

Abstract
We have compared the power of several allele-sharing statistics for “nonparametric” linkage analysis of X-linked traits in nuclear families and extended pedigrees. Our rationale was that, although several of these statistics have been implemented in popular software packages, there has been no formal evaluation of their relative power. Here, we evaluate the relative performance of five test statistics, including two new test statistics. We considered sibships of sizes two through four, four different extended pedigrees, 15 different genetic models (12 single-locus models and 3 two-locus models), and varying recombination fractions between the marker and the trait locus. We analytically estimated the sample sizes required for 80% power at a significance level of .001 and also used simulation methods to estimate power for a sample size of 10 families. We tried to identify statistics whose power was robust over a wide variety of models, with the idea that such statistics would be particularly useful for detection of X-linked loci associated with complex traits. We found that a commonly used statistic, Sall, generally performed well under various conditions and had close to the optimal sample sizes in most cases but that there were certain cases in which it performed quite poorly. Our two new statistics did not perform any better than those already in the literature. We also note that, under dominant and additive models, regardless of the statistic used, pedigrees with all-female siblings have very little power to detect X-linked loci.
Introduction
Statistical methods for linkage analysis are usually divided into “parametric” and “nonparametric” methods. Although there is ongoing debate about the appropriateness of this division and about which methods are best under what circumstances (e.g., see Abreu et al. 1999; Sham et al. 2000), it seems fair to say that the nonparametric, or “allele-sharing,” statistics have been particularly important in mapping studies of complex traits. In this article, we consider some of the issues involved in applying allele-sharing statistics to detect X-linked loci involved in complex traits.
Although parametric methods have been used extensively for X-chromosomal linkage analysis (e.g., see Nyholt et al. 1998; Vallada et al. 1998; Xu et al. 1998), there has been limited development and use of nonparametric methods for detection of X linkage. Weeks et al. (1995) developed an X-linked version of the affected-pedigree-member method, based on identity-by-state sharing. Cordell et al. (1995) extended the sib-pair method of Risch (1990a, 1990b, 1990c) and Holmans (1993) to the X chromosome, and Dupuis and Van Eerdewegh (2000) developed a similar theory for the pseudoautosomal region. Two of the most popular software packages for nonparametric linkage analysis—GENEHUNTER (Kruglyak et al. 1996) and Allegro (Gudbjartsson et al. 2000)—have implemented X-chromosome statistics. However, these statistics are not actually defined or described in the literature, and GENEHUNTER dropped the X-linkage option after version 1.3. Since there is certainly evidence suggesting the involvement of X-chromosome loci in complex traits (e.g., see Hamer et al. 1993; Nyholt et al. 1998; DeLisi et al. 2000; Liu et al. 2001), we saw a clear need for a more complete study of allele-sharing statistics for X linkage in general pedigrees.
In this article, we first describe the X-chromosome allele-sharing statistics implemented in Allegro and in GENEHUNTER version 1.3. We then use analytic methods and simulation to compare the power of those statistics, as well as that of two new statistics that we propose. We consider a number of different pedigree types and genetic models. Our goal is to find a statistic whose power is robust over a wide variety of situations, since one does not expect to know much, in advance, about the trait model when studying a complex trait. Our methodological approach is very similar to that used in our recent study of autosomal allele-sharing statistics (Sengul et al. 2001).
Subjects and Methods
Pedigrees
We studied both nuclear families and extended pedigrees. The nuclear families consisted of parents with unknown trait phenotypes and two to four affected siblings of all possible sex combinations. We considered four different extended-pedigree structures, which obviously does not account for all possibilities but which allowed us to investigate the effects of several important pedigree features. The extended pedigrees consisted of affected individuals (shaded symbols in figs. 1 and 2) and individuals with unknown trait phenotypes (unshaded symbols in figs. 1 and 2). The first extended pedigree, which we call the “MMT” pedigree (fig. 1A), would normally be considered inconsistent with X-linked inheritance, since it contains a male-to-male transmission, in which there are affected males in two consecutive generations. However, this pedigree is perfectly compatible with a model that includes X linkage along with phenocopies and/or heterogeneity. The second extended pedigree, which we call “NMMT,” is similar, except that it does not include male-to-male transmission (fig. 1B). We also considered two different pedigrees involving affected sibships that are first cousins through sisters; pedigree “CSFF” contains only female affected individuals (fig. 1A), and pedigree “CSMF” contains both male and female affected individuals (fig. 1B). Figures 1 and 2 show unique numerical labels for each founder allele at a hypothetical marker; we use these in discussing identity-by-descent (IBD)–sharing configurations below.
Figure 1.
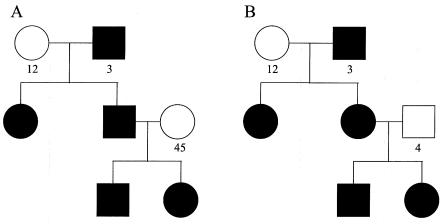
MMT pedigree (A) and NMMT pedigree (B). The numbers denote unique founder alleles; unshaded symbols denote unknown phenotypes.
Figure 2.
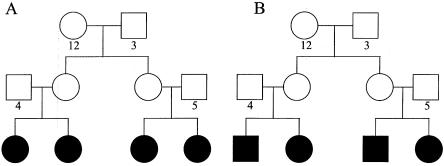
CSFF pedigree (A) and CSMF pedigree (B). Notation is as in figure 1.
Genetic Models
We considered single-locus two-allele dominant, additive, and recessive models with full and reduced penetrance and with varying levels of phenocopies. Table 1 lists all of the single-locus models, with the female penetrances denoted by f0, f1, and f2 and with the male penetrances denoted by g0 and g1. In all cases, the phenocopy rate is the same for males and females (g0 = f0 for dominant and additive models; g0 = f0 = f1 for recessive models). All models have allele frequency, q, equal to 0.1. The population trait frequencies vary between ∼5% and ∼10% for males (Kmale) and between ∼0.5% and ∼20% for females (Kfemale). Table 1 also presents the relative risks to sibs of a proband, for male-male pairs (λmm), male-female pairs (λmf), and female-female pairs (λff), computed by the formulae presented by Cordell et al. (1995).
Table 1.
Single-Locus Genetic Models
| Model | q | f0 | f1 | f2 | g0 | g1 | Kmale | Kfemale | λmm | λmf | λff | Description |
| Dominant: | ||||||||||||
| 1 | .1 | 0 | .5 | .5 | 0 | .5 | .050 | .095 | 5.5 | 3.1 | 4.1 | No phenocopies, reduced penetrance |
| 2 | .1 | .005 | 1 | 1 | .005 | 1 | .105 | .194 | 5.1 | 3.0 | 4.0 | 5% of cases are phenocopies, full penetrance |
| 3 | .1 | .005 | .5 | .5 | .005 | .5 | .055 | .099 | 4.7 | 2.8 | 3.8 | 5% of cases are phenocopies, reduced penetrance |
| 4 | .1 | .05 | .5 | .5 | .05 | .5 | .095 | .136 | 2.0 | 1.6 | 2.3 | Half of cases are phenocopies, reduced penetrance |
| Additive: | ||||||||||||
| 5 | .1 | 0 | .25 | .5 | 0 | .5 | .050 | .050 | 5.5 | 3.3 | 4.4 | No phenocopies, reduced penetrance |
| 6 | .1 | .005 | .5 | 1 | .005 | 1 | .105 | .104 | 5.1 | 3.0 | 4.1 | 5% of cases are phenocopies, full penetrance |
| 7 | .1 | .005 | .25 | .5 | .005 | .5 | .055 | .054 | 4.7 | 2.9 | 3.8 | 5% of cases are phenocopies, reduced penetrance |
| 8 | .1 | .05 | .25 | .5 | .05 | .5 | .095 | .091 | 2.0 | 1.5 | 1.7 | Half of cases are phenocopies, reduced penetrance |
| Recessive: | ||||||||||||
| 9 | .1 | 0 | 0 | .5 | 0 | .5 | .050 | .005 | 5.5 | 5.5 | 55.0 | No phenocopies, reduced penetrance |
| 10 | .1 | .005 | .005 | 1 | .005 | 1 | .105 | .015 | 5.1 | 3.9 | 24.9 | 5% of cases are phenocopies, full penetrance |
| 11 | .1 | .005 | .005 | .5 | .005 | .5 | .055 | .010 | 4.7 | 3.0 | 14.4 | 5% of cases are phenocopies, reduced penetrance |
| 12 | .1 | .05 | .05 | .5 | .05 | .5 | .095 | .055 | 2.0 | 1.2 | 1.4 | Half of cases are phenocopies, reduced penetrance |
The phenocopy rates in the single-locus models can be interpreted as including heterogeneity effects, and the single-locus models are also good approximations to certain interaction models that produce small marginal effects. However, we also did a limited investigation of explicit two-locus models. Model “H-rare” is a rare-allele dominant heterogeneity model. At each locus, the disease allele has frequency 0.01; penetrance is 0 for anyone with no disease alleles and is 1.0 for anyone with one or more disease alleles. Model “H-common” is a common-allele dominant heterogeneity model; it has the same penetrance matrices as does model “H-rare”, but the disease-allele frequency at each locus is 0.07. Finally, model “Additive” is an additive model. The disease-allele frequency at each locus is 0.05, there is a phenocopy rate of 0.2 for females and males, and each disease allele at either locus adds an additional 0.2 to the penetrance, so that, for example, a female who is homozygous for the disease allele at both loci has penetrance 1.0.
Allele-Sharing Statistics
We considered five different allele-sharing statistics for testing X linkage. All of the statistics can be applied to any pedigree type. Each statistic measures IBD among affected relatives, but somewhat differently. Our notation for the IBD-sharing configurations is that used by Whittemore and Halpern (1994); founder alleles are given unique numbers (as in figs. 1 and 2), and then the IBD-sharing configurations of family members are described in terms of which founder alleles are inherited by each person. Configurations are understood to include inheritance patterns that are equivalent up to permutation of the founder-allele labels; for example, if we have a mother and father with genotypes labeled 12 and 34 at an autosomal marker, then the IBD-sharing configurations among two offspring may be either (13 13), (13 14), or (13 24), and it is understood that the (13 13) configuration also includes (14 14), (23 23), and (24 24).
One of the allele-sharing statistics that we considered is the commonly used autosomal statistic Sall (Whittemore and Halpern 1994). For autosomal loci, Sall is calculated by forming all possible sets consisting of one allele from each affected individual and then summing the numbers of nontrivial permutations of those sets. This definition can be extended straightforwardly to the X chromosome; for example, if we have two females and a male, with alleles (by descent) labeled 12, 13, and 1, we can form the sets (1 1 1), (1 3 1), (2 1 1), and (2 3 1). Alternatively, males can be treated as homozygous, which has some minor computational advantages and which results in exactly the same statistic. Both Allegro and GENEHUNTER version 1.3 implement Sall for the X chromosome.
All of the other statistics that we consider in this article are “pairs” statistics, formed by scoring allele sharing between each pair of affected individuals in the pedigree and then adding that score over all pairs. The commonly used autosomal statistic Spairs (e.g., see McPeek 1999) is of this type; it defines the pairwise allele-sharing score for each pair simply as the number of alleles (zero, one, or two) shared IBD. GENEHUNTER version 1.3 and Allegro both implement X-chromosome statistics called “Spairs,” but we found that the two statistics are not the same: GENEHUNTER treats males as homozygous on the X chromosome, and Allegro treats them as hemizygous, which results in pairwise allele-sharing scores (and, thus, versions of Spairs) that are not equivalent; for example, if a male-male pair shares an allele IBD, Allegro treats the sharing configuration as (1 1) and gives it a score of 1, whereas GENEHUNTER treats the sharing configuration as (11 11) and gives it a score of 2. Table 2 shows all of the pairwise allele-sharing scores used by each version of Spairs: the Allegro statistic (SApairs), the GENEHUNTER statistic (SGHpairs), and our two new statistics, SX50 and SX75 (which are defined below). If we assume that there is no inbreeding, male-male and male-female pairs can share only zero alleles or one allele IBD. Female-female pairs can share zero, one, or two alleles IBD, although a female-female sibling pair can share only either one allele or two alleles. The rationale for our new statistics, SX50 and SX75, is as follows: Allegro and GENEHUNTER assign very different relative scores to complete sharing between females (13 13) versus complete sharing between males (1 1): Allegro gives much greater weight to female-female sharing, and GENEHUNTER gives much greater weight to male-male sharing. Our two new statistics give equal scores to male-male and female-female sharing, but the two differ in the scores (0.5 vs. 0.75) that they give to male-female (1 13) sharing.
Table 2.
Pairwise Allele-Sharing Scores Used by Different Statistics
| Relative Pair and Configuration | SApairs | SGHpairs | SX75 | SX50 |
| Female-female: | ||||
| (13 13) | 2 | 1 | 1 | 1 |
| (13 23) | 1 | .5 | .5 | .5 |
| (13 24) | 0 | 0 | 0 | 0 |
| Male-male: | ||||
| (1 1) | 1 | 2 | 1 | 1 |
| (1 2) | 0 | 0 | 0 | 0 |
| Male-female: | ||||
| (1 13) | 1 | 1 | .75 | .5 |
| (2 13) | 0 | 0 | 0 | 0 |
As an alternative to the definitions above, all of the statistics that we considered can be described as scoring functions that assign a score, S, to each possible IBD-sharing configuration, φ, that the entire pedigree takes on (Whittemore and Halpern 1994). Thus, for a single pedigree, the statistic is written as
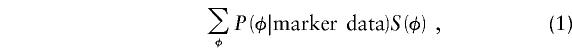 |
where the conditional probabilities of the IBD-sharing configurations, P(φ|marker data), can be computed by whatever method is desired (e.g., by software such as GENEHUNTER). To construct a linkage test, the statistic for each pedigree is normalized by subtracting the null hypothesis mean and dividing by the null hypothesis SD. The normalized statistics are summed over pedigrees, and the sum is asymptotically normally distributed under reasonable regularity conditions. The scores for different pedigrees can also be weighted before they are summed, to reflect different amounts of information contained in pedigrees of different sizes and types. Because of the normalization, additive and multiplicative constants are irrelevant to the definitions of the statistics—that is, any statistics that result in identical normalized coefficients would have identical power; for example, Kruglyak et al. (1996) gave an alternative definition of Sall that is equivalent to Whittemore and Halpern’s definition to a linear transformation.
Table 3 lists the IBD-sharing configurations and scoring functions of all the statistics for an affected sib trio. The scoring functions are normalized as described above, which allows them to be compared directly. Note that a positive normalized score for a given IBD-sharing configuration means that the configuration is considered to be evidence in favor of linkage, whereas a negative normalized score means that the configuration is considered to be evidence against linkage. Table 4 gives the configurations and scoring functions for a family of four affected siblings. We do not present a table for sibling pairs; since each type of sibling pair (male-male, male-female, and female-female) has only two possible IBD-sharing configurations, all of the statistics are equivalent to each other for sibling pairs. Tables 5–8 give the normalized scoring functions for the extended pedigrees that we considered. For each pedigree, the order of the individuals in the IBD-sharing configuration is from first generation to last and from left to right, in each generation (see figs. 1 and 2). The IBD-sharing configurations in each table are ordered from greatest sharing to least, although this ordering is not necessarily well defined, since different ways of quantifying IBD sharing are possible.
Table 3.
Normalized Sibship Statistics for Three Affected Siblings
|
Normalized Coefficient for Statistic |
|||||
| IBD Configurationa | SApairs | SGHpairs | Sall | SX75 | SX50 |
| Three males: | |||||
| (1 1 1) | 1.73 | 1.73 | 1.73 | 1.73 | 1.73 |
| (1 1 2) | −.58 | −.58 | −.58 | −.58 | −.58 |
| Two males–one female: | |||||
| (1 1 13) | 1.73 | 1.63 | 1.70 | 1.71 | 1.63 |
| (2 2 13) | −.58 | .00 | −.24 | −.34 | .00 |
| (1 2 13) | −.58 | −.82 | −.73 | −.69 | −.82 |
| One male–two females: | |||||
| (1 13 13) | 1.73 | 1.67 | 1.71 | 1.71 | 1.73 |
| (1 13 23) | −.58 | −.33 | −.43 | −.43 | −.58 |
| (2 13 13) | −.58 | −1.00 | −.85 | −.85 | −.58 |
| Three females: | |||||
| (13 13 13) | 1.73 | 1.73 | 1.73 | 1.73 | 1.73 |
| (13 13 23) | −.58 | −.58 | −.58 | −.58 | −.58 |
The mother's allele labels are 1/2, and the father's allele label is 3.
Table 4.
Normalized Sibship Statistics for Four Affected Siblings
|
Normalized Coefficient for Statistic |
|||||
| IBD Configurationa | SApairs | SGHpairs | Sall | SX75 | SX50 |
| Four males: | |||||
| (1 1 1 1) | 2.45 | 2.45 | 2.62 | 2.45 | 2.45 |
| (1 1 1 2) | .00 | .00 | −.24 | .00 | .00 |
| (1 1 2 2) | −.82 | −.82 | −.56 | −.82 | −.82 |
| Three males–one female: | |||||
| (1 1 1 13) | 2.45 | 2.32 | 2.57 | 2.42 | 2.32 |
| (2 2 2 13) | .00 | .77 | .20 | .35 | .77 |
| (1 1 2 13) | .00 | −.26 | −.33 | −.12 | −.26 |
| (1 2 2 13) | −.82 | −.77 | −.59 | −.81 | −.77 |
| Two males–two females: | |||||
| (1 1 13 13) | 2.45 | 2.26 | 2.51 | 2.41 | 2.33 |
| (1 1 13 23) | .00 | .52 | .16 | .27 | .33 |
| (1 2 13 13) | .00 | −.52 | −.48 | −.27 | −.33 |
| (2 2 13 13) | −.82 | −.52 | −.48 | −.80 | −.33 |
| (1 2 13 23) | −.82 | −.87 | −.69 | −.80 | −1.00 |
| One male–three females: | |||||
| (1 13 13 13) | 2.45 | 2.32 | 2.50 | 2.40 | 2.45 |
| (1 13 13 23) | .00 | .26 | .04 | .16 | .00 |
| (2 13 13 13) | .00 | −.77 | −.66 | −.48 | .00 |
| (2 13 13 23) | −.82 | −.77 | −.66 | −.80 | −.82 |
| Four females: | |||||
| (13 13 13 13) | 2.45 | 2.45 | 2.55 | 2.45 | 2.45 |
| (13 13 13 23) | .00 | .00 | −.11 | .00 | .00 |
| (13 13 23 23) | −.82 | −.82 | −.70 | −.82 | −.82 |
The mother's allele labels are 1/2, and the father's allele label is 3.
Table 5.
IBD Configurations and Normalized Coefficients for the MMT Pedigree
|
Normalized Coefficient for Statistic |
||||
| IBD Configuration | SApairs, SX50 | SGHpairs | Sall | SX75 |
| (3 13 1 4 14) | 1.34 | 1.39 | 1.45 | 1.37 |
| (3 13 1 4 15) | .45 | .28 | .23 | .34 |
| (3 13 2 4 24) | −.45 | −.28 | −.38 | −.34 |
| (3 13 2 4 25) | −1.34 | −1.39 | −1.30 | −1.37 |
Table 6.
IBD Configurations and Normalized Coefficients for the NMMT Pedigree
|
Normalized Coefficient for Statistic |
|||||
| IBD Configuration | SApairs | SGHpairs | Sall | SX75 | SX50 |
| (3 13 13 3 34) | 1.58 | 1.61 | 1.75 | 1.63 | 1.60 |
| (3 13 23 3 34) | .95 | 1.30 | 1.62 | 1.18 | 1.07 |
| (3 13 13 3 14) | .32 | .38 | −.17 | .28 | .53 |
| (3 13 13 1 14) | .32 | −.23 | −.49 | .06 | .00 |
| (3 13 13 1 34) | .32 | −.23 | −.49 | .06 | .00 |
| (3 13 23 3 24) | −.95 | −.23 | −.42 | −.62 | −.53 |
| (3 13 23 2 34) | −.95 | −1.15 | −.81 | −1.07 | −1.07 |
| (3 13 23 2 24) | −1.58 | −1.45 | −1.00 | −1.52 | −1.60 |
Table 7.
IBD Configurations and Normalized Coefficients for the CSFF Pedigree
|
Normalized Coefficient for Statistic |
||
| IBD Configuration | SApairs, SGHpairs, SX75, SX50 | Sall |
| (14 14 15 15) | 2.56 | 2.86 |
| (14 14 15 35) | .37 | .15 |
| (14 14 35 35) | −.37 | −.33 |
| (14 34 15 35) | −.37 | −.49 |
| (14 14 25 35) | −1.10 | −.81 |
| (14 34 25 35) | −1.10 | −.89 |
Table 8.
IBD Configurations and Normalized Coefficients for the CSMF Pedigree
|
Normalized Coefficient for Statistic |
|||||
| IBD Configuration | SApairs | SGHpairs | Sall | SX75 | SX50 |
| (1 14 1 15) | 2.56 | 2.41 | 2.88 | 2.52 | 2.49 |
| (1 14 1 35) | .37 | .83 | .37 | .60 | .66 |
| (1 14 3 15) | .37 | −.12 | −.20 | .12 | .06 |
| (1 34 1 35) | −.37 | −.12 | −.31 | −.36 | .06 |
| (1 14 3 35) | −.37 | −.44 | −.43 | −.36 | −.55 |
| (3 14 3 25) | −1.10 | −.44 | −.54 | −.84 | −.55 |
| (1 14 2 35) | −1.10 | −1.07 | −.77 | −1.08 | −1.16 |
| (1 34 2 35) | −1.10 | −1.38 | −.88 | −1.32 | −1.16 |
Power Computations
We computed power for a test of a single marker, assuming perfect IBD information, with varying recombination fractions (θ = 0.0, 0.05, 0.10, and 0.20) between the marker and the trait locus. (The effect that increasing the recombination fraction has on power is very similar to the effect that is produced by reducing the marker informativeness.) For the two-locus models, we used only θ = 0.0. We used analytic methods to calculate power for all the recombination fractions and, in addition, simulation methods for θ = 0.0. The analytic calculations assume normality and thus fail to account for the fact that some statistics have skewed distributions for small sample sizes and, therefore, higher power; however, since that higher power is accompanied by higher false-positive rates, the analytic calculation provides a “fairer” comparison among the statistics, at least asymptotically. We report results primarily from our analytic comparison, but we also comment on what our simulation studies found about increased power and false-positive rates for the more skewed statistics.
For our analytic calculation, we first calculated the probability of each IBD-sharing configuration under each alternative hypothesis, conditional on the set of affected relatives. For the single-locus models, this was done by MENDEL (Lange et al. 1988); for the two-locus models, we wrote a program to simulate families, selected those with the right set of affected individuals, and scored the IBD-sharing configurations. We then used these IBD-sharing–configuration probabilities to compute the mean and variance of each statistic, on the basis of the normalized coefficients. This procedure is discussed in more detail by Sengul et al. (2001). The sample size (number of families) needed to obtain 80% power with a significance level of .001 by a Z-test was computed as follows: sample size = (zασ0 + zβσa )2/(μa− μ0 )2, where zα = 3.0902, zβ = 0.8416, and where μ0 (σ0) and μa (σa) are, respectively, the means (SDs) under the null and the alternative hypotheses. This approach assumes normality of the statistics but allows them to have different variances under the null and alternative hypotheses.
For our simulation studies of the power for small sample sizes, we simulated 10,000 data sets of 10 pedigrees. For the single-locus models, this was done by FASTSLINK (Ott 1989; Weeks et al. 1990), and, for the two-locus models, this was done by our simulation program (described briefly in the preceding paragraph). These simulations were done conditional on the given disease phenotypes. We simulated one marker with distinct founder alleles (perfect IBD information), in complete linkage with the trait locus. We then computed the empirical power and false-positive rate, using the .001 nominal significance cutoff from the normal distribution.
For each genetic model and each pedigree type, we also derived the optimal statistic, by using a grid search to numerically maximize the aforementioned sample-size formula over all possible statistics of the form shown in equation (1). We included these optimal statistics in all of our analytic power studies. This allowed us to potentially identify models and/or pedigrees for which none of the statistics that we evaluated performed well.
Results
Tables 9–11 show the results of our analytic sample-size calculations for the single-locus models, and table 12 shows the results for the two-locus models. For each pedigree type, statistics that are equivalent (i.e., have the same normalized coefficients) are grouped together.
Table 9.
Estimated Sample Sizes for the Extended Pedigrees under Single-Locus Models
|
Estimated Sample Sizeb |
||||||||||||
| Dominant Model |
Additive Model |
Recessive Model |
||||||||||
| Pedigree and Statistic(s)a | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| MMT: | ||||||||||||
| Optimal test | ∞ | 21,975 | 6,334 | 351 | 89 | 89 | 89 | 120 | 8 | 8 | 9 | 27 |
| Sall | ∞ | 23,008 | 6,587 | 358 | 93 |
93 | 92 |
120 | 9 |
10 | 10 |
29 |
| SApairs, SX50 | ∞ | 24,075 | 6,855 | 353 | 100 | 99 | 98 | 122 | 11 | 11 | 11 | 31 |
| SGHpairs | ∞ | 22,666b |
6,489 |
354 | 94 | 93 | 93 | 120 | 10 | 10 | 11 | 30 |
| SX75 | ∞ | 23,106 | 6,602 | 352 |
96 | 95 | 94 | 120 | 10 | 11 | 11 | 30 |
| NMMT: | ||||||||||||
| Optimal test | 13 | 14 | 14 | 23 | 31 | 32 | 33 | 52 | 19 | 20 | 20 | 40 |
| Sall | 13 |
14 |
14 |
24 |
32 |
33 |
33 |
53 |
982 | 974 | 963 | 615 |
| SApairs | 23 | 24 | 24 | 37 | 40 | 41 | 42 | 64 | 51 |
52 |
52 |
78 |
| SX50 | 20 | 20 | 21 | 32 | 37 | 38 | 39 | 58 | 73 | 74 | 75 | 101 |
| SGHpairs | 16 | 17 | 17 | 28 | 35 | 35 | 36 | 54 | 146 | 147 | 149 | 169 |
| SX75 | 18 | 19 | 19 | 30 | 35 | 36 | 37 | 56 | 81 | 82 | 83 | 110 |
| CSFF: | ||||||||||||
| Optimal test | 32 | 32 | 33 | 51 | 31 | 32 | 33 | 74 | 7 | 7 | 8 | 40 |
| Sall | 35 |
35 |
36 |
53 |
31 |
32 |
33 |
74 |
7 |
8 | 8 | 41 |
| SApairs, SX50, SGHpairs, SX75 | 39 | 39 | 40 | 57 | 34 | 34 | 35 | 75 | 8 | 8 | 8 | 43 |
| CSMF: | ||||||||||||
| Optimal test | 9 | 9 | 9 | 16 | 9 | 9 | 9 | 19 | 7 | 8 | 9 | 33 |
| Sall | 9 |
9 |
10 | 16 |
9 | 9 |
9 |
19 |
7 |
8 |
9 |
40 |
| SApairs | 10 | 10 | 10 | 17 | 10 | 10 | 10 | 20 | 8 | 9 | 10 | 45 |
| SX50 | 10 | 10 | 10 | 17 | 10 | 10 | 10 | 20 | 8 | 9 | 10 | 37 |
| SGHpairs | 10 | 10 | 10 | 17 | 10 | 10 | 10 | 20 | 9 | 9 | 10 | 35 |
| SX75 | 10 | 10 | 10 | 17 | 9 | 10 | 10 | 20 | 8 | 9 | 10 | 39 |
Statistics are grouped together when they have identical normalized coefficients.
A sample size that is the unique maximum among statistics (excluding optimal test) is in boldface italic; a sample size that is the unique minimum among statistics (excluding optimal test) is underlined.
Table 10.
Estimated Sample Sizes for Three Affected Siblings under Single-Locus Models[Note]
|
Estimated Sample Size |
||||||||||||
| Dominant Model |
Additive Model |
Recessive Model |
||||||||||
| Pedigree and Statistic(s) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Three males: | ||||||||||||
| All statistics | 11 | 11 | 11 | 22 | 11 | 11 | 11 | 22 | 11 | 11 | 11 | 22 |
| Two males–one female: | ||||||||||||
| Optimal test | 13 | 14 | 14 | 28 | 13 | 13 | 14 | 32 | 11 | 12 | 14 | 48 |
| Sall | 13 | 14 | 14 | 28 | 13 | 14 | 14 | 32 | 11 | 12 | 14 | 54 |
| SX75 | 13 | 14 | 14 | 28 | 13 | 13 |
14 | 32 | 11 | 12 | 14 | 56 |
| SApairs | 13 | 14 | 14 | 29 | 13 | 14 | 14 | 34 | 11 | 12 | 14 | 64 |
| SX50, SGHpairs | 14 | 14 | 15 | 29 | 13 | 14 | 15 | 33 | 12 | 13 | 14 | 50 |
| One male–two females: | ||||||||||||
| Optimal test | 20 | 21 | 22 | 52 | 24 | 25 | 26 | 72 | 11 | 11 | 12 | 46 |
| Sall, SX75 | 21 | 22 | 23 | 54 | 24 | 25 | 26 |
72 |
11 | 11 | 12 | 47 |
| SApairs, SX50 | 20 |
21 |
22 |
52 |
24 | 25 | 27 | 74 | 11 | 11 | 12 | 46 |
| SGHpairs | 22 | 23 | 24 | 56 | 24 | 26 | 27 | 73 | 11 | 12 | 12 | 49 |
| Three females: | ||||||||||||
| All statistics | 220 | 224 | 227 | 309 | 237 | 243 | 248 | 429 | 11 | 11 | 11 | 34 |
Note.— Notation is as described in the footnotes to table 9.
Table 11.
Estimated Sample Sizes for Four Affected Siblings under Single-Locus Models[Note]
|
Estimated Sample Size |
||||||||||||
| Dominant Model |
Additive Model |
Recessive Model |
||||||||||
| Pedigree and Statistic(s) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Four males: | ||||||||||||
| Optimal test | 10 | 10 | 10 | 17 | 10 | 10 | 10 | 17 | 10 | 10 | 10 | 17 |
| Sall | 10 |
10 |
10 |
17 |
10 |
10 |
10 |
17 |
10 |
10 |
10 |
17 |
| SApairs, SX50, SGHpairs, SX75 | 11 | 11 | 11 | 18 | 11 | 11 | 11 | 18 | 11 | 11 | 11 | 18 |
| Three males–one female: | ||||||||||||
| Optimal test | 11 | 11 | 11 | 19 | 11 | 11 | 11 | 20 | 10 | 11 | 11 | 23 |
| Sall | 11 |
11 |
12 | 19 |
11 |
11 |
12 | 20 |
10 |
11 |
11 |
26 |
| SApairs | 12 | 12 | 13 | 20 | 12 | 12 | 13 | 21 | 11 | 12 | 12 | 29 |
| SX50, SGHpairs | 12 | 12 | 13 | 20 | 12 | 12 | 13 | 21 | 12 | 12 | 13 | 24 |
| SX75 | 12 | 12 | 12 | 20 | 12 | 12 | 12 | 21 | 11 | 12 | 12 | 26 |
| Two males–two females: | ||||||||||||
| Optimal test | 14 | 14 | 14 | 25 | 15 | 15 | 16 | 29 | 10 | 10 | 10 | 23 |
| Sall | 14 |
15 | 15 |
25 |
15 |
15 |
16 | 29 | 10 |
11 | 11 | 24 |
| SApairs | 16 | 16 | 16 | 26 | 17 | 17 | 17 | 32 | 11 | 11 | 11 | 25 |
| SX50 | 15 | 15 | 16 | 26 | 16 | 16 | 16 | 30 | 12 | 12 | 12 | 25 |
| SGHpairs | 16 | 16 | 16 | 27 | 16 | 16 | 17 | 29 | 12 | 13 | 13 | 26 |
| SX75 | 15 | 16 | 16 | 26 | 16 | 16 | 17 | 30 | 11 | 11 | 12 | 25 |
| One male–three females: | ||||||||||||
| Optimal test | 22 | 24 | 25 | 53 | 32 | 33 | 34 | 65 | 10 | 10 | 10 | 19 |
| Sall | 25 | 26 | 27 |
55 | 32 |
33 |
34 |
66 | 11 | 11 | 11 | 20 |
| SApairs, SX50 | 25 | 26 | 28 | 55 | 35 | 36 | 37 | 70 | 11 | 11 | 11 | 20 |
| SGHpairs | 28 | 29 | 30 | 60 | 33 | 34 | 35 | 66 | 12 | 12 | 12 | 22 |
| SX75 | 26 | 27 | 29 | 57 | 33 | 34 | 35 | 66 | 11 | 11 | 12 | 21 |
| Four females: | ||||||||||||
| Optimal test | 334 | 337 | 340 | 404 | 280 | 282 | 284 | 347 | 10 | 10 | 10 | 18 |
| Sall | 358 |
360 |
362 |
412 |
282 |
284 |
286 |
348 |
10 |
10 |
11 |
18 |
| SApairs, SX50, SGHpairs, SX75 | 384 | 385 | 386 | 429 | 291 | 292 | 293 | 349 | 11 | 11 | 11 | 19 |
Note.— Notation is as described in the footnotes to table 9.
Table 12.
Estimated Sample Sizes for Selected Pedigrees under Two-Locus Disease Models[Note]
|
Estimated Sample Size |
|||
| Pedigree and Statistic(s) | H-Rare Model | H-Common Model | Additive Model |
| NMMT pedigree: | |||
| Optimal test | 27 | 64 | 1,927 |
| Sall | 28 |
67 |
2,175 |
| SApairs | 47 | 102 | 2,772 |
| SX50 | 42 | 92 | 2,421 |
| SGHpairs | 36 | 82 | 2,249 |
| SX75 | 38 | 86 | 2,380 |
| CSFF pedigree: | |||
| Optimal test | 20 | 170 | 596 |
| Sall | 22 |
192 |
617 |
| SApairs, SX50, SGHpairs, SX75 | 26 | 222 | 620 |
| CSMF pedigree: | |||
| Optimal test | 12 | 45 | 2,691 |
| Sall | 13 |
45 |
2,910 |
| SApairs | 15 | 50 | 2,819 |
| SX50 | 16 | 51 | 2,905 |
| SGHpairs | 16 | 51 | 3,085 |
| SX75 | 15 | 49 | 2,888 |
Note.— Notation is as described in the footnotes to table 9.
Single-Locus Models
Extended pedigrees
For the MMT pedigree, Sall was the best statistic overall (table 9). It gave sample sizes close to optimal for the additive and recessive models, and it was reasonably good for the dominant models as well. Dominant models required very large sample sizes for this pedigree, regardless of the statistic used, because the observed male-to-male transmission becomes increasingly unlikely as the model becomes more dominant and more penetrant. SApairs and SX50 generally performed poorly for this pedigree. For the recessive models, the simulation results basically agreed with the analytical results, showing that Sall had the best power (and also had the correct false-positive rate). For the additive and dominant models, the simulated sample size of 10 had low power, regardless of the statistic.
For the NMMT pedigree, Sall was the best, with close to optimal sample sizes under the dominant and additive models, whereas SApairs was the worst (table 9). In contrast, the situation was reversed under the recessive models; Sall was the worst, with quite large sample sizes (>600 families), whereas SApairs was the best. The sample sizes for Sall were very close to optimal for the dominant and additive models. However, for the recessive models, we observed large sample-size differences between the optimal tests and the best statistic, SApairs. The simulation results for the NMMT pedigree were consistent with the analytical results. All statistics except Sall had the expected false-positive rate, whereas Sall's false-positive rate was elevated by a factor of 3–4.
For the CSFF pedigree, Sall was the best statistic for all models and had nearly optimal sample-size requirements (table 9), whereas the other four statistics were equivalent to each other. The simulation results also indicated that Sall was the best statistic, although Sall had an elevated false-positive rate of 0.004–0.007, whereas the other statistics had a less elevated false-positive rate, 0.002–0.004.
For the CSMF pedigree, Sall had consistently smaller sample-size requirements than did the other statistics (table 9), except under recessive model 12, which has reduced penetrance and a high phenocopy rate. For model 12, SGHpairs was the best, whereas SApairs was the worst. However, the simulation results showed that, for recessive model 12, Sall was the best, closely followed by SGHpairs, whereas SApairs was the worst. This difference may have been due to the false-positive rates in the simulation; Sall had an elevated false-positive rate in the range 0.005–0.006, whereas the other statistics had false-positive rates in the range 0.002–0.004.
Nuclear families
For the nuclear families, the sample-size differences between the best and worst statistics within a given sex configuration were quite small (tables 10 and 11)—often the two sample sizes differed by only one (except for the four-female sibships, under certain models). For nuclear families with only two affected siblings, all statistics have identical sample-size requirements within any model, because all statistics produce the same normalized coefficients.
Table 10 shows the sample sizes for three-sibling families, for four different sex combinations. All five statistics are identical to each other for three-female and three-male combinations, since there are only two possible IBD-sharing configurations (table 3). The best statistics for each sex combination performed the same as did the optimal test, except for recessive model 12. All statistics performed almost identically, in most conditions, for two-males–one-female and one-male–two-females combinations. For the two-males–one-female combination, SGHpairs and SX50 performed best for model 12, whereas SApairs performed worst. For the one-male–two-females combination, SGHpairs was the worst statistic more often than were the other statistics. Our simulations indicated that Sall had the lowest power for the two-males–one-female combination (for a sample size of 10), in most models. For the one-male–two-females combination, SApairs and SX50 were the best in most models, whereas SGHpairs was the worst; however, as in the analytical results, there were no dramatic power differences within each family type. Across all sex combinations, all statistics had similar elevated false-positive rates of ∼0.003 or ∼0.004, with a range of 0.002–0.005.
For four siblings, Sall was the best statistic more often than were the other statistics (table 11). For the three-males–one-female combination, recessive model 12 was again the exception, with SGHpairs and SX50 being the best statistics under model 12. SApairs, SGHpairs, SX75, and SX50 were equivalent to each other for four-female and four-male sibships, since they produced the same normalized coefficients (table 4). The best statistics in each sex combination performed very closely to the optimal test in most cases, except for the dominant models for four-female sibships. The simulation results show that Sall was the best in most models, and SGHpairs was the worst more often than were the other statistics; however, for the three-males–one-female combination, SGHpairs was the best under model 12, whereas SApairs was the worst, a finding that is consistent with the analytical sample-size estimates. Across all sex combinations, all statistics had similar elevated false-positive rates, of ∼0.004 or ∼0.005, with a range of 0.002–0.006.
Note that, under dominant and additive models, all-female sibships (of sizes two to four) had relatively large sample-size requirements for all statistics (as compared to the sample-size estimates for the other sex combinations). In contrast, all-female sibships under recessive models had low sample-size requirements, similar to those of the other sex combinations. In addition, the sample-size requirements increase as the number of females in the sibship increases, under the dominant and additive models, for all five statistics.
Two-Locus Models
Under the two-locus models, only the NMMT, CSFF, and CSMF pedigrees were studied (table 12). For the NMMT pedigree, Sall was consistently the best statistic, and SApairs was the worst, just as for the single-locus models. For the CSFF pedigree also, the two-locus results mirrored the single-locus results. For the CSMF pedigree, Sall was again the best for the heterogeneity models, but it did not perform as well for the additive models.
Discussion
In an attempt to find a robust allele-sharing statistic for X-linked traits that performs well across a variety of genetic models and pedigree structures, we have compared the relative sample-size requirements of five different allele-sharing statistics for X-linked traits. We have considered four different extended pedigrees, as well as all possible sex combinations in nuclear families with two, three, and four affected siblings. As might be expected, we have found that which statistic is best depends on both the true underlying mode of inheritance and the sex and distribution of the affected individuals within the pedigree.
For the extended pedigrees, Sall was found to be the most powerful and the closest to optimal for the majority of the genetic models (whether single locus or two locus). Similarly, for autosomal traits, Sall performs quite well across a large number of models (Feingold et al. 2000). However, this result does depend on the pedigree structure and genetic model, since, under recessive models for the NMMT pedigree, Sall has dramatically less power than do the other statistics (table 9). Sall is most powerful when there is sharing of alleles in common among the whole group of affected individuals, which is unlikely under recessive models for the NMMT pedigree. Similarly, for autosomal traits, Davis et al. (1997) found that, in situations in which the disease allele is likely to have entered the pedigree more than once (i.e., a recessive disease with a fairly common allele), Sall is much worse than Spairs. However, we did not, as we might have expected, observe this effect when Sall was applied under the two-locus models; for example, in our simulations of the CSFF pedigree under the “H-common” model, 49% of families were segregating disease alleles at both loci, and yet Sall was still the most powerful statistic. On the basis of this observation, we suggest that further exploration of the behavior of Sall is warranted in both the autosomal and the X-linked contexts. We also have seen that Sall is not the best statistic under dominant models for the MMT pedigree (table 9), but in this situation the sample-size requirements become extremely large as the genetic models become “stronger” and more inconsistent with the observed male-to-male transmission in this pedigree. For the recessive models for the NMMT pedigree, the optimal sample sizes were much smaller than the sample sizes obtained when any of the five statistics were used, which indicates that there is room for improved statistics for recessive models (table 9). This is not surprising, since previous studies have shown that most standard statistics do not have good power for autosomal recessive traits and that special recessively oriented statistics do much better (e.g., see Feingold and Siegmund 1997; Feingold et al. 2000; Sengul et al. 2001).
For the nuclear families, we observed only minor power differences among the five statistics, within any given sex configuration (tables 10 and 11); however, under dominant and additive models, the all-female sibships had quite low power, no matter which statistic we used. This result has been observed before, since Cordell et al. (1995) have shown that, under genetic models in which the female dominance variance is small, the power for full sisters is much less than that for other pairs of relatives. Clearly, any two female sibs share either one or two alleles IBD, since they always share the father's allele in common. If the disease allele has entered the sibship only once through the father, then the female sibship will contain no information for linkage. This effect is pronounced under the dominant and additive models, since one disease allele is enough to induce elevated penetrance. We do not observe such a dramatic effect when there is a least one male in the sibship, since the presence of at least one affected male makes it much more likely that the disease allele has entered the family through the mother rather than through the father. Under the recessive models, no matter which sex combinations the sibships have, all statistics result in comparably high power, since they are all based on the same information: the alleles that have been passed on from the mother. Similar but less-pronounced effects help explain the results for the CSFF pedigree (table 9), in which the sample sizes under the dominant and additive models are quite a bit larger than those under most of the recessive models.
Even though, in most cases, SGHpairs is the worst statistic, it is the most powerful statistic when male siblings outnumber female siblings (such as in the two-males–one-female and the three-males–one-female combinations) under the recessive model with a high phenocopy rate and reduced penetrance (model 12; see tables 10 and 11). Similarly, we also have observed that, in the CSMF pedigree, SGHpairs is the best statistic for model 12 (table 9). Under recessive models with high phenocopy rates, most female cases are phenocopies. Therefore, for such models, a statistic that scores female sharing less and that scores male sharing more would perform the best. SGHpairs performs well because it assigns to alleles shared between males a score higher than that assigned by any of the other statistics (table 2).
To complement and verify our analytical power computations, we also performed simulation-based power studies. Although the analytical power computations assume normality and a fixed type I–error rate, 0.001, the simulation studies do not make such assumptions and also permit us to generate empirical type I–error rates. Our simulation results indicate that, for the NMMT, CSFF, and CSMF pedigrees, Sall always has a larger type I–error rate than do the other statistics. This means that the simulation results showing that Sall generally has high power should be interpreted with some caution. This issue has been discussed in more detail by other authors (e.g., McPeek 1999). In the present study, however, the analytical results also show Sall to be the best statistic in most cases, so we can be fairly confident about this general qualitative result.
Sengul et al. (2001) examined the effect of increasing the distance between the marker and the trait locus in their study of linkage statistics for autosomal traits, and they found that the relative power of the statistics changed as θ increased. Therefore, in addition to our results, which assumed a fully informative marker perfectly linked (θ = 0.0) to the trait locus, we also computed estimated sample sizes at several different larger values of θ (i.e., 0.05, 0.10, and 0.20). We found no change in the relative ranking of the statistics for X-linked traits. Also, note that our results are based on a fully informative marker, but, if the marker is not fully informative, the result is very similar to that caused by an increase in θ. This issue is discussed in more detail by Sengul et al. (2000).
Cordell et al. (1995) investigated the power to detect X linkage in various types of affected relative pairs, as functions of the relative risks (λ's). The λ's for sibling pairs for the models that we considered are given in table 1. Our general results regarding which types of pedigrees are most powerful are consistent with those reported by Cordell et al. (1995). Those authors found that, when the female dominance variance is small (as in our additive and dominant models), sister-sister pairs have much lower power than do other types of relative pairs. We found, as discussed above, that this result is also true for larger sibships that consist entirely of females. When there is substantial dominance variance (as in our recessive models), sibships of sisters become roughly as useful as sibships that include brothers. This is because the sister-sister relative risk, λff, includes components due to both additive and dominance variances, whereas both the sister-brother relative risk, λmf, and the brother-brother relative risk, λmm, are functions of the additive variance only. A similar effect is seen in comparisons of the power that siblings and unilineal relatives have for the mapping of autosomal loci (e.g., see Feingold and Siegmund 1997). Comparing, across sex groups, first-cousin pairs whose parents are sisters, they found that male-male first-cousin pairs are more powerful than female-female first-cousin pairs. This would lead us to the prediction that the CSMF pedigree should have more power to detect linkage than does the CSFF pedigree, because the CSMF pedigree contains both male-male and female-female first-cousin pairs, whereas the CSFF pedigree contains only female-female first-cousin pairs. Our results (table 9) are consistent with this prediction.
It is important to note that affected relatives of the same general relationship (e.g., full siblings) can provide very different power to detect linkage, depending on their sex. This suggests that, for optimal power, one should pay close attention to proper weighting when combining statistics across families with different sex combinations. One could obtain an overall statistic by combining statistics after assigning appropriate weights to different sex pairs and/or pedigree configurations. The optimal weights depend on the genetic model. Although it is not difficult to derive the weights for any given model, the real challenge is to find weights that have robust power—that is, that have reasonably high power for a variety of models. We hope to explore such weighting issues in the future, building on the framework already developed for autosomal traits (e.g., see Sham et al. 1997; Teng and Siegmund 1997). One way to circumvent the weighting problem is to do parametric linkage analysis instead, which essentially imposes a particular set of weights based on the specified model. There have been some reports of good power obtained by parametric analysis of the autosomes for complex traits, by use of either fairly simple genetic models or “best of several models” approaches (e.g., see Abreu et al. 1999; Sham et al. 2000). It will be important to study how these approaches compare to nonparametric methods for X-linked loci.
Finally, this study has used a limited set of trait models and a limited set of pedigrees. Since the actual number of trait models and pedigrees that might be of interest is enormous, we cannot claim to have considered all important possibilities. Future work to study more pedigree types and more models would be useful.
Acknowledgments
This research was supported by the University of Pittsburgh and by National Institutes of Health grant AG16989.
References
- Abreu PC, Greenberg DA, Hodge SE (1999) Direct power comparisons between simple LOD scores and NPL scores for linkage analysis in complex diseases. Am J Hum Genet 65:847–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell HJ, Kawaguchi Y, Todd JA, Farrall M (1995) An extension of the maximum lod score method to X-linked loci. Ann Hum Genet 59:435–449 [DOI] [PubMed] [Google Scholar]
- Davis S, Goldin LR, Weeks DE (1997) SimIBD: a powerful robust nonparametric method for detecting linkage in general pedigrees. In: Pawlowitzki IH, Edwards JH, Thompson EA (eds) Genetic mapping of disease genes. Academic Press, London, pp 189–204 [Google Scholar]
- DeLisi LE, Smith AB, Razi K, Stewart J, Wang Z, Sandu HK, Philibert RA (2000) Investigation of a candidate gene for schizophrenia on Xq13 previously associated with mental retardation and hypothyroidism. Am J Med Genet 96:398–403 [DOI] [PubMed] [Google Scholar]
- Dupuis J, Van Eerdewegh P (2000) Multipoint linkage analysis of the pseudoautosomal regions, using affected sibling pairs. Am J Hum Genet 67:462–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold E, Siegmund D (1997) Strategies for mapping heterogeneous recessive traits by allele-sharing methods. Am J Hum Genet 60:965–978 [PMC free article] [PubMed] [Google Scholar]
- Feingold E, Song KK, Weeks DE (2000) Comparison of allele-sharing statistics for general pedigrees. Genet Epidemiol 19 Suppl 1:S92–S98 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A (2000) Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13 [DOI] [PubMed] [Google Scholar]
- Hamer DH, Hu S, Magnuson VL, Hu N, Pattatucci AML (1993) A linkage between DNA markers on the X chromosome and male sexual orientation. Science 261:321–327 [DOI] [PubMed] [Google Scholar]
- Holmans P (1993) Asymptotic properties of affected-sib-pair linkage analysis. Am J Hum Genet 52:362–374 [PMC free article] [PubMed] [Google Scholar]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- Lange K, Weeks D, Boehnke M (1988) Programs for pedigree analysis: MENDEL, FISHER, and dGENE. Genet Epidemiol 5:471–472 [DOI] [PubMed] [Google Scholar]
- Liu J, Nyholt DR, Magnussen P, Parano E, Pavone P, Geschwind D, Lord C, Iversen P, Hoh J, the Autism Genetic Resource Exchange Consortium, Ott J, Gilliam TC (2001) A genomewide screen for autism susceptibility loci. Am J Hum Genet 69:327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPeek MS (1999) Optimal allele-sharing statistics for genetic mapping using affected relatives. Genet Epidemiol 16:225–249 [DOI] [PubMed] [Google Scholar]
- Nyholt DR, Dawkins JL, Brimage PJ, Goadsby PJ, Nicholson GA, Griffiths LR (1998) Evidence for an X-linked genetic component in familial typical migraine. Hum Mol Genet 7:459–463 [DOI] [PubMed] [Google Scholar]
- Ott J (1989) Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci USA 86:4175–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risch N (1990a) Linkage strategies for genetically complex traits. I. Multilocus models. Am J Hum Genet 46:222–228 [PMC free article] [PubMed] [Google Scholar]
- ——— (1990b) Linkage strategies for genetically complex traits. II. The power of affected relative pairs. Am J Hum Genet 46:229–241 [PMC free article] [PubMed] [Google Scholar]
- ——— (1990c) Linkage strategies for genetically complex traits. III. The effect of marker polymorphism on analysis of affected relative pairs. Am J Hum Genet 46:242–253 [PMC free article] [PubMed] [Google Scholar]
- Sengul H, Weeks DE, Feingold E (2001) A survey of affected-sibship statistics for nonparametric linkage analysis. Am J Hum Genet 69:179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham PC, Lin MW, Zhao JH, Curtis D (2000) Power comparison of parametric and nonparametric linkage tests in small pedigrees. Am J Hum Genet 66:1661–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham PC, Zhao JH, Curtis D (1997) Optimal weighting scheme for affected sib-pair analysis of sibship data. Ann Hum Genet 61:61–69 [DOI] [PubMed] [Google Scholar]
- Teng J, Siegmund D (1997) Combining information within and between pedigrees for mapping complex traits. Am J Hum Genet 60:979–992 [PMC free article] [PubMed] [Google Scholar]
- Vallada HP, Vasques L, Curtis D, Zatz M, Kirov G, Lauriano V, Gentil V, Murray RM, McGuffin P, Owen M, Gill M, Craddock N, Collier DA (1998) Linkage analysis between bipolar affective disorder and markers on chromosome X. Psychiatr Genet 8:183–186 [DOI] [PubMed] [Google Scholar]
- Weeks DE, Ott J, Lathrop GM (1990) SLINK: a general simulation program for linkage analysis. Am J Hum Genet Suppl 47:A204 [Google Scholar]
- Weeks DE, Valappil TI, Schroeder M, Brown DL (1995) An X-linked version of the affected-pedigree-member method of linkage analysis. Hum Hered 45:25–33 [DOI] [PubMed] [Google Scholar]
- Whittemore AS, Halpern J (1994) A class of tests for linkage using affected pedigree members. Biometrics 50:118–127 [PubMed] [Google Scholar]
- Xu J, Meyers D, Freije D, Isaacs S, Wiley K, Nusskern D, Ewing C, et al (1998) Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet 20:175–179 [DOI] [PubMed] [Google Scholar]