

Abstract
Background
The arginine of the D/E/NRY motif in Rhodopsin family G protein-coupled receptors (GPCRs) is conserved in 96% of these proteins. In some GPCRs, this arginine in transmembrane 3 can form a salt bridge with an aspartic acid or glutamic acid in transmembrane 6. The Drosophila melanogaster GPCR Trapped in endoderm-1 (Tre1) is required for normal primordial germ cell migration. In a mutant form of the protein, Tre1sctt, eight amino acids RYILIACH are missing, resulting in a severe disruption of primordial germ cell development. The impact of the loss of these amino acids on Tre1 structure is unknown. Since the missing amino acids in Tre1sctt include the arginine that is part of the D/E/NRY motif in Tre1, molecular dynamics simulations were performed to explore the hypothesis that these amino acids are involved in salt bridge formation and help maintain Tre1 structure.
Results
Structural predictions of wild type Tre1 (Tre1+) and Tre1sctt were subjected to over 250 ns of molecular dynamics simulations. The ability of the model systems to form a salt bridge between the arginine of the D/E/NRY motif and an aspartic acid residue in transmembrane 6 was analyzed. The results indicate that a stable salt bridge can form in the Tre1+ systems and a weak salt bridge or no salt bridge, using an alternative arginine, is likely in the Tre1sctt systems.
Conclusions
The weak salt bridge or lack of a salt bridge in the Tre1sctt systems could be one possible explanation for the disrupted function of Tre1sctt in primordial germ cell migration. These results provide a framework for studying the importance of the arginine of the D/E/NRY motif in the structure and function of other GPCRs that are involved in cell migration, such as CXCR4 in the mouse, zebrafish, and chicken.
Keywords: GPCR, Tre1, Molecular dynamics, Germ cell migration, Salt bridge formation
Background
G protein-coupled receptors (GPCRs) are the largest class of membrane proteins, accounting for 2% of genes in the human genome [1-3]. In general, GPCRs are responsible for modulating signals from the extracellular environment and transducing these stimuli into intracellular signaling cascades and cellular responses. GPCRs are involved in a wide range of cellular processes including cell movement, neurotransmission and olfaction, and can also be involved in disease progression with roles in metastasis, angiogenesis, cell proliferation and inflammation [4,5]. Since GPCRs are involved in maintaining homeostasis as well as disease progression, GPCRs are an important group of proteins to study, informing basic cellular and molecular biology as well as pharmaceutical applications.
Although there are many different genes encoding for GPCRs, all GPCRs share a common structure. GPCRs have seven transmembrane α-helices (TM1-TM7) connected by three intracellular and three extracellular loops. There are five main families of human GPCRs (Rhodopsin, Secretin, Glutamate, Adhesion and Frizzled/Taste2) [1-3], and this classification holds true for GPCRs in other bilateral species [6].
GPCRs are inherently difficult to crystallize due to their transmembrane nature and the fact that individual GPCRs are typically expressed at low levels within cells. GPCRs, like other transmembrane proteins, require a membrane-like environment to remain in a properly folded conformation. The required presence of a membrane makes the overexpression and subsequent purification of GPCRs challenging. The first GPCR crystal structure, bovine rhodopsin, was determined in 2000 [7], with nearly seven years passing before a crystal structure for the second GPCR was published. To date there are 16 GPCRs crystallized, all representing the Rhodopsin family of GPCRs [8]. Additionally, six of these proteins (bovine opsin, bovine rhodopsin, human A2A-adenosine receptor, turkey β1-adrenergic receptor, human β2-adrenergic receptor, and rat neurotensin receptor NTSR1) have been crystallized in active-like states [8].
Due to the difficulties of GPCR purification and crystallization, protein structure prediction programs and molecular dynamics (MD) simulations are frequently used to investigate the structures of GPCRs. There are currently three computational techniques available to generate a three-dimensional structural prediction of a protein: homology modeling, threading, and ab initio modeling. Homology modeling builds a three-dimensional structure by first identifying an evolutionarily related homologous protein with a known structure to use as a template. The program then aligns the amino acid sequence of the protein of interest to the amino acid sequence of the chosen template and finally builds the model [9-11]. The relatively low number of GPCR crystal structures is a major limitation to homology modeling. A lack of diverse structures means that a majority of GPCRs will still lack a homologous protein to use as a template. It is possible to build a highly accurate model when the template protein and the protein sequence of interest share 50% or more sequence identity [9,10]. However, when the sequence identity is below 30%, the protein structure prediction will likely more closely resemble the template structure than the native structure of the protein [12]. The sequence identity between crystallized GPCRs and other known GPCRs is often below 30% [13]. Due to the prevalence of low sequence identity, it is suggested that both sequence identity and structural information be used when choosing the template protein [13].
Threading, similar to homology modeling, is a template-based approach to structure prediction. The first step in threading is to search for evolutionary relatives to the protein sequence of interest. This is commonly accomplished with Position-Specific Iterative Basic Local Alignment Search Tool (PSI-BLAST) [14]. PSI-BLAST generates a sequence profile, which is used by a secondary structure predictor, like PSIPRED [15], to determine the secondary structure of the protein sequence of interest. Both the secondary structure and the sequence profile from PSI-BLAST are used in a threading algorithm to identify template proteins from the Protein Data Bank that have similar protein folds to the sequence of interest. Templates used in threading may show no evolutionary relationship [11]. The use of multiple templates, creating a chimeric GPCR, has been shown to provide a more accurate model than using a single protein template [13,16,17]. Multiple templates can be used in both homology modeling and threading.
Ab initio modeling builds a three-dimensional protein model from sequence information alone, without using a template structure, based upon the assumption that the protein structure will assume the lowest free energy conformation [9]. Ab initio modeling can work well for proteins with less than 120 amino acids [11]. Although there are three different ways to build a protein structure prediction, some current modeling programs use a combination of approaches to predict a structure [11]. The accuracy of the final model is linked to the template(s) chosen, and some approaches to generating a protein structural prediction work better on certain proteins or parts of proteins than others [18,19].
With only 16 distinct GPCR proteins crystallized, it can be difficult to find a suitable template(s) to use with the modeling software. Part of this challenge has been alleviated by the availability of web servers specifically designed for modeling GPCRs, such as GPCR-ModSim [20] and GPCR-Iterative Threading ASSEmbly Refinement (GPCR-ITASSER) [21-23]. GPCR-ModSim is a server that allows investigators to model GPCRs using MODELLER [9,20,24] and GPCR-ModSim users have the option of choosing whether to align their GPCR sequence with inactive-like crystallized GPCRs or active-like crystallized GPCRs. GPCR-ModSim aligns the sequence and shows the percent identity with the available templates. The user can then choose which template to use and GPCR-ModSim generates a homology model using MODELLER. Once a homology model is generated, the user has the option of submitting it for MD simulations in a solvated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine lipid bilayer [20].
GPCR-ITASSER is another web server that allows for protein structure prediction [21-23]. GPCR-ITASSER takes the initial GPCR sequence and identifies evolutionary relatives using PSI-BLAST and secondary structures using PSIPRED. The results from PSI-BLAST and PSIPRED are used by the Local Meta-Threading Server (LOMETS) to find potential templates in the Protein Data Bank. Any sequence without a matched template is modeled using an ab initio helix-modeling program. Additional restraints to the protein structure are incorporated through the use of the online database GPCRRD (GPCR Research Database), which contains experimental restraints from other GPCR databases and literature [21-23]. The ab initio modeling, results from threading, and restraints from the GPCRRD are all used to assemble and build a structural model. This structural model is refined using Fragment-Guided Molecular Dynamics [22] to give the final model.
In this study, the GPCR-ModSim [20] and the GPCR-ITASSER [21] web servers were used to predict protein structures of the GPCR Trapped in endoderm-1 (Tre1). Tre1 is a Rhodopsin family GPCR required for proper Drosophila melanogaster primordial germ cell migration [25-27]. In a mutant form of the protein, Tre1sctt, primordial germ cell migration is severely disrupted. The primordial germ cells scatter across the posterior half of the embryo rather than populating the two gonads. The molecular lesion in tre1sctt RNA is a point mutation that results in an in-frame deletion of eight amino acids, RYILIACH [27]. Two of these amino acids (RY) are part of the highly conserved D/E/NRY motif in TM3 of Rhodopsin family GPCRs. The D/E/NRY motif is thought to act as a micro-switch in the activation mechanism of Rhodopsin family GPCRs [3,28], and the arginine is conserved in 96% of Rhodopsin family GPCRs [29]. The arginine of the D/E/NRY motif (R3.50 following Ballesteros-Weinstein nomenclature [30]) can form a salt bridge with TM6 in numerous GPCRs [31-42], and while the exact role of the salt bridge is unknown [43], it is clear that the arginine is very important for Tre1+ function [27]. The first position of the D/E/NRY motif is also highly conserved. It is an acidic residue (aspartic acid (D) or glutamic acid (E)) in 86% of GPCRs [29]. In some GPCRs, the aspartic acid or glutamic acid can interact with the neighboring arginine to form an intrahelical salt bridge in addition to the interaction with TM6 [36,44]. Since an acidic residue in the first position of the motif is not present in Tre1 (NRY motif), an intrahelical salt bridge with the arginine does not form. This could make the interhelical salt bridge with TM6 more important.
Protein structure predictions of putative inactive structures of both Tre1+ and Tre1sctt were generated with GPCR-ModSim [20] and GPCR-ITASSER [21]. The NAMD simulation package [45] was used to perform MD simulations on both Tre1+ and Tre1sctt embedded in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) lipid bilayer, and MD simulations were run on four different model systems for over 250 ns each. The proteins were embedded in a POPE lipid bilayer since phosphoethanolamine is the most abundant phospholipid in Drosophila cell membranes [46]. The NRY motif of Tre1 was studied by examining the possibility of salt bridge formation between the arginine of the NRY motif and an aspartic acid residue of TM6. One of the wild type Tre1 model systems shows potential for a strong salt bridge to form between R134 of the NRY motif and D266 of TM6. The distances between the residues are favorable for salt bridge formation and could indicate that the salt bridge promotes interhelical stabilization of the Tre1 GPCR. The lack of similar interactions in the mutant model systems with an alternative arginine residue as well as in vivo data with Tre1sctt[27] suggests that the arginine of the NRY motif is important to the function and maintenance of an inactive structure that allows for subsequent activation of the Tre1 GPCR.
Results
Protein structure prediction
The amino acid sequences for Tre1+ and Tre1sctt were used for protein structure predictions using GPCR-ModSim [20] and GPCR-ITASSER [21]. Both GPCR-ModSim and GPCR-ITASSER are web servers for GPCR protein structure prediction, however the web servers differ in the approach taken to generate a protein structure prediction. The GPCR-ModSim server automates the process of using the homology modeling program MODELLER to model GPCRs [20], while GPCR-ITASSER uses multiple threading programs as well as the GPCR Research Database to predict protein structures [21]. These two web servers were used in this study to generate four independent protein structure predictions, two each for Tre1+ and Tre1sctt. While MODELLER is an established program that has been used to predict GPCR structures for simulations before, this is the first study to use structural predictions from GPCR-ITASSER.
Tre1+ and Tre1sctt were modeled to the seven inactive-like GPCRs on the GPCR-ModSim web server. From the multiple sequence alignment generated, it was clear that any one of the seven available GPCR crystal structures could be used as a template to model Tre1+ and Tre1sctt. However, GPCR-ModSim allows only one template to be chosen, and for both Tre1+ and Tre1sctt the template chosen was squid rhodopsin (PDB ID: 2Z73). Not only did squid rhodopsin show the highest total sequence identity to Tre1+ and Tre1sctt (17.4% for Tre1+ and 16.7% for Tre1sctt) (Table 1), but also squid rhodopsin seemed to be the best choice from earlier work using the web server I-TASSER. The I-TASSER web server uses threading to generate a structural prediction of a protein and allows a user to submit a sequence with or without selecting a template to use [11]. The structural prediction from I-TASSER using Tre1+ sequence and no selected template looked most similar to the structural prediction when squid rhodopsin was chosen as a template (data not shown). Therefore, squid rhodopsin was selected as the most appropriate template. Using squid rhodopsin as a template, ten models each for Tre1+ and Tre1sctt were generated using MODELLER. The two models chosen for further study using MD simulations were selected based on the lowest Discrete Optimized Protein Energy (DOPE-HR) score [47] and are named mtre1 (Tre1+) and msctt (Tre1sctt).
Table 1.
Sequence alignments in GPCR-ModSim indicate squid rhodopsin has the greatest identity to Tre1+ and Tre1sctt
| Template |
Tre1+ |
Tre1sctt |
|---|---|---|
| Total % identities | Total % identities | |
| 1U19 – bovine rhodopsin |
14.3 |
14.1 |
| 2RH1 – human β2-adrenergic receptor |
15.1 |
14.6 |
| 2VT4 – turkey β1-adrenergic receptor |
14.8 |
14.3 |
| 2Z73 – squid rhodopsin * |
17.4 |
16.7 |
| 3EML – human A2A-adenosine receptor |
14.0 |
13.5 |
| 3ODU – human chemokine receptor 4 |
16.3 |
14.8 |
| 3PBL – human D3 dopamine receptor | 16.1 | 15.9 |
* Denotes the template chosen for homology modeling.
The second set of independent protein structure predictions for Tre1+ and Tre1sctt were built using GPCR-ITASSER [21]. The amino acid sequences for Tre1+ and Tre1sctt were submitted to GPCR-ITASSER and used in the local threading server to find template proteins. Both Tre1+ and Tre1sctt were modeled to Substance P, human β2-adrenergic receptor, bovine rhodopsin and human A2A adenosine receptor. In addition, Tre1+ was modeled to turkey β1-adrenergic receptor and human β2-adrenergic receptor-Gs protein complex, while Tre1sctt was modeled to squid rhodopsin. Even though an active GPCR (human β2-adrenergic receptor-Gs protein complex) was a template for the Tre1+ model, it is thought that this Tre1+ model reflects an inactive conformation of the protein as the resulting model from GPCR-ITASSER is a consensus of restraints from six templates, all but one being inactive. The two best models, based on GPCR-ITASSER confidence scores, were chosen for further study in MD simulations. The models generated by GPCR-ITASSER are named gtre1 (Tre1+) and gsctt (Tre1sctt).
The four models, mtre1, msctt, gtre1, and gsctt, selected for further study are shown in Figure 1A. At first glance, all four of the models look similar, but there are distinct differences. Namely, helices 5 and 6 (yellow and gold chains) are roughly the same length as the other five helices in mtre1 and msctt (arrowheads in Figure 1B). In gtre1 and gsctt, helices 5 and 6 are extended relative to the other five helices. Helical extensions of helices 5 and 6 are present in the crystal structure of squid rhodopsin (PDB ID: 2Z73) [48] which was used as a template structure for mtre1, msctt, and gsctt. These differences change the architecture of intracellular loop 3. Additionally, intracellular loop 2 (green) has different structures in mtre1 and gtre1 (arrows in Figure 1B). In mtre1, this loop region is unstructured. In contrast, there is a short helix in intracellular loop 2 in gtre1. These differences in intracellular loop structure can be attributed to the template structure(s) used to generate mtre1 and gtre1. Intracellular loop 2 is of interest since it is the location of some of the residues missing in Tre1sctt.
Figure 1.

Three-dimensional models for Tre1+ and Tre1sctt. mtre1 and msctt are the models generated with GPCR-ModSim and gtre1 and gsctt are the models generated with GPCR-ITASSER. mtre1 and gtre1 are models for Tre1+ and msctt and gsctt are models for Tre1sctt. The N-termini are colored blue and the C-termini are colored red. (A) The best models chosen from the GPCR-ModSim and GPCR-ITASSER. (B) A closer view of helices 5 and 6 (yellow and gold) denoted by arrowheads, and intracellular loop 2 (green) shown by arrows. In the gtre1 and gsctt models, helices 5 and 6 are extended compared to the other 5 helices. Intracellular loop 2 is unstructured in mtre1 but contains a short helix in gtre1. (C) The resulting models after 262 ns (mtre1), 258 ns (msctt), 270 ns (gtre1) or 276 ns (gsctt) of MD simulations with the protein embedded in a POPE lipid bilayer. The differences in helical length of helices 5 and 6 and the structure of intracellular loop 2 between the initial structures generated by GPCR-ModSim and GPCR-ITASSER are still present after the MD.
Building biologically relevant model systems
As GPCRs exist in a membrane environment, the four different protein structure predictions were inserted into a solvated POPE lipid bilayer using the Membrane Builder in the CHARMM-GUI [49,50]. The final solvated membrane systems are named the same as the structural predictions, mtre1, msctt, gtre1 and gsctt. Each system was subjected to over 250 ns of MD and an example of the mtre1 system after MD is shown in Figure 2.
Figure 2.

Protein structure prediction of mtre1 embedded in a POPE lipid bilayer. The mtre1 model system shown here is after 262 ns of molecular dynamics. The extracellular surface of the bilayer is on top and the intracellular surface of the bilayer is on the bottom. mtre1 is depicted as ribbons, with the N-terminus colored blue and the C-terminus colored red. The POPE bilayer is represented as sticks and is colored green. Water and ions are depicted as grey lines.
Experimental work has shown that at 310 K a POPE lipid in a POPE lipid bilayer maintains a surface area of 59.75 – 60.75 Å2[51]. To confirm the correct surface areas per lipid were maintained in mtre1, msctt, gtre1 and gsctt, the Voronoi Tesselation and Monte Carlo (VTMC) integration method [52] was used. This method allows for calculation of the surface area per lipid in membrane-lipid systems. VTMC calculates the surface area per boundary and non-boundary lipids. Non-boundary lipids are described as those lipids not interacting with atoms of the protein. It is important to make the distinction between lipid types (boundary versus non-boundary) since lipids interacting with atoms of the protein will have a decreased surface area per lipid. The results of the VTMC analysis are shown in Table 2 and confirm that the non-boundary lipids in each of the model systems maintained the correct surface area per lipid, ranging from 59.0 – 60.2 Å2.
Table 2.
Voronoi Tessellation Monte Carlo integration method confirms the model systems have maintained a fluid-phase bilayer
| Model system |
Average surface area (Å2/lipid) |
|
|---|---|---|
| Boundary lipid | Non-boundary lipid | |
| mtre1 |
45.2 |
59.8 |
| msctt |
45.1 |
59.0 |
| gtre1 |
45.4 |
60.1 |
| gsctt | 47.9 | 60.2 |
Average surface areas per lipid were calculated using the Voronoi Tessellation Monte Carlo integration method [52] and calculated at the end of each simulation for all model systems. A boundary lipid is a lipid that contacts an atom from the protein and a non-boundary lipid is a lipid that makes no contact with protein atoms. Experimental values of surface area per non-boundary POPE lipid is 59.75 – 60.75 Å2[51]. Computationally determined surface area per non-boundary POPE lipid using CHARMM36 is 59.2 Å2[66].
Global movements of the model systems
Root mean squared deviation (RMSD) was computed over the course of the simulation for the Cα atoms of the transmembrane regions of the proteins to measure structural stability (Figure 3). As seen from the curves in Figure 3, the RMSD values did not change significantly from the starting structure and each curve began to stabilize after 150 ns of dynamics. This suggests that the systems have equilibrated. Further confirmation of structural stability after 150 ns can be seen from the curves of the RMSD values calculated for the complete proteins computed over the course of the simulation (Additional file 1: Figure S1).
Figure 3.

RMSD of the transmembrane regions shows the protein structures have equilibrated. Root mean squared deviation (RMSD) was calculated for the Cα atoms of the transmembrane domains and is plotted over simulation time. The transmembrane regions of the protein models do not change much compared to the starting structures over the course of simulations, as the final RMSD values are between 2.0 and 3.5 Å.
Root mean squared fluctuation (RMSF) was also used as a way to qualitatively characterize the protein dynamics. Here, RMSF describes the fluctuations of each Cα atom of the amino acid residues in the proteins averaged over the simulation time, beginning at 150 ns (Figure 4). The general fluctuations of specific regions of the proteins are similar between each of the Tre1+ and Tre1sctt models. It is clear from all four plots that the regions of the protein with the least amount of movement are the transmembrane regions. Intracellular loop 3 shows the greatest fluctuations, which is expected since it is the longest loop in Tre1. Of all the model systems, intracellular loop 3 of gsctt has the highest RMSF values. The high degree of fluctuation seen in intracellular loop 3 of gsctt could be due to its architecture, as intracellular loop 3 is more helical in gsctt than it is in mtre1, msctt, and gtre1. In gsctt, intracellular loop 3 begins as a helical extension of TM5, and then it has an unstructured loop region, followed by another helical region that connects with TM6. The high RMSF values in intracellular loop 3 in gsctt come from the residues in intracellular loop 3 that are in the unstructured loop. It is possible that the more rigid α-helical segments in intracellular loop 3 in gsctt prevent some of the other, unstructured residues in the loop from making important contacts with other parts of the protein. This could cause higher RMSF values.
Figure 4.

RMSF of each model system shows transmembrane regions move less than the loop regions. The root mean squared fluctuation (RMSF) for the mtre1, msctt, gtre1 and gsctt model systems beginning after 150 ns of dynamics are shown. The N- and C-termini are not included in the plots since the termini had high RMSF values and made it difficult to see the fluctuations in the other regions of the protein. The black bars denote the regions of the protein that are within the lipid bilayer. In most model systems, intracellular loop 3 (IL3) shows the greatest fluctuations.
A third qualitative assessment of the simulations was an all-to-all RMSD calculation on the transmembrane Cα atoms of the Tre1 protein, shown in Figure 5 as heat maps. Heat maps of all-to-all RMSD calculations show the number of different states the protein has visited during the course of the simulation. The darker diagonal blocks in each plot show when the Tre1 protein (Tre1+ or Tre1sctt) explores conformations that are structurally very similar. Darker off-diagonal blocks suggest that the protein revisits a conformation over the course of the simulation, although in this case, low RMSD alone is not sufficient to guarantee that two, noncontiguous in time, structures are similar [53]. In the heat maps shown here, each simulation samples two or more conformational substates. The quality of sampling is comparable to the sampling obtained at 250 ns in previous MD simulations of GPCRs [53-55].
Figure 5.

Heat maps of all-to-all RMSD calculations show sampling of conformational substates during MD simulations. Transmembrane Cα atoms in mtre1, msctt, gtre1 and gsctt model systems were used in these calculations. The all-to-all RMSD calculation computes the RMSD of all pairs of frames in a trajectory. This all-to-all RMSD calculation was performed using the VMD plug-in RMSD Trajectory Tool [71]. The RMSD scale is different for each model system and is located to the right of each plot.
The final protein structures after the dynamics in a lipid bilayer are shown in Figure 1C (the lipids, water and ions are not shown). In general, the transmembrane regions of the protein structures did not change drastically from the initial structures. Some of the loops have changed considerably, but the distinctions between the initial structures generated by GPCR-ModSim and GPCR-ITASSER noted previously are still present. To quantitatively determine changes in secondary structure over the course of the simulations, the Dictionary of Secondary Structure of Proteins [56,57] was used (Additional file 2: Figure S2). These plots confirm the seven transmembrane helices remain stable throughout the simulation, and the termini and loops are regions of change. While the significance of the differences between the GPCR-ModSim and GPCR-ITASSER models remain to be understood, it is interesting that even after over 250 ns of MD, some of the structural differences between the model systems were not resolved. This could mean that the structural models generated by the different modeling programs represent different protein conformations of Tre1.
Studies of the NRY motif of Tre1
From previous genetic studies, it is known that the arginine of the NRY motif (R3.50 following Ballesteros-Weinstein nomenclature [30]) in Tre1 is critical to the function of this GPCR [27]. Other than the critical nature of the arginine to Tre1 function, very little is known about the potential structural roles for this amino acid. It is possible that the arginine of the NRY motif in TM3 is involved in forming a salt bridge with an aspartic acid residue in TM6. A similar salt bridge in other GPCRs is thought to be important for holding GPCRs in inactive or activated states [32-42]. If there is a salt bridge in Tre1 between the arginine of the NRY motif and an aspartic acid residue in TM6, loss of this arginine could remove this salt bridge and impair function, which would be consistent with experimental observations that germ cell migration requires this arginine in Tre1 for function [27]. It is also possible that an alternative arginine just downstream of the deleted amino acids in Tre1sctt could be used to form a salt bridge with TM6. This alternative salt bridge could explain why the tre1sctt allele does not appear to be a complete loss-of-function allele of the tre1 gene. In Tre1+ the sequence around the arginine is NRYILIACHSR*Y. In Tre1sctt, the amino acids RYILIACH are missing and the remaining sequence is NSR*Y. The arginine of the NRY motif in Tre1+ is numbered as R134. The arginine R* in Tre1sctt, the alternative arginine, is numbered as R135, meaning this alternative arginine is located one residue from where the original arginine is located in Tre1+. Therefore, this alternative arginine could be close enough to form a salt bridge in the Tre1sctt protein. To test this hypothesis, the potential for salt bridge formation was evaluated in all model systems. Here, a salt bridge is defined as a noncovalent interaction between the carboxylate group of aspartic acid (D266 in Tre1+ and D258 in Tre1sctt) and the guanidium group of arginine (R134 in Tre1+ and R135 in Tre1sctt). As aspartic acid residues have two oxygen atoms that could be involved in a salt bridge, and arginine residues have two nitrogen atoms that could be involved in a salt bridge, the distance between both of the oxygen atoms of the aspartic acid in TM6 and both of the nitrogen atoms of the arginine in TM3 were calculated and plotted over the simulation time (Figure 6). Interatomic distances of 3.2 Å or less were considered favorable for salt bridge formation and such distances were seen in mtre1, gtre1, and msctt. The atoms studied in gsctt were never close enough to form a salt bridge. As shown in Figure 7B, the nitrogen atoms of R135 in gsctt are not oriented towards the oxygen atoms of the aspartic acid residue in TM6 as they are in the gtre1 system (Figure 7A). The MD simulation of gtre1 shows interatomic N-O distances of 3.2 Å or less for 3 of the 4 possible N-O pairs throughout most of the simulation (NH1-OD1, NH2-OD1, NH2-OD2). N-O distances in mtre1 were consistently greater than 3.2 Å except for ~75 ns towards the end of the simulation (NH2-OD2). The differences in interatomic N-O distances between mtre1 and gtre1 could be due to different conformations of Tre1 being represented or due to inherent differences in the initial protein structure predictions. msctt does not appear to be able to form a stable salt bridge using the alternative arginine, R135, as correct interatomic N-O distances are only transiently seen over the course of the simulation. To ensure the differences in interatomic N-O distances were not due to differences in the distance between TM3 and TM6 carbon backbones, the Cα-Cα distances were measured between the arginine (R134 or R135) in TM3 and aspartic acid (D266 or D258) in TM6 in all systems. Plots of the Cα-Cα distances over the course of the simulation showed that the distances between the residues studied here were similar (Additional file 3: Figure S3).
Figure 6.

Distance calculations suggest a salt bridge could form in mtre1, msctt and gtre1 model systems. Distances between oxygen atoms of the carboxylate group of aspartic acid (D) (OD1, OD2) and nitrogen atoms of the guanidium group of arginine (R) (NH1, NH2) are plotted. N-O distances of 3.2 Å (dotted lines) or less are capable of forming a salt bridge. The N-O distances are close enough to form a salt bridge transiently in mtre1 and msctt. There is potential for a salt bridge to be consistently present in the gtre1 model system. The N-O distances are too great in gsctt to form a salt bridge at any point during the simulation.
Figure 7.
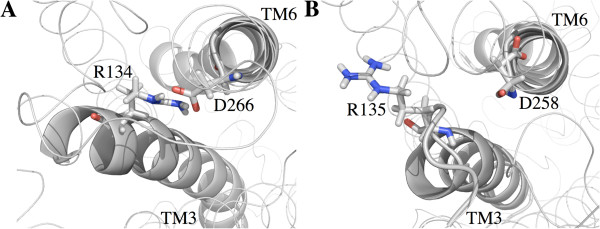
Orientation of the arginine in gsctt is not correct for salt bridge formation. Intracellular views of TM3 and TM6 are shown. The arginine and aspartic acid residues studied for formation of a salt bridge are shown as sticks, with the nitrogen atoms labeled blue and the oxygen atoms labeled red. TM3 and TM6 helices are represented as gray ribbons. (A) The gtre1 model system at 270 ns. In gtre1, R134 and D266 are close enough to form a salt bridge. Nitrogen – oxygen distances range from 2.4 – 4.9 Å. (B) The gsctt model system at 276 ns. The nitrogen atoms of R135 and the oxygen atoms of D266 are not facing each other, which prevents a salt bridge from forming between these residues. Nitrogen – oxygen distances range from 10.3 – 14.8 Å.
Discussion
The Tre1 GPCR is an important component of primordial germ cell migration in Drosophila [25-27]. In a severe partial loss-of-function allele of the tre1 gene, tre1sctt, proper primordial germ cell migration is disrupted. The Tre1sctt protein is missing eight amino acids, RYILIACH, from the junction of the third transmembrane domain and second intracellular loop [27]. This study was performed to analyze how the loss of the amino acids RYILIACH may affect Tre1 structure.
Protein structure predictions were generated for Tre1+ and Tre1sctt using GPCR-ModSim [20] and GPCR-ITASSER [21]. The four resulting structures were inserted into a POPE lipid bilayer and subjected to over 250 ns of MD simulations each. Interesting insights into the structures of Tre1+ and Tre1sctt were gained from this study. First, as shown by the RMSD versus time plots and the RMSF plots (Figures 3 and 4), Tre1+ and Tre1sctt behave similarly. The RMSD values for both Tre1+ and Tre1sctt protein structure predictions begin to stabilize around 150 ns of each MD run. Also, as shown by Figure 4, the general fluctuations of specific regions of the proteins are similar between Tre1+ and Tre1sctt.
The only primary sequence difference between Tre1+ and Tre1sctt is that Tre1sctt is missing the eight amino acids RYILIACH. Of the eight amino acids, the arginine is the most conserved residue, being present in 96% of Rhodopsin family GPCRs [29]. The arginine and the tyrosine are part of a highly conserved D/E/NRY motif in Rhodopsin family GPCRs [3,28]. The D/E/NRY motif is thought to have roles as a micro-switch, being involved in holding the GPCR in an active or inactive state by forming a salt bridge with an aspartic acid residue or a glutamic acid residue in TM6 [31-42]. Interestingly, the arginine is also the most critical residue of the amino acids RYILIACH for proper primordial germ cell migration in Drosophila. When the arginine is substituted by an alanine, a severe loss-of-function germ cell phenotype is observed [27]. The germ cell phenotype from the arginine substitution is indistinguishable from the phenotype when the amino acids RYILIACH are missing. Based upon this knowledge, it was hypothesized that a salt bridge involving the conserved D/E/NRY motif is present in Tre1+ and absent in Tre1sctt. A salt bridge with the D/E/NRY motif could be important for maintaining Tre1+ in a conformation required for efficient ligand binding. The lack of salt bridge in Tre1sctt could alter the protein conformation such that the ligand cannot recognize the receptor. It is also possible that an alternative salt bridge using a nearby arginine could be formed in Tre1sctt. If an alternative salt bridge forms, it could be involved in restoring some function of the GPCR.
The ability of the four model systems to form a salt bridge (Figure 6) was examined. Wild-type systems, mtre1 and gtre1, confirm that it is possible for a salt bridge to form between the arginine of the NRY motif (R134) and an aspartic acid (D266) in TM6. The salt bridge analysis using the mutant systems, msctt and gsctt, present a different picture. While it is possible for a salt bridge to form between the alternative arginine (R135) and the aspartic acid of TM6 (D258) in the msctt system, the salt bridge would not be very stable. Distances favorable for salt bridge formation were not consistently present during the simulation (Figure 6). It is clear from Figure 6 that no salt bridge would be expected to form in gsctt. It is possible that the salt bridge seen in gtre1 promotes interhelical stabilization of the protein, and this stabilization could be important for proper function of Tre1. The inability to form a stable salt bridge could disrupt Tre1sctt protein structure making it unable to properly receive its ligand, or could alter the confirmation of Tre1sctt such that it cannot bind interacting proteins. An alternative explanation for the salt bridge analysis results is that the systems have not been sufficiently sampled (Additional file 4: Table S1 and Additional file 5: Table S2), and a salt bridge could still form in the Tre1sctt forms of the protein.
With the salt bridge analysis there are significant differences between the independent model systems for Tre1+ and Tre1sctt. While it is possible that the different model systems represent two different protein conformations of Tre1, it is also possible that these differences can be attributed to how the protein structure predictions were generated. mtre1 and msctt were built using GPCR-ModSim [20] which uses the homology modeling program MODELLER. gtre1 and gsctt were built using GPCR-ITASSER [21-23] which predicts protein structures through the use of threading. To further study Tre1 structure, a third independent structure of Tre1 could be built. For example, GPCR-Sequence Structure Feature Extractor (GPCR-SSFE) could be used to generate another starting structure. GPCR-SSFE is a database in addition to a homology modeling program that creates homology models of GPCRs using multiple templates and the program MODELLER [24,58]. The ability to use multiple templates is significant since the use of multiple templates with MODELLER has been shown to give more accurate homology models than using a single template [17].
Conclusions
In this study, the role of the arginine of the NRY motif in Tre1 was investigated. It is known from previous work that this arginine is critical to the proper function of the Tre1 GPCR in Drosophila primordial germ cell migration [27]. Whether or not it is important for Tre1 structure was unknown. The results presented here suggest that a salt bridge may form between this critical arginine and an aspartic acid in TM6 in Tre1.
GPCRs are a common class of proteins involved in cell migration. Similar to how Tre1 is involved in Drosophila primordial germ cell migration, another GPCR, CXCR4, is important for proper primordial germ cell migration in mouse, zebrafish and chicken [59-62]. Like Tre1, CXCR4 contains the highly conserved D/E/NRY motif. While a salt bridge with the arginine of the DRY motif was not present in the crystal structure of the human CXCR4 [63], it would be interesting to learn if the arginine of the DRY motif is important to the structure of mouse, zebrafish or chicken CXCR4 and what implications this would have on primordial germ cell migration. The importance of the arginine to both the function [27] and structure of Tre1 could also mean that the arginine of the DRY motif in CXCR4 is important for its structure and function.
Primordial germ cell migration is an important process to study as it serves as a model for cell migration. In many animals, the primordial germ cells are formed at a place distant to the presumptive gonads requiring the primordial germ cells to migrate to their target tissues. In order for the primordial germ cells to properly migrate to the presumptive gonads, the primordial germ cells are required to initiate migration, migrate through various tissues, evade or suppress cell death mechanisms and respond to directional cues. The study of primordial germ cell migration as a model for cell migration will help to better understand the mechanisms of cell movements, enabling the development of new techniques or approaches to treat cancer or other diseased states caused by improper cell migration.
Methods
Protein structure predictions
Protein structure predictions were generated for Tre1+ (GenBank ID: AAF46059) and Tre1sctt using GPCR-ModSim [20] and GPCR-ITASSER [21]. Using squid rhodopsin (PDB ID: 2Z73) as a template, ten homology models were generated for both Tre1+ and Tre1sctt by GPCR-ModSim. The best model (as judged by the lowest DOPE-HR score [47]) was chosen for further refinement of the loop regions. The DOPE-HR score is used to assess the quality of the models generated by examining the energy of the protein models. Five models were generated for each protein after loop refinement and again the best model was chosen based on DOPE-HR score.
Five models of both Tre1+ and Tre1sctt were generated by GPCR-ITASSER and the best model as judged by the confidence score (C-score) was chosen for this study. C-score is an estimation of the structural prediction and is based on the threading alignments from LOMETS and convergence during the structural refinements [11].
Building a system to reflect a Drosophila cellular membrane environment
The protein structure predictions (mtre1, msctt, gtre1 and gsctt) were embedded in a solvated (0.15 M NaCl) and pre-equilibrated POPE lipid bilayer using the Membrane Builder in the CHARMM-GUI [49,50]. For the mtre1 system, 101 Na+ and 112 Cl- ions were added to neutralize the system and the system contained 37,557 water molecules. The upper and lower leaflets of the membrane contained 141 and 137 POPE lipids, respectively. The mtre1 system had a total of 153,870 atoms. The msctt system contained 110 Na+ and 120 Cl- ions, 40,839 water molecules, 141 and 137 POPE lipids on upper and lower leaflets of the membrane, respectively, and 163,593 total atoms. The gtre1 system contained 69 Na+ and 80 Cl- ions, 26,139 water molecules, 140 and 137 POPE lipids on upper and lower leaflets of the membrane, respectively, and 119,427 total atoms. The gsctt system contained 68 Na+ and 78 Cl- ions, 26,102 water molecules, 139 and 140 POPE lipids on upper and lower leaflets of the membrane, respectively, and 119,423 total atoms.
Molecular dynamics simulations
MD simulations were performed using the NAMD 2.8 simulation package [45]. The CHARMM22 [64,65] and CHARMM36 [66] force fields were used for proteins and lipids, respectively, and water molecules were described using TIP3P [67]. All systems were simulated at 310 K. Temperature and pressure were held constant with Langevin dynamics [45] and the Nose-Hoover Langevin piston [68,69]. Particle-mesh Ewald was used to calculate electrostatic interactions [70] and a 12 Å cut-off for van der Waals interactions was used. Each system was simulated on three compute nodes, each containing one Intel(R) Xeon(R) X5650 CPU (6 cores at 2.67 GHz), two Nvidia C2070 graphical processing units (GPUs) and 24 GB of RAM connected by QDR QLogic Infiniband.
After building the systems with the Membrane Builder in the CHARMM-GUI, six short (25 or 100 ps) equilibrium simulations were performed to gradually equilibrate the systems. Details for the equilibrium simulations can be found in reference [49]. Briefly, positional harmonic restraints were used on the protein backbone, protein side chains and ions. Additional harmonic restraints were used on the water molecules, to prevent water molecules from entering the hydrophobic region of the membrane, and the lipid head groups, to keep the lipid head groups level with the Z-axis. The restraints were reduced at each subsequent equilibrium simulation. The first two simulations used the NVT (constant volume and temperature) ensemble and the last four equilibrium simulations used the NPAT (constant pressure, area and temperature) ensemble. A timestep of 1 fs was used for the first three equilibrium simulations, which were 25 ps each. The last three equilibrium simulations used a 2 fs timestep and were run for 100 ps each [49]. Production runs began after the systems were equilibrated and used an NPT (constant pressure and temperature) ensemble and a 2 fs timestep. Harmonic restraints were not used in the production runs. Production runs were 262 ns, 258 ns, 270 ns, and 276 ns for mtre1, msctt, gtre1 and gsctt, respectively.
Data analysis
Visual Molecular Dynamics 1.9 (VMD) [71] was used to visualize the trajectories and to perform the all-to-all RMSD calculations and the salt bridge analysis. The Voronoi Tesselation and Monte Carlo (VTMC) integration method was used to calculate the surface area per lipid in all model systems [52] to ensure the systems maintained a biologically relevant, fluid phase lipid bilayer. The Dictionary of Secondary Structure in Proteins [56,57] with the do_dssp interface supplied by GROMACS [72] was used to calculate the evolution of secondary structures over time for each model.
Amino acid numbering
Amino acid residues are labeled using the single-letter code for the amino acid followed by the absolute sequence number. For example, arginine 134 is labeled R134. Tre1sctt is missing eight amino acids compared to Tre1+; however, the absolute sequence number of amino acid residues studied in this protein is still used. For example, an aspartic acid residue in TM6 is labeled D266 in Tre1+ and D258 in Tre1sctt.
Statistical analyses
Like any set of data, MD simulations are prone to statistical errors. The errors can be from inaccuracies in the model or inadequate sampling. For this reason, it is important to report the statistical uncertainty of values determined from simulations. In order to calculate the statistical uncertainty of different values in a simulation, the number of independent samples within a single simulation needs to be known. It has been suggested that estimation of a value of interest based on less than 20 statistically independent samples is considered unreliable [54]. To calculate the number of independent samples within a simulation, the decorrelation time must be calculated.
To calculate the decorrelation time, this study used the structural histogram analysis and automated effective sample size methods developed by the Zuckerman lab [73,74], as well as the block covariance overlap method (BCOM) from the Grossfield lab [53]. The effective sample size gives the degrees to which a simulation has sampled the conformational space of the protein and BCOM is a method used to measure the extent of convergence of a simulation. All of these tools are available through the LOOS (Lightweight Object-Oriented Structure) analysis library [75]. Only transmembrane Cα atoms were used in the decorrelation time calculations.
The decorrelation times as estimated by the structural histogram analysis and by the automated effective sample size are shown in Additional file 4: Table S1 and the decorrelation times calculated using the BCOM are shown in Additional file 5: Table S2. The results from the structural histogram analysis, the automated effective sample size calculation and the blocked covariance overlap method indicate that the systems have insufficient sampling and have not converged. This means that statistics generated from the data are not sufficient to draw statistically meaningful conclusions. This is not surprising. Microsecond simulations (or longer) with other GPCRs did not show convergence using these same methods [53]. Since the systems in this study have not converged, the values presented in this study represent a more qualitative assessment of the simulations.
Abbreviations
GPCR: G protein-coupled receptor; Tre1: Trapped in endoderm-1; TM: Transmembrane; MD: Molecular dynamics; POPE: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine; GPCR-ITASSER: GPCR-Iterative Threading ASSEmbly Refinement; VTMC: Voronoi Tesselation and Monte Carlo integration method; RMSD: Root mean squared deviation; RMSF: Root mean squared fluctuation.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MMP, MHL, and CRC conceived the study. MMP performed the simulations and data analysis. MMP, MHL, and CRC wrote the manuscript. All authors read and approved the final manuscript.
Supplementary Material
RMSD of entire protein structures shows equilibration began at 150 ns. Description: Root mean squared deviation (RMSD) was calculated for each complete protein and is plotted over simulation time. The curves show the protein structures began to equilibrate after 150 ns.
Evolution of protein secondary structure over time. Description: The secondary structure of the proteins in each of the model systems was calculated and plotted over the simulation time with the do_dssp interface supplied by GROMACS [72]. Residues with the same secondary structure are in the same color. These plots show that the transmembrane regions of the proteins (blue) remain stable throughout the simulations.
Distances between Cα residues of TM3 and TM6 are similar in all model systems. Description: The distances between the Cα residues of R134 and D266 in Tre1+ and R135 and D258 in Tre1sctt were calculated and plotted over simulation time.
Approximate decorrelation times for the four different model systems. Description: τd1 is the decorrelation time as estimated from the plot of σ2 (t) with step sizes 2, 4 and 5 [73]. τd2 is the decorrelation time from the automated effective sample size calculation [74]. Both calculations are part of the LOOS analysis library [75]. a 10 bins were used, b 20 bins were used.
Assessing convergence of the different model systems using the blocked covariance overlap method. Description: BCOM is the blocked covariance overlap method and BBCOM is the bootstrapped blocked covariance overlap. t1 - t3 are decorrelation times from fitting the BCOM/BBCOM curve to: f(t) = k1e-t/t1 + k2e-t/t2 + k3e-t/t3 + 1[53]. The BCOM/BBCOM ratio decays to a final ratio of greater than 1 for each model system. This suggests that the systems have not yet converged. BCOM/BBCOM is part of the LOOS analysis library [75].
Contributor Information
Margaret M Pruitt, Email: mamdahl@iastate.edu.
Monica H Lamm, Email: mhlamm@iastate.edu.
Clark R Coffman, Email: ccoffman@iastate.edu.
Acknowledgements
We thank Iowa State University for providing funds for the computer system used in this research. Support for MMP and CRC came from the Roy J. Carver Charitable Trust. We would also like to thank Dr. Spencer Pruitt for technical assistance and insightful discussions.
References
- Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Gloriam DE, Fredriksson R, Schiöth HB. The G protein-coupled receptor subset of the rat genome. BMC Genomics. 2007;8:338. doi: 10.1186/1471-2164-8-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström MC, Schiöth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7(4):339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Cotton M, Claing A. G protein-coupled receptors stimulation and the control of cell migration. Cell Signal. 2009;21(7):1045–1053. doi: 10.1016/j.cellsig.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7(2):79–94. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Schiöth HB. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol Pharmacol. 2005;67(5):1414–1425. doi: 10.1124/mol.104.009001. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke C, Motoshima H, Fox B, Trong I, Teller D, Okada T, Stenkamp R, Yamamoto M, Miyano M. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein–coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53(1):531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294(5540):93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4(1):1–14. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Devries ME, Skolnick J. Structure modeling of all identified G protein-coupled receptors in the human genome. PLoS Comput Biol. 2006;2(2):e13. doi: 10.1371/journal.pcbi.0020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth C, Kleinau G, Krause G. Comparitive sequence and structural analyses of G-protein-coupled-receptor crystal structures and implications for molecular models. PLoS ONE. 2009;4(9):1–14. doi: 10.1371/journal.pone.0007011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292(2):195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Mobarec JC, Sanchez R, Filizola M. Modern homology modeling of G-protein coupled receptors: which structural template to use? J Med Chem. 2009;52(16):5207–5216. doi: 10.1021/jm9005252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokkar P, Mohandass S, Ramachandran M. Multiple templates-based homology modeling enhances structure quality of AT1 receptor: validation by molecular dynamics and antagonist docking. J Mol Model. 2010;17(7):1565–1577. doi: 10.1007/s00894-010-0860-z. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Progress and challenges in protein structure prediction. Curr Opin Struct Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Protein structure prediction: when is it useful? Curr Opin Struct Biol. 2009;19(2):145–155. doi: 10.1016/j.sbi.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez D, Bello X, Gutiérrez-de-Terán H. Molecular modelling of G protein-coupled receptors through the web. Molecular Informatics. 2012;31(5):334–341. doi: 10.1002/minf.201100162. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang Y. GPCRRD: G protein-coupled receptor spatial restraint database for 3D structure modeling and function annotation. Bioinformatics. 2010;26(23):3004–3005. doi: 10.1093/bioinformatics/btq563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liang Y, Zhang Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011;19(12):1784–1795. doi: 10.1016/j.str.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang Y. A novel side-chain orientation dependent potential derived from random-walk reference state for protein fold selection and structure prediction. PLoS ONE. 2010;5(10):e15386. doi: 10.1371/journal.pone.0015386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Kunwar P, Starz-Gaiano M, Bainton R, Heberlein U, Lehmann R. Tre1, a GPCR, directs transepithelial migration of Drosophila germ cells. Plos Biol. 2003;1(3):372–384. doi: 10.1371/journal.pbio.0000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunwar PS, Sano H, Renault AD, Barbosa V, Fuse N, Lehmann R. Tre1 GPCR initiates germ cell transepithelial migration by regulating Drosophila melanogaster E-cadherin. J Cell Biol. 2008;183:1–12. doi: 10.1083/JCB1831OIA1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamps AR, Pruitt MM, Herriges JC, Coffman CR. An evolutionarily conserved arginine is essential for Tre1 G protein-coupled receptor function during germ cell migration in Drosophila melanogaster. PLoS ONE. 2010;5(7):e11839. doi: 10.1371/journal.pone.0011839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Frimurer TM, Holst B, Rosenkilde MM, Schwartz TW. Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol Sci. 2009;30(5):249–259. doi: 10.1016/j.tips.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Mirzadegan T, Benkö G, Filipek S, Palczewski K. Sequence analyses of G-protein-coupled receptors: similarities to rhodopsin. Biochemistry. 2003;42(10):2759–2767. doi: 10.1021/bi027224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neuroscience. 1995;25:366–428. [Google Scholar]
- Chien EYT, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizaki Y, Maruyama K, Nakano-Tetsuka M, Saito Y. Distinct roles of the DRY motif in rat melanin-concentrating hormone receptor 1 in signaling control. Peptides. 2009;30:974–981. doi: 10.1016/j.peptides.2009.01.017. [DOI] [PubMed] [Google Scholar]
- Ballesteros J, Kitanovic S, Guarnieri F, Davies P, Fromme BJ, Konvicka K, Chi L, Millar RP, Davidson JS, Weinstein H, Sealfon SC. Functional microdomains in G-protein-coupled receptors. The conserved arginine-cage motif in the gonadotropin-releasing hormone receptor. J Biol Chem. 1998;273(17):10445–10453. doi: 10.1074/jbc.273.17.10445. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276(31):29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- Shapiro DA, Kristiansen K, Weiner DM, Kroeze WK, Roth BL. Evidence for a model of agonist-induced activation of 5-hydroxytryptamine 2A serotonin receptors that involves the disruption of a strong ionic interaction between helices 3 and 6. J Biol Chem. 2002;277(13):11441–11449. doi: 10.1074/jbc.M111675200. [DOI] [PubMed] [Google Scholar]
- Vogel R, Mahalingam M, Ludeke S, Huber T, Siebert F, Sakmar T. Functional Role of the “Ionic Lock” - an interhelical hydrogen-bond network in family a heptahelical receptors. J Mol Biol. 2008;360:648–655. doi: 10.1016/j.jmb.2008.05.022. [DOI] [PubMed] [Google Scholar]
- Zhu SZ, Wang SZ, Hu J, El-Fakahany EE. An arginine residue conserved in most G protein-coupled receptors is essential for the function of the m1 muscarinic receptor. Mol Pharmacol. 1994;45(3):517–523. [PubMed] [Google Scholar]
- Scheer A, Costa T, Fanelli F, DeBenedetti P, Mhaouty-Kodja S, Abuin L, Nenniger-Tosato M, Cotecchia S. Mutational analysis of the highly conserved arginine within the Glu/Asp-Arg-Tyr Motif of the alpha1b-adrenergic receptor: effects on receptor isomerization and activation. Mol Pharmacol. 2000;57:219–231. [PubMed] [Google Scholar]
- Scheer A, Fanelli F, Costa T, De Benedetti PG, Cotecchia S. Constitutively active mutants of the alpha 1B-adrenergic receptor: role of highly conserved polar amino acids in receptor activation. EMBO J. 1996;15(14):3566–3578. [PMC free article] [PubMed] [Google Scholar]
- Greasley PJ, Fanelli F, Rossier O, Abuin L, Cotecchia S. Mutagenesis and modelling of the alpha(1b)-adrenergic receptor highlight the role of the helix 3/helix 6 interface in receptor activation. Mol Pharmacol. 2002;61(5):1025–1032. doi: 10.1124/mol.61.5.1025. [DOI] [PubMed] [Google Scholar]
- Capra V, Veltri A, Foglia C, Crimaldi L, Habib A, Parenti M, Rovati GE. Mutational analysis of the highly conserved ERY motif of the thromboxane A2 receptor: alternative role in G protein-coupled receptor signaling. Mol Pharmacol. 2004;66(4):880–889. doi: 10.1124/mol.104.001487. [DOI] [PubMed] [Google Scholar]
- Greasley PJ, Fanelli F, Scheer A, Abuin L, Nenniger-Tosato M, DeBenedetti P, Cotecchia S. Mutational and computational analysis of the alpha1b-adrenergic receptor: involvement of basic and hydrophobic residues in receptor activation and G protein coupling. J Biol Chem. 2001;276(49):46485–46494. doi: 10.1074/jbc.M105791200. [DOI] [PubMed] [Google Scholar]
- Fanelli F, De Benedetti PG. Computational modeling approaches to structure − function analysis of G protein-coupled receptors. Chem Rev. 2011;111(9):R438–PR535. doi: 10.1021/cr100437t. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into 2-adrenergic receptor function. Science. 2007;318(5854):1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones H, Harwood J, Bowen I, Griffiths G. Lipid composition of subcellular membranes from larvae and prepupae of Drosophila melanogaster. LIPIDS. 1992;27(12):984–987. doi: 10.1007/BF02535576. [DOI] [PubMed] [Google Scholar]
- Shen M-Y, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Kouyama T. Crystal structure of squid rhodopsin. Nature. 2008;453(7193):363–367. doi: 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- Jo S, Kim T, Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE. 2007;2(9):e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophysj. 2009;97(1):50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappolt M, Hickel A, Bringezu F, Lohner K. Mechanism of the Lamellar: inverse hexagonal phase transition examined by high resolution x-ray diffraction. Biophys J. 2003;84:3111–3122. doi: 10.1016/S0006-3495(03)70036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Ogushi F, Sugita Y. Analysis of lipid surface area in protein-membrane systems combining Voronoi Tessellation and Monte Carlo integration methods. J Comput Chem. 2012;33(3):286–293. doi: 10.1002/jcc.21973. [DOI] [PubMed] [Google Scholar]
- Romo TD, Grossfield A. Block covariance overlap method and convergence in molecular dynamics simulation. J Chem Theory Comput. 2011;7(8):2464–2472. doi: 10.1021/ct2002754. [DOI] [PubMed] [Google Scholar]
- Grossfield A, Zuckerman DM. Quantifying uncertainty and sampling quality in biomolecular simulations. Ann Rep Comput Chem. 2009;5:23–48. doi: 10.1016/S1574-1400(09)00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romo TD, Grossfield A, Pitman MC. Concerted interconversion between ionic lock substates of the beta(2) adrenergic receptor revealed by microsecond timescale molecular dynamics. Biophys J. 2010;98(1):76–84. doi: 10.1016/j.bpj.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolym. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Joosten R, Te Beek T, Krieger E, Hekkelman M, Hooft R, Schneider R, Sander C, Vriend G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39:D411–D419. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth CL, Kreuchwig A, Kleinau G, Krause G. GPCR-SSFE: a comprehensive database of G-protein-coupled receptor template predictions and homology models. BMC Bioinforma. 2011;12:185. doi: 10.1186/1471-2105-12-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ara T, Nakamura Y, Egawa T, Sugiyama T, Abe K, Kishimoto T, Matsui Y, Nagasawa T. Impaired colonization of the gonads by primordial germ cells in mice lacking a chemokine, stromal cell-derived factor-1 (SDF-1) Proc Natl Acad Sci USA. 2003;100(9):5319–5323. doi: 10.1073/pnas.0730719100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doitsidou M, Reichman-Fried M, Stebler J, Koprunner M, Dorries J, Meyer D, Esguerra C, Leung T, Raz E. Guidance of primordial germ cell migration by the chemokine SDF-1. Cell. 2002;111:647–659. doi: 10.1016/S0092-8674(02)01135-2. [DOI] [PubMed] [Google Scholar]
- Knaut H, Werz C, Geisler R, Consortium TTS, Nüsslein-Volhard C. A zebrafish homologue of the chemokine receptor Cxcr4 is a germ-cell guidance receptor. Nature. 2003;421(6920):279–282. doi: 10.1038/nature01338. [DOI] [PubMed] [Google Scholar]
- Molyneaux KA, Zinszer H, Kunwar P, Schaible K, Stebler J, Sunshine M, O’Brien W, Raz E, Littman D, Wylie C, Lehmann R. The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development. 2003;130(18):4279–4286. doi: 10.1242/dev.00640. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science. 2010;330(6007):1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A, Bashford D, Bellott M, Dunbrack R, Evanseck J, Field M, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau F, Mattos C, Micknick S, Ngo T, Nguyen D, Prodhom B, Reiher W, Roux B, Schlenkrich M, Smith J, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuezera J, Yin D, Karplus M. All-Atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- MacKerell AD Jr. Empirical force fields for biological macromolecules: overview and issues. J Comput Chem. 2004;25(13):1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell Jr AD, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W, Chandrasekhar J, Madura J, Impey R, Klein M. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- Feller S, Zhang Y, Pastor R, Brooks B. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys. 1995;103(11):4613–4621. doi: 10.1063/1.470648. [DOI] [Google Scholar]
- Nosé S. A unified formulation of the constant temperature molecular dynamics methods. J Chem Phys. 1984;81(1):511. doi: 10.1063/1.447334. [DOI] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J Chem Phys. 1992;98(12):10089–10092. [Google Scholar]
- Humphry W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, Shirts M, Smith J, Kasson P, Van der Spoel D, Hess B, Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinform. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman E, Zuckerman DM. On the structural convergence of biomolecular simulations by determination of the effective sample size. J Phys Chem B. 2007;111(44):12876–12882. doi: 10.1021/jp073061t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bhatt D, Zuckerman DM. Automated sampling assessment for molecular simulations using the effective sample size. J Chem Theory Comput. 2010;6(10):3048–3057. doi: 10.1021/ct1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romo T, Grossfield A. LOOS: an extensible platform for the structural analysis of simulations. Conf IEEE EMBS. 2009;2009:2332–2335. doi: 10.1109/IEMBS.2009.5335065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RMSD of entire protein structures shows equilibration began at 150 ns. Description: Root mean squared deviation (RMSD) was calculated for each complete protein and is plotted over simulation time. The curves show the protein structures began to equilibrate after 150 ns.
Evolution of protein secondary structure over time. Description: The secondary structure of the proteins in each of the model systems was calculated and plotted over the simulation time with the do_dssp interface supplied by GROMACS [72]. Residues with the same secondary structure are in the same color. These plots show that the transmembrane regions of the proteins (blue) remain stable throughout the simulations.
Distances between Cα residues of TM3 and TM6 are similar in all model systems. Description: The distances between the Cα residues of R134 and D266 in Tre1+ and R135 and D258 in Tre1sctt were calculated and plotted over simulation time.
Approximate decorrelation times for the four different model systems. Description: τd1 is the decorrelation time as estimated from the plot of σ2 (t) with step sizes 2, 4 and 5 [73]. τd2 is the decorrelation time from the automated effective sample size calculation [74]. Both calculations are part of the LOOS analysis library [75]. a 10 bins were used, b 20 bins were used.
Assessing convergence of the different model systems using the blocked covariance overlap method. Description: BCOM is the blocked covariance overlap method and BBCOM is the bootstrapped blocked covariance overlap. t1 - t3 are decorrelation times from fitting the BCOM/BBCOM curve to: f(t) = k1e-t/t1 + k2e-t/t2 + k3e-t/t3 + 1[53]. The BCOM/BBCOM ratio decays to a final ratio of greater than 1 for each model system. This suggests that the systems have not yet converged. BCOM/BBCOM is part of the LOOS analysis library [75].