels of target genes were calculated relative to the housekeeping gene β-actin with the formula 2(Ct (β-actin)- Ct (target gene)).
Table 1.
Primer sequences used for RT-PCR
| HSP70 |
Forward |
AGGTGGATTAGAGGCTCTTT |
| Reverse |
AACCTAGGACTTGATTGCAGA |
|
| TNFα |
Forward |
ACAAGCCCGTAGCCCACGTC |
| Reverse |
AGGAGCACGTAGTCGGGGCA |
|
| IL6 |
Forward |
GTCTCGAGCCCACCAGGAACG |
| Reverse |
AAGCCTCCGACTTGTGAAGTGGT |
|
| IL1β |
Forward |
GAGCCCGTCCTCTGTGACTCGT |
| Reverse |
AGGCCCAAGGCCACAGGGATT |
|
| CXCL1 |
Forward |
TCGCCAATGAGCTGCGCTGT |
| Reverse |
CAAGGCAAGCCTCGCGACCAT |
|
| TLR2 |
Forward |
GGGAAGGCCATTCTGCCCAGG |
| Reverse |
CGGAGGTTCACACAGGCTCGC |
|
| β-actin |
Forward |
GGCCAACCGTGAAAAGATGA |
| Reverse | GGACAACACAGCCTGGATGG |
Statistical analysis
To analyze the effects of LPS, Vt and their interaction, general estimated equations (GEE) were performed. With GEE effects of separate parameters as well as their interaction can be calculated, taking repeated measures over time in the same animals and baseline values as covariates into account when appropriate. A statistically significant interaction implies that the effect of high (vs low) Vt ventilation over time differs between LPS and non-LPS-treated animals. T-tests were performed to evaluate the effect of high Vt ventilation in LPS-treated animals on mRNA expression which were normally distributed. Spearman correlations were calculated for relations since parameters were distributed non-parametrically. Data are shown as mean ± SEM, exact P values are given if >0.001. A P-value < 0.05 was considered statistically significant.
Results
Baseline characteristics were similar between the groups. Seven LPS-treated animals ventilated with high Vt died prematurely (4 after t=150 min; 1 after t=180 min; 2 after t=210 min).
Hemodynamic measurements
Hemodynamic data are shown in Figure 1. MAP (P<0.001), CVP (P<0.001) and HR (P<0.001) decreased by high Vt ventilation, but no effect of LPS was seen. Cardiac output decreased by the interaction between LPS and high Vt ventilation (P=0.001). Blood pH decreased by LPS (Figure 2A, P<0.001) but only LPS-treated animals ventilated with high Vt reached levels consistent with acidosis, a marker of hypoperfusion.
Figure 1.
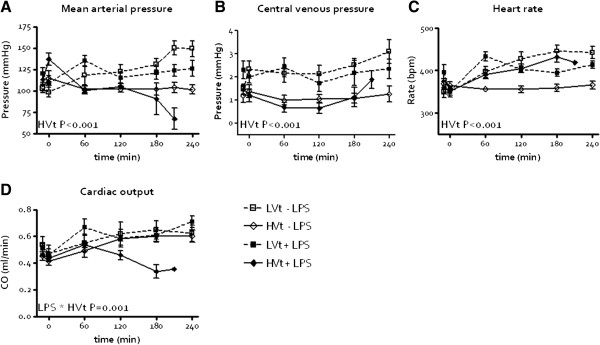
In vivo measurements in the course of time (LPS=lipopolysaccharide; LVt=low tidal volume (Vt) ventilation, HVt=high Vt ventilation). Mean arterial pressure (A), central venous pressure (B) and heart rate (C). Cardiac output (D) decreased by the interaction between LPS installation and HVt ventilation.
Figure 2.

Ventilator-induced lung injury induction in the course of time (LPS=lipopolysaccharide; LVt=low tidal volume (Vt) ventilation, HVt=high Vt ventilation). A. pH. B. PCO2. C. PaO2/FiO2. D. PO2. E. Mean airway pressure. F Pulmonary wet/dry ratio increased by LPS and HVt ventilation interaction.
Respiratory parameters
The PaCO2 increased by LPS (P<0.001) and decreased by high Vt ventilation (Figure 2B, P<0.001) but remained within the normal range in all cases. PaO2/FIO2 decreased by LPS (P=0.022) and high Vt ventilation (Figure 2C, P=0.011). The PaO2 decreased by LPS (P<0.01) and by high Vt ventilation (P = 0.028) but not by the interaction between LPS and high Vt ventilation (Figure 2D). In the high Vt ventilation + LPS group the lowest PaO2 was 62 mmHg at FiO2 1.0. Mean airway pressure slightly increased by high Vt ventilation (Figure 2E, P<0.001) but was not affected by LPS. Pulmonary wet/dry weight ratio increased by both LPS (P<0.001) and high Vt ventilation (Figure 2F, P<0.001) and an interaction was seen (P=0.001).
Myocardial function ex vivo
LPS decreased myocardial contractile function ex vivo with a decrease in LV systolic pressure (Figure 3A, P<0.001), LV developed pressure (Figure 3C, P=0.004) and LV +dP/dtmax (Figure 3D, P=0.015). LPS also decreased relaxation with an increase in LV –dP/dtmin (Figure 3E, P=0.001). An interaction of LPS with high Vt ventilation was seen for LV developed pressure (P=0.006), LV +dP/dtmax (P=0.006) and LV -dP/dtmin (P=0.007), so that high Vt augmented the effect of LPS. Cardiac pCa50 decreased by LPS (Figure 3F, P<0.001) but was not affected by high Vt ventilation. Affluent coronary flow (16.0 ± 1.1 ml/min) and cardiac wet/dry weight ratio (6.6 ± 0.1) were similar between the groups excluding differences in perfusion or myocardial edema as contributing factors to dysfunction.
Figure 3.
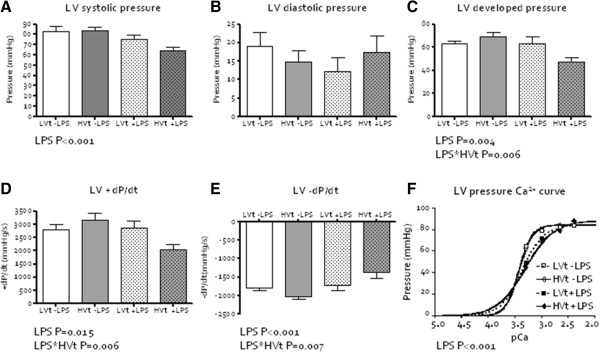
Myocardial function ex vivo (LPS=lipopolysaccharide; LVt=low tidal volume (Vt) ventilation, HVt=high Vt ventilation). A. Left ventricular (LV) systolic pressure. B. LV diastolic pressure. C. LV developed pressure decreased by the interaction between LPS installation and HVt ventilation. D. LV contractility, measured as +dP/dtmax, decreased by the interaction between LPS installation and HVt ventilation. E. LV relaxation, measured as –dP/dtmin, decreased by the interaction between LPS installation and HVt ventilation. F. LV pCa50.
Myocardial inflammation
Because we were unable to confirm the involvement of Ca2+ handling or sensitivity in our model, we searched for other explanations for the observed results. We therefore studied the well known pulmonary heat shock protein (HSP)70 response [24,25] and the myocardial toll-like receptor (TLR)2-induced pro-inflammatory response since the latter may be related [26]. Myocardial TNFα, IL6 and IL1β expressions were not affected by either LPS or high Vt ventilation. TLR2 mRNA expression increased by LPS (P=0.035) but not by high Vt ventilation. Myocardial CXCL1 mRNA expression increased by LPS (P=0.017), high Vt ventilation (P=0.004) and an interaction between LPS and high Vt was seen (P=0.013). CXCL1 (r=−0.60, P=0.002) and TLR2 (r=−0.54, P=0.008) mRNA expressions inversely correlated with LV systolic pressure across groups. TLR2 mRNA expression also correlated with LV –dP/dtmin (r=0.48, P=0.021). Pulmonary HSP70 mRNA expression increased by the interaction between LPS and high Vt ventilation (Table 2, P<0.001), whereas no effect on myocardial HSP70 mRNA expression was observed.
Table 2.
Median values and interquartile ranges for pulmonary and myocardial mRNA expression in arbitrary units (A.U.)
| |
LVt, LPS (−) |
HVt, LPS (−) |
LVt, LPS (+) |
HVt, LPS (+) |
|---|---|---|---|---|
| (N = 6) | (N = 6) | (N = 5) | (N = 6) | |
| Pulmonary mRNA expression (A.U.) | ||||
|
HSP70 |
17*10-3 |
2.5*10-3 |
3.8*10-3 |
16*10-3# |
| (8.7-26*10-3) |
(0.050-8.2*10-3) |
(3.4-6.2*10-3) |
(8.4-27*10-3) |
|
| Myocardial mRNA expression (A.U.) | ||||
|
TLR2 |
0.10*10-3 |
0.10*10-3 |
0.10*10-3 |
0.25*10-3 |
| (0–0.22*10-3) |
(0–0.25*10-3) |
(0.10-0.25*10-3) |
(0.18-0.55*10-3) |
|
|
TNFα |
0.10*10-3 |
0.15*10-3 |
0.20*10-3 |
0.45*10-3 |
| (0–0.38*10-3) |
(0.075-1.6*10-3) |
(0.05-0.35*10-3) |
(0.10-0.63*10-3) |
|
|
IL6 |
0 |
0.05*10-3 |
0.10*10-3 |
0.35*10-3 |
| (0–0.35*10-3) |
(0–3.1*10-3) |
(0–7.0*10-3) |
(0–4.7*10-3) |
|
|
IL1β |
0.45*10-3 |
1.3*10-3 |
1.6*10-3 |
1.9*10-3 |
| (0.075-3.9*10-3) |
(0.38-15*10-3) |
(0.65-2.6*10-3) |
(0.83-4.9*10-3) |
|
|
CXCL1 |
17*10-3 |
12*10-3 |
21*10-3 |
119*10-3 ## |
| (2.73-57.8*10-3) | (3.8-67*10-3) | (16-37*10-3) | (71-225*10-3) | |
Relative expression levels of target genes were calculated relative to the housekeeping gene β-actin with the formula 2(Ct (β-actin)- Ct (target gene)). LPS lipopolysaccharide, LVt low tidal volume (Vt) ventilation, HVt high Vt ventilation, HSP70 heat shock protein 70, TLR2 toll like receptor 2, TNF α tumor necrosis factor α, IL6 Interleukin 6, IL1β Interleukin 1β, CXCL1 chemokine (C-X-C motif) ligand 1. The interaction between LPS and HVt ventilation increased pulmonary HSP70 (#, P<0.001) and myocardial CXCL1 mRNA expression (##, P=0.013).
Discussion
In this study, we showed that injurious MV with high Vt in a model of LPS-induced pneumonia aggravated LPS-induced myocardial dysfunction ex vivo. In contrast with our hypothesis, we could not confirm alterations in Ca2+ sensitivity or calcium handling. Additional analyses suggested that myocardial dysfunction was potentially mediated through pulmonary release of HSP70 stimulating myocardial TLR2.
We used a two-hit model with intratracheal LPS installation and MV with Vt of 19 ml/kg to create VILI in line with previous studies [20,21]. This approach resulted in lung injury: pulmonary gas exchange deteriorated and the pulmonary wet-to-dry ratio increased, consistent with VILI. Mortality was high as in the LPS and high Vt group only one animal survived 4 hours, but this did not preclude ex vivo studies. Furthermore, myocardial function was impaired as demonstrated by a decreased CO in vivo without a fall in CVP, and a decreased LV systolic pressure, developed pressure and +dP/dtmax and increased –dP/dtmin ex vivo following LPS installation [5,21]. Moreover, we found that the LPS-induced myocardial dysfunction in vivo and ex vivo was aggravated by high Vt ventilation and development of VILI over time as shown by a further worsening of LV function parameters. It can be surmised that the mechanisms underlying VILI and myocardial dysfunction are comparable to those in sepsis, including myocardial inflammation [27]. In fact, MV may evoke an inflammatory response that is similar to that evoked by endotoxin, signifying a synergistic (i.e. double-hit) effect [28,29]. Injurious MV augments lung injury caused by intra-tracheal instillation of bacterial products [30,31]. Our results are in line with such a synergistic effect of MV in lungs and heart and we examined potential routes.
The causative mechanisms underlying the observed decrease in ex vivo myocardial function may be multifactorial. We hypothesized that high Vt ventilation caused myocardial dysfunction through a Ca2+ dependent manner. First, it can be postulated that distant organ failure after experimental injurious MV might be due to spillover of LPS from the injured lungs into the systemic circulation [31,32] which can then decrease myocardial function by inflammatory responses decreasing myocardial Ca2+ handling and sensitivity [12,13]. Although LPS-induced myocardial dysfunction in our study was associated with a decreased cardiac pCa50, high Vt ventilation did not affect cardiac pCa50, thereby excluding an effect on Ca2+ handling and sensitivity and rendering injurious MV-induced pulmonary release of LPS in the systemic circulation as a mediator unlikely. As we measured Ca2+ sensitivity in hearts with intact cell membranes, the Ca2+ sensitivity in this study should otherwise not be confused with myofilament sensitivity [23]. As we cannot distinguish between myofilament sensitivity and Ca2+ handling we cannot exclude the possibility that both parameters were altered. Second, acidosis may decrease myocardial function through a Ca2+-dependent mechanism [33]. Nonetheless, the acidosis we observed in vivo is less likely to be a cause of the observed dysfunction aggravated by injurious MV. Finally, hypoxia-induced cardiac dysfunction is also Ca2+-dependent as it effects the L-type Ca2+ channel [15-17]. However, we think that the level of hypoxia is also less likely to be cause of the observed dysfunction as there was no interaction between LPS and high Vt ventilation. Furthermore, inadequate delivery of O2 seems unlikely. In our experiments, hypoperfusion seems unlikely as a contributory factor because the coronary flow ex vivo was similar between groups. Myocardial edema may affect myocardial function but no difference in cardiac wet/dry weight ratio was found, thereby excluding edema as a causative factor [34].
Hence, we may conclude that the VILI-induced aggravation of myocardial dysfunction was most likely not mediated by a Ca2+-dependent pathway. In fact, our data suggest that a factor related to the severity of VILI contributed to myocardial inflammation and dysfunction, for instance via ventilator-induced lung-borne HSP70, was involved. Extracellular HSP70 can bind to myocardial TLR2 and may thereby cause an inflammatory response indicated by an increase in myocardial CXCL1 expression and Ca2+-independent myocardial dysfunction [26]. So, this route was subsequently studied when myocardial dysfunction appeared independent of Ca2+ handling in our experiments. We found that pulmonary but not myocardial HSP70 mRNA expression increased in severe VILI, after high Vt ventilation in combination with LPS exposure, as shown before [24,25]. We only may speculate about its involvement. In favor of our speculations is that it has been shown before that HSP70 may be actively released by cells under stress [35]. Indeed, injuring the lung with diesel exhaust, caused increases both pulmonary and systemic HSP70 expression [36]. Thus, HSPs may have a cytoprotective effect in VILI, but may also be involved in the activation of innate immunity when released into the systemic circulation. Explaining the high mRNA expression in the low Vt group in the absence of LPS is challenging as there is no explanation readily available. It has been discussed that the expression of HSP70 does not result from the transcription of a single gene, but is derived from what may be a complex interplay of several underlying genes [37]. It may thus be postulated that the HSP response following MV in the absence of LPS in our model is dependent upon both the degree of lung injury, the inflammatory response and their time course, so that we may have missed upregulation of HSP70 in the high Vt control group [38,39]. We have previously observed such a time course for TNF-α in a rat model of endotoxaemia-induced peritonitis [14]. This aspect warrants further investigation. Myocardial HSP70 mRNA expression was not increased, so that it is conceivable that pulmonary release of HSP70 had contributed to myocardial dysfunction. The myocardium of LPS-treated animals was probably more prone to bind lung-borne extracellular HSP70 by TLR2, since TLR2 was already upregulated by LPS in agreement with others [40]. We also found induction of CXCL1 after LPS and even more so after high Vt ventilation and CXCL1 mRNA expression inversely correlated with LV systolic pressure. Its exact role needs to be explored in further study. However, support for a Ca2+-independent role of CXC1 in myocardial dysfunction, as suggested by our study, comes from another report [26,41] and inhibition of CXCL1 decreases right ventricular failure in a model of pulmonary embolism [42]. In any case, myocardial expression of other inflammatory mediators was not increased, which might be due to a different time course of expression: TNF-α expression is increased 2 hours after LPS exposure, but not detectable anymore after 4 hours [14].
There are some limitations to the present study that may influence the interpretation of the main findings. First, mortality was high in the group of rats subjected to both high Vt ventilation and LPS. Only one animal completed the four-hour study protocol. This indicates that the time point at which the myocardial function was measured ex vivo as well as well when the organs were collected for analysis was not consistent among the animals in this group (the earliest being after 2.5 hrs of ventilation), but this does not invalidate our conclusions. Second, we were only able to measure mRNA expression levels and not the actual protein contents, but time may be needed for protein synthesis to become manifest after gene expression. Also, the concept that VILI-derived pulmonary HSP70 is directly responsible for myocardial inflammation and Ca2+-independent dysfunction warrants further investigation by mechanistic or intervention studies. We were unable to measure HSP70 in serum as its mechanistic role was not part of the primary hypothesis of our exploratory study. Our study was primarily designed as a proof of principle study to test the hypothesis that a Ca2+ dependent pathway was involved. When we refuted this hypothesis based on our findings, we explored alternative pathways in available tissue samples. Furthermore, measuring HSP70 in serum does not answer the question whether or not it is lung-borne, unless venous and arterial blood samples are taken and a pulmonary gradient can be measured.
In conclusion, our study shows that injurious ventilation with high Vt aggravates the effects of lung injury caused by intratracheal instillation of LPS on myocardial dysfunction, possibly through enhancing myocardial inflammation by lung-borne mediators, not involving Ca2+ handling and sensitivity.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LS designed the experimental set-up, performed the experiments and drafted and revised the manuscript. JWK advised in the experimental design and revised the manuscript. FBP, MCJK and ABJG conceived the study, designed the experimental set-up and revised the manuscript. All authors read and approved the final manuscript.
Contributor Information
Lonneke Smeding, Email: lonnekesmeding@gmail.com.
Jan Willem Kuiper, Email: j.w.kuiper@vumc.nl.
Frans B Plötz, Email: fb.plotz@tergooiziekenhuizen.nl.
Martin CJ Kneyber, Email: m.c.j.kneyber@umcg.nl.
AB Johan Groeneveld, Email: a.b.j.groeneveld@erasmusmc.nl.
References
- Kuiper JW, Groeneveld AB, Slutsky AS, Plotz FB. Mechanical ventilation and acute renal failure. Crit Care Med. 2005;33:1408–1415. doi: 10.1097/01.CCM.0000165808.30416.EF. [DOI] [PubMed] [Google Scholar]
- Slutsky AS, Tremblay LN. Multiple system organ failure. Is mechanical ventilation a contributing factor? Am J Respir Crit Care Med. 1998;157:1721–1725. doi: 10.1164/ajrccm.157.6.9709092. [DOI] [PubMed] [Google Scholar]
- Kuiper JW, Vaschetto R, Della CF, Plotz FB, Groeneveld AB. Bench-to-bedside review: Ventilation-induced renal injury through systemic mediator release–just theory or a causal relationship? Crit Care. 2011;15:228. doi: 10.1186/cc10282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper JW, Plotz FB, Groeneveld AJ, Haitsma JJ, Jothy S, Vaschetto R, Zhang H, Slutsky AS. High tidal volume mechanical ventilation-induced lung injury in rats is greater after acid instillation than after sepsis-induced acute lung injury, but does not increase systemic inflammation: an experimental study. BMC Anesthesiol. 2011;11:26. doi: 10.1186/1471-2253-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeding L, Lust E, Plotz FB, Groeneveld AB. Clinical implications of heart-lung interactions. Neth J Med. 2010;68:56–61. [PubMed] [Google Scholar]
- Manny J, Grindlinger G, Mathe AA, Hechtman HB. Positive end-expiratory pressure, lung stretch, and decreased myocardial contractility. Surgery. 1978;84:127–133. [PubMed] [Google Scholar]
- Nin N, Penuelas O, de Paula M, Lorente JA, Fernandez-Segoviano P, Esteban A. Ventilation-induced lung injury in rats is associated with organ injury and systemic inflammation that is attenuated by dexamethasone. Crit Care Med. 2006;34:1093–1098. doi: 10.1097/01.CCM.0000205663.92384.E7. [DOI] [PubMed] [Google Scholar]
- Brander L, Sinderby C, Lecomte F, Leong-Poi H, Bell D, Beck J, Tsoporis JN, Vaschetto R, Schultz MJ, Parker TG. et al. Neurally adjusted ventilatory assist decreases ventilator-induced lung injury and non-pulmonary organ dysfunction in rabbits with acute lung injury. Intensive Care Med. 2009;35:1979–1989. doi: 10.1007/s00134-009-1626-x. [DOI] [PubMed] [Google Scholar]
- Kneyber MC, Gazendam RP, Niessen HW, Kuiper JW, Dos Santos CC, Slutsky AS, Plotz FB. Mechanical ventilation during experimental sepsis increases deposition of advanced glycation end products and myocardial inflammation. Crit Care. 2009;13:R87. doi: 10.1186/cc7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowitz B, Roy S, Gatto LA, Habashi N, Nieman G. Lung injury induced by sepsis: lessons learned from large animal models and future directions for treatment. Expert Rev Anti Infect Ther. 2011;9:1169–1178. doi: 10.1586/eri.11.141. [DOI] [PubMed] [Google Scholar]
- Grandel U, Bennemann U, Buerke M, Hattar K, Seeger W, Grimminger F, Sibelius U. Staphylococcus aureus alpha-toxin and Escherichia coli hemolysin impair cardiac regional perfusion and contractile function by activating myocardial eicosanoid metabolism in isolated rat hearts. Crit Care Med. 2009;37:2025–2032. doi: 10.1097/CCM.0b013e31819fff00. [DOI] [PubMed] [Google Scholar]
- Tavernier B, Mebazaa A, Mateo P, Sys S, Ventura-Clapier R, Veksler V. Phosphorylation-dependent alteration in myofilament ca2+ sensitivity but normal mitochondrial function in septic heart. Am J Respir Crit Care Med. 2001;163:362–367. doi: 10.1164/ajrccm.163.2.2002128. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, del Nido PJ, Ibrahim AE, Poutias DN, Glynn P, Cao-Danh H, Cowan DB, McGowan FX Jr. Increased myocardial calcium cycling and reduced myofilament calcium sensitivity in early endotoxemia. Surgery. 1999;126:231–238. doi: 10.1016/S0039-6060(99)70160-1. [DOI] [PubMed] [Google Scholar]
- Smeding L, van der Laarse WJ, van Veelen TA, Lamberts RR, Niessen HW, Kneyber MC, Groeneveld AB, Plotz FB. Early myocardial dysfunction is not caused by mitochondrial abnormalities in a rat model of peritonitis. J Surg Res. 2012;176:178–184. doi: 10.1016/j.jss.2011.05.055. [DOI] [PubMed] [Google Scholar]
- Fearon IM, Palmer AC, Balmforth AJ, Ball SG, Mikala G, Schwartz A, Peers C. Hypoxia inhibits the recombinant alpha 1C subunit of the human cardiac L-type Ca2+ channel. J Physiol. 1997;500(Pt 3):551–556. doi: 10.1113/jphysiol.1997.sp022041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movafagh S, Morad M. L-type calcium channel as a cardiac oxygen sensor. Ann N Y Acad Sci. 2010;1188:153–158. doi: 10.1111/j.1749-6632.2009.05095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa AO, Movafagh S, Cleemann L, Morad M. Hypoxic regulation of cardiac Ca2+ channel: possible role of haem oxygenase. J Physiol. 2012;590:4223–4237. doi: 10.1113/jphysiol.2012.236570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijveldt RJ, Prins HA, van Kemenade FJ, Teerlink T, van Lambalgen AA, Boelens PG, Rauwerda JA, van Leeuwen PA. Low arginine plasma levels do not aggravate renal blood flow after experimental renal ischaemia/reperfusion. Eur J Vasc Endovasc Surg. 2001;22:232–239. doi: 10.1053/ejvs.2001.1444. [DOI] [PubMed] [Google Scholar]
- The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 2000;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- Herrera MT, Toledo C, Valladares F, Muros M, Az-Flores L, Flores C, Villar J. Positive end-expiratory pressure modulates local and systemic inflammatory responses in a sepsis-induced lung injury model. Intensive Care Med. 2003;29:1345–1353. doi: 10.1007/s00134-003-1756-5. [DOI] [PubMed] [Google Scholar]
- Villar J, Cabrera NE, Casula M, Valladares F, Flores C, Lopez-Aguilar J, Blanch L, Zhang H, Kacmarek RM, Slutsky AS. WNT/beta-catenin signaling is modulated by mechanical ventilation in an experimental model of acute lung injury. Intensive Care Med. 2011;37:1201–1209. doi: 10.1007/s00134-011-2234-0. [DOI] [PubMed] [Google Scholar]
- Lamberts RR, Vaessen RJ, Westerhof N, Stienen GJ. Right ventricular hypertrophy causes impairment of left ventricular diastolic function in the rat. Basic Res Cardiol. 2007;102:19–27. doi: 10.1007/s00395-006-0620-5. [DOI] [PubMed] [Google Scholar]
- van der Velden J, Papp Z, Zaremba R, Boontje NM, de Jong JW, Owen VJ, Burton PB, Goldmann P, Jaquet K, Stienen GJ. Increased Ca2+−sensitivity of the contractile apparatus in end-stage human heart failure results from altered phosphorylation of contractile proteins. Cardiovasc Res. 2003;57:37–47. doi: 10.1016/S0008-6363(02)00606-5. [DOI] [PubMed] [Google Scholar]
- Hoegl S, Boost KA, Flondor M, Scheiermann P, Muhl H, Pfeilschifter J, Zwissler B, Hofstetter C. Short-term exposure to high-pressure ventilation leads to pulmonary biotrauma and systemic inflammation in the rat. Int J Mol Med. 2008;21:513–519. [PubMed] [Google Scholar]
- Vreugdenhil HA, Haitsma JJ, Jansen KJ, Zijlstra J, Plotz FB, Van Dijk JE, Lachmann B, Van VH, Heijnen CJ. Ventilator-induced heat shock protein 70 and cytokine mRNA expression in a model of lipopolysaccharide-induced lung inflammation. Intensive Care Med. 2003;29:915–922. doi: 10.1007/s00134-003-1747-6. [DOI] [PubMed] [Google Scholar]
- Mathur S, Walley KR, Wang Y, Indrambarya T, Boyd JH. Extracellular heat shock protein 70 induces cardiomyocyte inflammation and contractile dysfunction via TLR2. Circ J. 2011;75:2445–2452. doi: 10.1253/circj.CJ-11-0194. [DOI] [PubMed] [Google Scholar]
- Villar J, Blanco J, Zhang H, Slutsky AS. Ventilator-induced lung injury and sepsis: two sides of the same coin? Minerva Anestesiol. 2011;77:647–653. [PubMed] [Google Scholar]
- Vaneker M, Joosten LA, Heunks LM, Snijdelaar DG, Halbertsma FJ, Van EJ, Netea MG, Van der Hoeven JG, Scheffer GJ. Low-tidal-volume mechanical ventilation induces a toll-like receptor 4-dependent inflammatory response in healthy mice. Anesthesiology. 2008;109:465–472. doi: 10.1097/ALN.0b013e318182aef1. [DOI] [PubMed] [Google Scholar]
- Villar J, Cabrera NE, Casula M, Flores C, Valladares F, Az-Flores L, Muros M, Slutsky AS, Kacmarek RM. Mechanical ventilation modulates TLR4 and IRAK-3 in a non-infectious, ventilator-induced lung injury model. Respir Res. 2010;11:27. doi: 10.1186/1465-9921-11-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury AM, McCaig LA, Yao LJ, Veldhuizen RA, Lewis JF. Host response to intratracheally instilled bacteria in ventilated and nonventilated rats. Crit Care Med. 2004;32:2502–2507. doi: 10.1097/01.CCM.0000148010.08296.9A. [DOI] [PubMed] [Google Scholar]
- Nahum A, Hoyt J, Schmitz L, Moody J, Shapiro R, Marini JJ. Effect of mechanical ventilation strategy on dissemination of intratracheally instilled Escherichia coli in dogs. Crit Care Med. 1997;25:1733–1743. doi: 10.1097/00003246-199710000-00026. [DOI] [PubMed] [Google Scholar]
- Verbrugge SJ, Sorm V, Van V, Mouton JW, Gommers D, Lachmann B. Lung overinflation without positive end-expiratory pressure promotes bacteremia after experimental Klebsiella pneumoniae inoculation. Intensive Care Med. 1998;24:172–177. doi: 10.1007/s001340050541. [DOI] [PubMed] [Google Scholar]
- Mitchell JH, Wildenthal K, Johnson RL Jr. The effects of acid–base disturbances on cardiovascular and pulmonary function. Kidney Int. 1972;1:375–389. doi: 10.1038/ki.1972.48. [DOI] [PubMed] [Google Scholar]
- Laine GA. Change in (dP/dt)max as an index of myocardial microvascular permeability. Circ Res. 1987;61:203–208. doi: 10.1161/01.RES.61.2.203. [DOI] [PubMed] [Google Scholar]
- Mambula SS, Stevenson MA, Ogawa K, Calderwood SK. Mechanisms for Hsp70 secretion: crossing membranes without a leader. Methods. 2007;43:168–175. doi: 10.1016/j.ymeth.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kido T, Bai N, Yatera K, Suzuki H, Meredith A, Mukae H, Rosenfeld ME, van Eeden SF. Diesel exhaust inhalation induces heat shock protein 70 expression in vivo. Inhal Toxicol. 2011;23:593–601. doi: 10.3109/08958378.2011.595843. [DOI] [PubMed] [Google Scholar]
- Somji S, Todd JH, Sens MA, Garrett SH, Sens DA. Expression of the constitutive and inducible forms of heat shock protein 70 in human proximal tubule cells exposed to heat, sodium arsenite, and CdCl(2) Environ Health Perspect. 1999;107:887–893. doi: 10.1289/ehp.99107887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copland IB, Kavanagh BP, Engelberts D, McKerlie C, Belik J, Post M. Early changes in lung gene expression due to high tidal volume. Am J Respir Crit Care Med. 2003;168:1051–1059. doi: 10.1164/rccm.200208-964OC. [DOI] [PubMed] [Google Scholar]
- Hirsch J, Hansen KC, Sapru A, Frank JA, Chalkley RJ, Fang X, Trinidad JC, Baker P, Burlingame AL, Matthay MA. Impact of low and high tidal volumes on the rat alveolar epithelial type II cell proteome. Am J Respir Crit Care Med. 2007;175:1006–1013. doi: 10.1164/rccm.200605-621OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang M, Fan J. Pattern recognition receptor-dependent mechanisms of acute lung injury. Mol Med. 2010;16:69–82. doi: 10.2119/molmed.2009.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JH, Mathur S, Wang Y, Bateman RM, Walley KR. Toll-like receptor stimulation in cardiomyoctes decreases contractility and initiates an NF-kappaB dependent inflammatory response. Cardiovasc Res. 2006;72:384–393. doi: 10.1016/j.cardiores.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Zagorski J, Gellar MA, Obraztsova M, Kline JA, Watts JA. Inhibition of CINC-1 decreases right ventricular damage caused by experimental pulmonary embolism in rats. J Immunol. 2007;179:7820–7826. doi: 10.4049/jimmunol.179.11.7820. [DOI] [PubMed] [Google Scholar]