

Abstract
HIV-1 envelope protein gp120 has been implicated in neurotoxin production by monocytic cells, namely macrophages and microglia, and the pathogenesis of HIV-1 associated neurocognitive disorders (HAND). We previously showed in cerebrocortical cell cultures from rodents containing microglia, astrocytes and neurons, that overall inhibition of p38 MAPK signaling abrogated the neurotoxic effect of HIV-1 gp120. However, the time course of p38 MAPK activation and the contribution of this kinase in the various cell types remained unknown. In this study, we found that for HIV gp120-induced neurotoxicity to occur, active p38 MAPK is required in monocytic lineage cells, namely macrophages and microglia, and neuronal cells. In cerebrocortical cell cultures HIV-1 gp120 stimulated a time-dependent overall increase of active p38 MAPK and the activated kinase was primarily detected in microglia and neurons. Interestingly, both increased activation of p38 MAPK and neuronal death in response to gp120 were prevented by prior depletion of microglia, or in the presence of CCR5 ligand CCL4 or of p38 MAPK inhibitors. In human monocytic THP-1 cells and primary monocyte-derived macrophages (MDM), HIV gp120 stimulated production of neurotoxins was abrogated by prior introduction into the cells of a dominant-negative p38 MAPK mutant or p38 MAPK siRNA. In addition, the neurotoxic effects of cell-free supernatants from gp120-stimulated monocytic THP-1 cells were prevented in microglia-depleted cerebrocortical cells pretreated with a pharmacological inhibitor of p38 MAPK. Thus, p38 MAPK signaling was critical upon exposure to HIV gp120 for both the neurotoxic phenotype of monocytic cells and subsequent toxin-initiated neuronal apoptosis.
Introduction
Patients infected with HIV-1 can develop neurological complications in addition to AIDS, including motor and cognitive dysfunctions termed HIV-1-associated neurocognitive disorders (HAND) [1]. In the central nervous system (CNS), invading infected mononuclear phagocytes, namely monocytes and macrophages, and resident microglia may be acting as a reservoir for HIV-1 [2–4]. By secreting viral proteins that are thought to also stimulate uninfected microglia and macrophages to release neurotoxins, those infected immune cells are presumed to be ultimately responsible for the development of HAND [4,5]. Development of HAND has been correlated to the neuropathological diagnoses of HIV encephalitis (HIVE) characterized by widespread reactive astrogliosis, myelin pallor, decreased synaptic and dendritic density, selective neuronal loss, and the infiltration and accumulation of monocytic cells, including blood-derived macrophages and resident microglia [6,7]. In fact, loss of neuronal processes and synapses and activation of both uninfected and infected microglia appears to constitute the best correlate to CNS impairment after HIV-1 infection [8,9]. However, the molecular mechanism of how HIV-1 triggers macrophage/microglial activation and neurotoxicity are not completely understood.
Several in vitro and in vivo studies have shown that the envelope glycoprotein gp120 of various HIV-1 strains produce injury and apoptosis in both primary human and rodent neurons [10–17]. HIV-1/gp120 coreceptors, in particular chemokine receptors CXCR4 and CCR5, are besides CD4 the first sites of host-virus interaction. Although CCR5 and CXCR4 are present not only on macrophages and microglia but also on neurons and astrocytes [16], HIV-1/gp120 requires CD4 receptors, which are only located on cells of immune lineage, to efficiently engage coreceptors and infect target cells [4,5]. However, interaction between gp120 and CXCR4/CCR5, independent of CD4, has been shown to cause activation of the G protein-coupled receptor and contribute to intracellular Ca2+ accumulation and signaling [18]. Thus, direct interaction of gp120 with neuronal chemokine receptors [17,18] may contribute to neuronal injury, while activation of macrophage HIV co-receptors and subsequent release of neurotoxins, such as excitotoxins, chemokines and/or pro-inflammatory cytokines, presumably provide the predominant trigger for neuronal injury and death, as previously suggested [3,12,15,19,20]. In support to the latter, depletion or inactivation of microglia in mixed neuronal-glial cell cultures completely abrogates HIV-1 gp120-induced neuronal death [15,21]. Furthermore, we have previously shown that both CCR5 and CXCR4 can mediate the neurotoxic effect of gp120 depending on the co-receptor usage of the virus strain from which the envelope protein originated [16].
In addition, β-chemokines and natural CCR5 ligands CCL3 (MIP-1α), CCL4 (MIP-1β), and CCL5 (RANTES) have been reported to be major suppressors of HIV-1 infection and disease progression [22]. These reports suggested that suppression occurs through steric hindrance, receptor internalization, and/or via CCR5-mediated protective signaling against HIV. In support of this, our group and others have found that the CCR5 ligands CCL4 and CCL5 block neuronal death caused by HIV-1 gp120 in vitro [14,15].
Macrophages or microglia are principal targets for both β-chemokines and gp120 variants, but the mechanism in which gp120-induced chemokine receptor signaling in macrophages/microglia results in a neurotoxic phenotype is not well-defined. However, previous studies have shown that possible mechanisms of HIV-1 neuropathogenesis involve the perturbation of intracellular signaling by HIV-1 gp120 and subsequent release of neurotoxic factors from activated macrophages and microglia [3,12,15,19,23–25]. Intracellular signaling pathways such as those of Src family kinase Lyn [20], PI3K20, Akt [16], the focal adhesion-related proline-rich tyrosine kinase Pyk2 [20,25], phosphatidylcholine phospholipase C (PC-PLC) [26], proteins of the MAPK family [15,16,25,27] and the transcription factor p53 [21,27] have been implicated as potential pathways responsible for gp120-induced macrophage activation and neurotoxicity. Of these, MAPKs are involved in several diverse biological activities including differentiation, proliferation, apoptosis and inflammation [28]. The stress-activated p38 MAPK has been shown to be responsible for neuronal death, microglial/macrophage activation and proinflammatory cytokine production in several CNS disorders [15,16,29–32]. In fact, we previously showed that inactivating p38 MAPK with p38-specific pharmacological inhibitor SB203580 or dominant negative p38α (p38αAF) in mixed neuronal-glial cultures prevented neuronal death triggered by HIV-1 gp120 while others reported that blocking p38 MAPK activity also ameliorated glutamate toxicity in neuron-rich cell cultures [15,16,32]. However, the molecular mechanism of p38 MAPK activation and its cell-specific responses downstream of chemokine receptor signaling in gp120-mediated neurotoxicity is still unknown.
For this purpose, we investigated the neurotoxic effects of HIV-1 gp120 on cell type-specific p38 MAPK signaling pathways using both primary mixed neuronal-glial cerebrocortical cultures from rodents and human mononuclear/monocytic THP-1 cells or primary human monocyte-derived macrophages (MDM) as models for macrophages and microglia. We found that HIV-1 gp120 activated p38 MAPK primarily in neurons and microglia, but not in astrocytes. We also found that depletion of microglia from mixed cultures and pretreatment with neuroprotective CCL4 reduced the activation of neuronal p38 MAPK after gp120 exposure. In addition, monocyte/macrophage-like THP-1 cells and MDM also required p38 MAPK for gp120-induced neurotoxin production. However, inhibiting p38 MAPK activation in neurons prevented death induced by neurotoxic media derived from gp120-stimulated THP-1 cells, whereas absence of HIV coreceptors in cerebrocortical neurons and astrocytes failed to protect the cell cultures from gp120-induced neurotoxicity of the monocytic cells. Our results suggested that p38 MAPK signaling is responsible for both the neurotoxic phenotype of macrophages/microglia and the initiation of neuronal apoptosis upon exposure to HIV gp120.
Materials and Methods
Reagents
Recombinant gp120 from different HIV-1 strains, SF2 (dual-tropic) or SF162 (CCR5-preferring), were obtained from NIH AIDS Research and Reference Reagent Program. Recombinant human chemokines were purchased from R&D Systems (Minneapolis, MN). A specific p38 MAPK inhibitor, SB203580, was obtained from Calbiochem (San Diego, CA). Chemokines and HIV gp120 were reconstituted in 0.1 % bovine serum albumin (BSA) at 100 X the final concentration and controls received BSA vehicle alone (0.001 % final concentration). Kinase inhibitor was dissolved in DMSO at 1,000 X the final concentration (0.1–10 μM) and was added to the cultures for 15 min prior to treatment with 200 pM or 1 nM HIV-1 gp120 strains.
Preparation of cerebrocortical, mixed neuronal-glial cell cultures
Rat mixed neuronal/glial cerebrocortical cultures were prepared from embryos of Sprague-Dawley rats at day 15 to 17 of gestation, as previously described by our group [15,16]. In brief, cells were cultured in 35 mm dishes with poly-L-lysine-coated glass coverslips (1.87 × 106 cells per dish) or poly-L-lysine-coated clear-bottom 96 well plates for imaging (BD Falcon; 0.087 × 106 cells per well) and D10C medium containing 80 % DMEM with high glucose (Invitrogen), 10 % FBS (Hyclone), 10 % F12 (Hyclone), 3 % 1 M Hepes (Omega), 200 mM L-glutamine (Invitrogen), and 100 U/ml penicillin with 100 μg/ml streptomycin (Invitrogen). Cell populations consisted of ~30 % neurons, ~70 % astrocytes and ~0.1 to 1 % microglia [16] and, generally, were used after 17 days in vitro when the majority of neurons were considered to be fully differentiated and susceptible to NMDA toxicity. For most experiments, rat cultures were transferred into pre-warmed Earle’s balanced salt solution (EBSS) containing 1.8 mM Ca2+ and 5 μM glycine without Mg2+. For some experiments in which the contribution of microglia to neurotoxicity was investigated, microglia were depleted by pre-treatment of the cultures overnight with 7.5 mM L-leucine methyl ester [15]. Importantly, rodents express CXCR4 and CCR5 homologues, which are capable of interacting with HIV-1 via gp120 binding [15,16]. Murine cerebrocortical cell cultures from CCR5/CXCR4 double knockout embryos were prepared as previously published by our group [16] and are similar to their above described counterpart derived from rat embryos with the exception that they comprise ~ 4–5 % microglia. All experiments involving animals have been approved by the Institutional Animal Care & Use Committee of the Sanford-Burnham Medical Research Institute.
Isolation and preparation of monocyte-derived macrophages
The preparation of human primary monocyte-derived macrophages (MDM) using a Ficoll gradient was performed as previously described [33,34]. In brief, whole blood from consenting healthy donors was obtained through the Normal Blood Donor Service of The Scripps Research Institute’s (TSRI) at Scripps Health Green Hospital, La Jolla, CA (P.I.: Daniel R. Salomon, M.D., TSRI, La Jolla CA; IRB Protocol: # 09-5252). Following an initial centrifugation step of the heparinized blood at 200 × g for 20 min, buffy coat cells were isolated using density gradients of Ficoll-paque (ρ 1.073; GE Health Life Sciences, Piscataway, NJ) and transferred into 75 cm2 cell culture flasks [35]. After allowing adherence in RPMI 1640 containing 2 mM glutamine, antibiotics (Penicillin/Streptomycin) and 10 % human AB serum (RPMI-ABS) at 37°C, 6% CO2, in humidified atmosphere, non adherent cells were removed by rigorous washing with warm RPMI 1640 (37°C) without supplements. Adherent monocytes were then cultured for 7 days in the above serum-containing medium to allow for differentiation into MDM [35]. For harvest and further experimentation the cells were detached by treatment with phosphate buffered saline (PBS) containing EDTA (Sigma, 0.2 g/l) for 5 to 10 min at 37°C and scraping with a rubber policeman. After washing three times in PBS cells were reseeded at 0.25 × 106 cells/0.5 ml medium per well in 24 well plates. Cell viability was monitored with Trypan Blue exclusion and usually exceeded 95 %.
Cell culture of human mononuclear THP-1 cells
Human mononuclear THP-1 cells, a model for monocyte lineage cells, including microglia and macrophages [12,36], were maintained in medium containing 90 % RPMI (Gibco), 10 % FBS (Hyclone), 2 mM L-glutamine (Sigma), and a combination of 100 U/ml penicillin with 100 μg/ml streptomycin (P/S, Sigma) at 37°C with 5 % CO2 and are usually split 1:3 to 1:4 once to twice a week after reaching approximately 1 × 106 cells per milliliter. For siRNA experiments, THP-1 cells were plated at 0.2 × 106 cells/ml after reaching a density of 0.3–0.4 × 106 cells/ml. Upon reaching a density of 0.6–0.7 × 106 cells/ml, cells were collected for nucleofection of siRNA duplexes using the Amaxa nucleofector system (Walkersville, MD) and the supplier’s protocol.
Assessment of neuronal death
Neuronal apoptotic death and loss was analyzed as previously described [16]. In brief, the number of neurons were quantified by immunostaining for the neuron-specific marker microtubule-associated protein 2 (MAP-2), and apoptotic nuclei were identified morphologically after staining nuclei with the DNA dye Hoechst 33342. Alternatively, apoptosis was quantified by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL; Apoptosis Detection System/Fluorescein, Promega, Madison, WI) to detect fragmented nuclear DNA in apoptotic nuclei as seen in Fig. 9. Neuronal survival was calculated from the percentage of neurons remaining after subtraction of those that had undergone apoptosis. Three to eight independent experiments were performed for each treatment.
Mononuclear cell-mediated neurotoxicity
In order to obtain conditioned cell culture supernatants, human mononuclear THP-1 cells or primary MDM were incubated for 24 h or 4 days in the presence or absence of recombinant viral envelope gp120 of HIV strains SF2 and SF162 in the same media used to culture rat cerebrocortical cells (THP-1 cells) or RPMI-ABS (MDM). The controls without gp120 received 0.001 % BSA (vehicle) only, or CCL4/MIP-1β or CCL5/RANTES (both at 20 nM). For some experiments, THP-1 cells were transduced using adenoviral expression vectors (AdV) encoding a dominant-negatively interfering mutant of p38 MAPK or GFP as control 2 days before the exposure to HIV-1 gp120 or BSA control in THP-1 culture media or RPMI-ABS in the case of MDM [16]. All AdV were kindly provided by Dr. J. Han, The Scripps Research Institute, La Jolla, CA [37], and amplified and titrated using standard procedures. Following a 1 or 4 day stimulation period, cell-free supernatants were transferred at a final 10 % concentration to the mixed rat neuronal-glial cultures depleted of microglia (as described in the Tissue culture section above) maintained in D10C medium as readout for neurotoxicity [12,36]. If not stated otherwise, neuronal survival was assessed after 72 h of incubation, as described above. In addition, cell lysates were collected as described below for immunoblotting.
siRNA nucleofection
MAPK14 (p38α MAPK) siRNA or non-targeting siRNA duplexes were obtained from Dharmacon/Thermo Fisher Scientific (Lafayette, CO) and Ambion/Applied Biosystems (Austin, TX). Silencer MAPK14 siRNA (ID# 1217 and 1312) duplexes from Ambion are referred to as “sip38α-1” and “sip38α-2,” respectively, in this manuscript. ON-TARGETplus MAPK14 siRNA (J-003512-21) from Dharmacon is referred to as “sip38α-3” in this manuscript. The siRNA duplexes were transfected into THP-1 cells at a density of 1 × 106 cells/ml using Amaxa (Walkersville, MD) nucleofector kits according to the manufacturer’s instructions. Briefly, 48 h after nucleofection with 2 μg siRNA, THP-1 cells were treated for 24 h with 200 pM HIV-1 gp120SF2 or gp120SF162. Supernatants or conditioned media from these treatments were then collected and used at 10% final concentration on microglia-depleted rat cerebrocortical cultures as previously described for neurotoxicity analysis. The mononuclear THP-1 cells were collected and analyzed for p38 MAPK activation via Western blot.
Immunofluorescence staining and deconvolution microscopy
After treatment and washing with PBS, rat cerebrocortical cultures were fixed for 25 min with 4 % PFA at 4°C and subsequently permeabilized with either 0.2 % Triton X-100 for 5 min at room temperature (for staining of MAP-2 or TUNEL alone) or 100 % methanol for 10 min at −20°C (for staining of phosphorylated p38 with or without MAP-2). Primary antibodies included mouse anti-MAP-2 (1:500, Sigma M4403; St. Louis, MO), chicken anti-MAP-2 (1:5000, Abcam ab5392; Cambridge, MA), rabbit anti-ACTIVE p38 (1:1000, Promega V1211; Madison, WI), and mouse anti-CD11b (1:150, Serotec MCA275; Raleigh, NC). Secondary antibodies included goat anti-rabbit or anti-mouse or anti-chicken IgGs Alexa 594/488/647 (1:2000, Invitrogen; Carlsbad, CA) and horse anti-mouse IgG Texas Red (1:150, Vector Labs; Burlingame, CA). Controls were included in which primary antibodies were either omitted or replaced with irrelevant IgG of the same subclass. Nuclear DNA was stained with Hoechst H33342. Deconvolution microscopy was performed as described earlier with filters for DAPI, FITC, CY3 and CY5 with the only modification that a constraint iterative algorithm was used for deconvolution (Slidebook software, Intelligent Imaging Innovations; Denver, CO) [16].
Cell Lysates
After washing with PBS, cerebrocortical cells or mononuclear THP-1 cells were harvested by adding 1 X Cell Lysis Buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 % Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin; Cell Signaling; Beverly, MA) supplemented with 5 mM NaF (Serine/Threonine protein phosphatase inhibitor) and Complete Protease Inhibitor Cocktail (Roche; Indianapolis, IN) on ice for 10 min. The lysed samples were transferred to microcentrifuge tubes, sonicated 4 times 5 seconds, and then cleared by centrifugation (13,200 rpm, 10 min) at 4°C. Lysate total protein concentrations were determined using the BCA protein assay kit (Pierce/Thermo Fisher Scientific; Rockford, IL). Alternatively, in some experiments THP-1 cells or MDM were directly dissolved at 0.25 × 106 cells/150 μl in SDS-containing gel loading-buffer (187.5 mM Tris-HCl (pH 6.8 at 25°C), 6% (w/v) SDS, 30% glycerol and 0.03% (w/v) bromophenol blue; Cell Signaling Technology, Beverly, MA).
Immunocomplex Kinase Assay for Activity of p38 MAPK or SAPK/JNK
The kinase assay for phosphorylated p38 MAPK and JNK were performed using the kinase assay kits from Cell Signaling Technology following the supplier’s instructions. In brief, immobilized anti-phospho-p38 MAPK monoclonal antibody was used to immunoprecipitate active p38 MAPK from cell lysate (200–400 μg total protein), followed by an in vitro kinase assay using ATF-2 as a substrate. ATF-2 phosphorylation was detected by western blotting using phospho-ATF-2 antibody (1:1000; Cell Signaling). For SAPK/JNK kinase activity, c-Jun fusion protein linked to agarose beads was used to pull down JNK enzyme from cell extracts (200–400 μg total protein); and phospho-c-Jun antibody was measured via western blotting (1:1000; Cell Signaling). All protein kinase assays were standardized for total cellular protein amount and lysate volume used in the initial immunoprecipitation step.
Immunoblotting
For Western blotting analysis of whole cell lysates or substrates of kinase assays, 4 X LDS sample buffer and 10 X reducing agent (Invitrogen; Carlsbad, CA) were added to samples containing equal amounts of total protein (10, 30 or 50 μg for lysates; 2 μg kinase substrate) and heated for 5 min at 100°C. Alternatively, cell lysates in SDS-containing gel loading-buffer were made 41 mM in dithiothreitol and heated. Samples were then electrophoretically separated on SDS-PAGE gels (NUPAGE; Invitrogen) under reducing conditions and subsequently electro-transferred to PVDF membranes (Bio-Rad; Hercules, CA). After blocking, membranes were incubated with gentle agitation overnight at 4°C with specific primary antibodies against ACTIVE p38 (1:100 (for cerebrocortical cells) – 1000 (for THP-1 cells and MDM); Promega), total p38 (1:1000, Cell Signaling) and β-actin (1:20,000; Ambion). The membranes were then incubated with secondary antibodies conjugated with horseradish-peroxidase for 30 min and exposed to SuperSignal Pico chemoluminescent detection kit (Pierce). Immunoblotting results were analyzed using Adobe Photoshop and densitometric measurements normalized against β-actin expression levels.
Statistical analysis
The data are expressed as mean values ± SEM for three-nine independent experiments. Statistical analysis was performed applying one-way ANOVA followed by Fisher’s PLSD post hoc test or student’s t-test using the StatView software package (version 5.0.1, SAS Institute Inc.). The significance level was set at p < 0.05. The immunoblotting and immunofluorescence images are representative samples of the at least three independent experiments performed.
Results
Kinetics of p38 MAPK activity in cerebrocortical cells cultures upon exposure to HIV gp120
We have previously shown that envelope protein gp120 derived from CCR5-preferring, CXCR4-preferring as well as dual-tropic HIV-1 strains can induce neurotoxicity and that this toxicity is equally mediated by the respective HIV coreceptors [16]. In order to extend the analysis of the HIV-1 coreceptor signaling pathways responsible for mediating neuronal death we investigated the kinetics and involvement of different MAPK pathways in induction of neuronal death by gp120. We used in vitro non-radioactive immunocomplex kinase assays for the stress-activated kinases (SAPK) p38 MAPK and, for comparison, SAPK/c-Jun N-terminal kinase (JNK) with p-ATF2 and p-cJun as substrate readouts, respectively. Cerebrocortical cell cultures were incubated for various time periods with HIV gp120 or vehicle as control as described for neurotoxicity assays [15,16]. Subsequently, the cells were transferred onto ice, quickly washed once with ice-cold PBS and lysed. The active kinases were precipitated from the cell lysate and exposed to assay substrates in the presence of ATP. Phosphorylated substrates were then visualized by Western blotting and quantified by densitometry. Baseline and HIV-1 gp120-induced activities of the protein kinases were assessed for each time point. In response to HIV-1 gp120SF2 exposure activation of p38 MAPK occurred early, in a short-lived peak at 5 min, followed by a temporary drop below baseline at 1 h. After the lowered kinase activity at 1 h, p38 kinase activity showed a sustained increase that was most pronounced above baseline at 3 h and 24 h (Fig. 1A). Thus, despite some variability between different batches of primary cerebrocortical cell cultures, which is to be expected, we observed a consistent pattern at early and late time-points of initial, temporary up- and down-regulation with an overall increase in p38 MAPK activity after exposure to gp120 (Fig. 1A). Altogether, the data indicated an overall trend towards increased kinase activity for p38 MAPK after gp120 exposure over a 24 h period (Fig. 1B, trend line). For comparison, the activity of JNK, which was monitored in the same way, showed a less pronounced deviation from the baseline over 24 h, except for the 5 min time point. Fig. 1C shows representative Western blot data for p38 MAPK (p-ATF2) and JNK (p-cJun) kinase assays at the significant time points of 5 min, 1 h, 3 h, and 24 h. In addition, direct immunoblotting for phospho-p38 MAPK was also used to detect the active kinase within rat cerebrocortical cultures (Fig. 1D and E). Increased phosphorylation of p38 MAPK occurred in response to gp120 from CXCR4-, CCR5- and CCR5/CXCR4-preferring, dual-tropic HIV-1 strains (IIIB, SF162 and SF2, respectively; Fig. 1D). Representative Western blot data showing increased phosphorylated p38 MAPK after 200 pM gp120SF2 treatment for 24 h is shown in Fig. 1E. Based on the finding that the activity of p38 MAPK deviated from baseline upon exposure to HIV gp120 most consistently at 5 min, 1 h, 3 h and 24 h, we focused in subsequent experiments primarily on p38 MAPK activity at those time periods.
FIGURE 1.

Kinetics of p38 MAPK and JNK activation in mixed neuronal-glial cerebrocortical cell cultures upon exposure to neurotoxic HIV envelope protein gp120. HIV-1 gp120SF2 causes significant deviation of p38 MAPK activity from baseline control condition at 5 min, 1 h, 3 h, and 24 h with an overall increase (A, and B for trend line for p38 MAPK activity). In contrast, activity of JNK is only significantly elevated at 5 min (A and C). Cerebrocortical cultures were incubated with dual-tropic gp120SF2 (200 pM) for the indicated time periods prior to immunoprecipitation and kinase assay analysis. The kinase activity of p38 MAPK and JNK using ATF-2 and c-Jun as substrates, respectively, were each detected by immunoblotting following the supplier’s protocol as mentioned in the Materials and Methods section. The kinase activity in untreated controls was measured for each time point and defined as the 100 % baseline value (A). Error bars represent the S.E.M. of four to nine independent experiments (A and B). Representative western blots of p-ATF2 (p-p38) and p-cJun (p-JNK) kinase assay results were shown after exposure to gp120 at 5 min, 1 h, 3 h and 24 h (C). In addition, recombinant gp120 of CXCR4-preferring (IIIB), dual-tropic (SF2) and CCR5-preferring (SF162) HIV-1 all activate p38 MAPK in cerebrocortical cells at 24 h (D). Examples of active p-p38 MAPK, p38 MAPK and actin Western blots used for densitometry analysis 24 h after gp120SF2 treatment are shown in (E). Equal amounts of cellular protein (10 μg/lane) were analyzed by SDS PAGE and Western blotting (D and E). The two bands detected as total p38 MAPK represent the α- (lower) and β- (upper) isoform of the kinase while the p-p38 MAPK is mostly of the α-isoform (E). Error bars represent the S.E.M. of three independent experiments (D). * = p < 0.05 compared with control (A, B and D).
Active p38 MAPK is required for HIV-1 gp120-induced neuronal death in mixed neuronal-glial cerebrocortical cell cultures in a dose-dependent fashion
After observing that activation of p38 MAPK to less than twice the baseline activity sufficed to mediate neuronal death, we assessed the dose-dependence of the neuroprotective effect that pharmacological inhibition of p38 MAPK showed in earlier studies. Therefore, we exposed primary rat cerebrocortical cultures to increasing doses of the p38 MAPK-specific pharmacological inhibitor, SB203580, prior to HIV-1 gp120 exposure. SB203580 acts as a competitive ATP-binding inhibitor which interrupts the kinase activity of p38 MAPK [38,39]. We treated the cultures with the compound at 0.1, 1.0 and 10 μM concentrations 15 min prior to treatment with 200 pM dual-tropic viral envelope protein gp120 from HIV-1 SF2 strain for 24 h. Controls received the vehicles for SB203580 (DMSO) and gp120 (PBS/BSA, 0.001 % final concentration) [15,16]. Subsequently, we assessed neuronal survival as readout for neurotoxicity by fluorescence microscopy using a combination of immunolabeling of neurons for MAP-2 in combination with nuclear DNA staining by H33342 as described in the Materials and Methods section. As expected, HIV gp120 reduced neuronal survival by about 23 ± 3.3 % in the absence of any p38 MAPK inhibitor whereas all the tested SB203580 concentrations lacked neurotoxicity (Fig. 2). Pretreatment of cultures with 10 μM SB203580 completely abrogated reduction in neuronal survival after gp120 treatment (p < 0.0001 compared to gp120; Fig. 2), confirming our previous studies [15,16]. Using 1.0 μM SB203580 to inhibit p38 MAPK activity, significantly increased neuronal survival after gp120 treatment to a slightly lesser extent than 10 μM (p < 0.02, compared to gp120; Fig. 2). Pretreatment of cultures with 0.1 μM SB203580 was found to result in neuronal survival that was significantly lower than in the control (p < 0.02) but significantly higher than in gp120 without kinase inhibitor (p < 0.05; Fig. 2). These results indicated only a partial protection against gp120 toxicity at concentrations lower than 1.0 μM and thus a causal, dose-dependent contribution of active p38 MAPK to gp120-induced neuronal injury and death.
FIGURE 2.
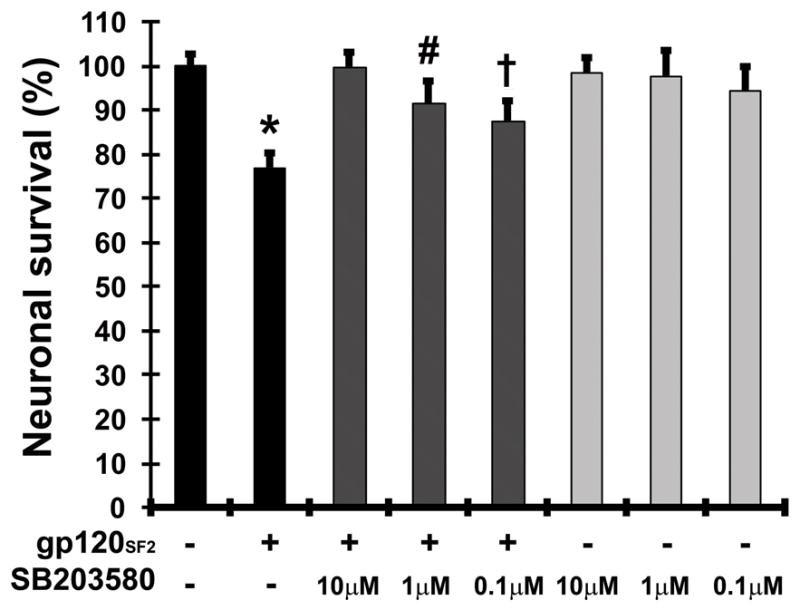
Inhibition of p38 MAPK prevents gp120-induced neuronal apoptosis in a dose-dependent fashion. Rat cerebrocortical cultures were incubated for 24 h with or without gp120SF2 (200 pM) in the presence or absence of 0.1, 1 or 10 μM of p38 MAPK inhibitor SB203580. Neuronal survival was assessed using fluorescence microscopy after fixation, permeabilization and immunolabeling of neurons for MAP-2 in combination with nuclear DNA staining by H33342. Error bars represent the S.E.M. of three to six independent experiments. * = p < 0.001 compared to control value; # = p < 0.02 compared to value for gp120; and † = p < 0.01 compared to value for control and gp120 (ANOVA with Fisher’s PLSD post hoc test).
Neuroprotective β-chemokine CCL4 reduces HIV-1 gp120-induced p38 MAPK activation
Previously, we found that the presence of CCL4, the natural ligand for CCR5, abrogated the neurotoxicity of gp120 in cerebrocortical cells [16,32]. Using the same experimental approach as in the previously reported neurotoxicity studies and the above discussed kinase assay, we found that treatment with CCL4 during exposure to the viral envelope protein suppressed the increase of p38 MAPK activity at 3 h and 24 h but not at 5 min (Fig. 3). This finding suggested that an increased activity of p38 MAPK at 3 and 24 h, but not 5 min, reflected a neurotoxic process. Whereas under protective conditions, such as the presence of the CCR5 ligand, p38 kinase activity remained close to background levels except for the early 5 min time point. Compared to the 1, 3 and 24 h time points, the activity of p38 MAPK at 7 h and 15 h was more variable and on average closer to baseline. That finding was in line with the observations for the same time period made during the experiments shown in Figure 1A. Since CCR5 is present in neuronal and glial cells, the protective ligand CCL4 could possibly have prevented an increase of p38 MAPK activity by interaction with neurons and/or glia [16].
FIGURE 3.

Presence of the neuroprotective chemokine CCL4 reduces the activation of p38 MAPK in the presence of neurotoxic gp120 in rat cerebrocortical cultures at 3 h and 24 h. Note that the activity of p38 MAPK at 7 h and 15 h was more variable and on average closer to baseline as it was to be expected for this time period from the experiments shown in Figure 1A. Neuroprotective CCL4 (20 nM) was added immediately prior to HIV-1 gp120 (200 pM) incubation [15,16]. p38 MAPK activity was detected by immunoblotting and evaluated using densitometry of p-ATF-2 as described in Materials and Methods section. The kinase activity in BSA vehicle-treated controls was measured for each time point and defined as the 100 % baseline value. As a separate control, activity of p38 MAPK for treatment with CCL4 alone was assessed at 5 min, 1 h, 3 h and 24 h only. Error bars represent the S.E.M. of four independent experiments. * = p < 0.05 compared with control.
HIV-1 gp120 activates p38 MAPK predominantly in neurons and microglia, but not in astrocytes
While all cells within the rat cerebrocortical cultures express p38 MAPK protein (Fig. 4), the spatial distribution of p38 MAPK activity induced by exposure to HIV-1 gp120 had not been defined. In order to localize activated p38 MAPK, we performed immunostaining on cultures treated for 24 h with 200 pM HIV-1 gp120 or vehicle control. These cultures were co-stained for active p38 MAPK, the neuronal marker (MAP-2), microglia marker (CD11b), and/or DNA dye Hoechst H33342. Phosphorylated or activated p38 MAPK was primarily found within neurons and microglia at 24 h post-gp120 stimulation within rat cerebrocortical cell cultures (Fig. 4A). As previously shown, these cultures consisted of about 70–75 % astrocytes, 25–30 % neurons and less than 1 % microglia [16]. Therefore, most of the active p38 MAPK within our rat cerebrocortical cultures appeared to reside in neurons. In fact, about 20 % of the cerebrocortical cell cultures consisted of neurons positive for active/phosphorylated p38 MAPK (pp38) with or without gp120 treatment at 5 min (21.3 ± 1.7% vs. 21.1 ± 2.0%), 1 h (21.1 ± 1.6% vs. 20.3 ± 1.8%), 3 h (22.9 ± 2.0% vs. 23.0 ± 1.8%) and 24 h (21.8 ± 2.1% vs. 17.2 ± 1.7%), (Fig. 4B). A reduction in the number of pp38-positive neurons at 24 h compared to control was observed (Fig. 4B), correlating to a loss of neurons after 24 h post-gp120 as seen in Fig. 2. Overall, immunofluorescence staining suggested that at any time between 71–85 % of neurons were pp38-positive regardless of gp120 treatment whereas pp38-positive microglia were detected as early as 10 min into exposure to gp120 but not vehicle control (data not shown).
FIGURE 4.

HIV-1 gp120 activates p38 MAPK in rat cerebrocortical neurons and microglia. Activated p38 MAPK primarily localizes to rat neurons and microglia after treatment with HIV-1 gp120SF162; (A) immunofluorescence staining for p38 MAPK, neuronal MAP-2 and microglial CD11b after 24 h treatment. Rat cerebrocortical cultures were treated with 0.001 % BSA (Control) or 200 pM HIV-1 gp120SF162 (HIV-1 gp120), fixed with 100 % methanol, and then immunolabeled with p-p38 (green), neuronal marker MAP-2 (white), microglia marker CD11b (red) and DNA stain Hoechst H33342 (blue). Virtually the same results were obtained when cerebrocortical cells were treated with gp120SF2 (not shown). Neurons showing activated p38 MAPK (pp38) immunostaining were quantified after 5 min, 1 h, 3 h, and 24 h after treatment with gp120 via microscopy (B). The number of neurons positive for activated p38 MAPK were reduced at 24 h post-gp120 treatment. Cell lysates were analyzed for p38 MAPK activation after removal of neurons from culture with 300 μM NMDA treatment for 20 min two days prior to incubation with 200 pM gp120SF2 for 24 h (G Control or G gp120) using p38 MAPK kinase assay (C). Without neurons, baseline p38 MAPK activity is significantly lower than that from complete cultures; and gp120 treatment does not deviate from control treatment without neurons (C). Cell lysates were also analyzed by immunoblotting after 20 min 300 μM NMDA pre-treatment two days earlier to deplete cultures of neurons or following overnight pre-treatment with 7.5 mM LME to ablate microglia or without pretreatment (control) using anti-active p38 MAPK (pp38), anti-p38 MAPK, anti-synaptophysin, anti-GFAP and anti-actin antibodies (D). Densitometry of basal expression levels of active p38 MAPK/total p38 MAPK in culture after NMDA pre-treatment (Glia Only) or LME pre-treatment (No Microglia) or Control (Neurons + Glia) from Western blot data is shown in (E). Note that equal amounts of total protein were loaded for each sample and thus the Glia-Only sample contains relatively more glial proteins than the other samples. Error bars represent the S.E.M of three independent experiments (C and E). Scale bar in images, 10 μm. For all significance analysis, * = p < 0.05 compared to control.
We also performed p38 MAPK kinase assays on rat cerebrocortical cultures depleted of neurons after 20 min pretreatment with 300 μM N-methyl-D-aspartic acid (NMDA) 2 days prior to exposure to HIV gp120. These experiments showed that neurons contributed 47.6 ± 1.1 % of total protein found within the cerebrocortical cell lysates (n = 30; data not shown). Cultures consisting of neurons and glia (N + G) or neuron-depleted glia-only cultures (G) were assayed for p38 MAPK kinase activity after 5 min, 1 h, 3 h, and 24 h treatments with gp120 (Fig. 4C). Glia-only cultures had a significantly reduced basal p38 MAPK activity (* p < 0.05 compared to N+G Control values) and no difference in p38 MAPK activity between control and gp120-treated cultures was witnessed after depletion of neurons (Fig. 4C).
In fact, immunoblotting experiments indicated that removal of neurons reduced the basal levels of activated p38 MAPK by 77.8 ± 4.5 % compared to control cerebrocortical culture (Neurons + Glia) (Fig. 4D and 4E). Depletion of microglia (LME), on the other hand, did not detectably change the amount of active or total p38 MAPK at basal levels (Fig. 4D and 4E).
Microglial depletion reduces HIV-1 gp120-induced p38 MAPK activation in cerebrocortical neurons
In previous studies, depletion of microglia from cerebrocortical cultures with L-leucine methyl ester (LME) was found to abrogate the neurotoxicity of gp120 [15,16]. Using the same experimental approach as in the previously reported neurotoxicity studies, we found that prior depletion of microglia before exposure to the viral envelope protein also suppressed the increase of p38 MAPK activity at 3 h and 24 h but not at 5 min (Fig. 5). Hence, the increased activity of p38 MAPK in response to HIV-1 gp120 from 3 h onward is dependent on the presence of microglia. Our observation also suggested that the early peak of p38 MAPK activity at 5 min occurred independently of microglia, and thus through a direct interaction of gp120 with astrocytes and/or neurons. Moreover, and similar to the findings in the experiments with CCL4, this peak at 5 min was apparently not associated with neurotoxicity.
FIGURE 5.

Depletion of microglia from mixed rat cerebrocortical cultures reduces the activation of p38 MAPK in the presence of neurotoxic gp120 at 3 h and 24 h. Depletion of microglia was accomplished by overnight pre-treatment with 7.5 mM L-leucine methyl-ester (LME) and was followed by treatment with gp120SF2 for 5 min, 1 h, 3 h, and 24 h and collection of cell lysates for p38 MAPK kinase assay as shown in schematic diagram (A). Kinase assay results were detected by immunoblotting and evaluated using densitometry of p-ATF-2 as described in the legend to Fig. 1. The kinase activity in BSA vehicle-treated controls (with or without LME pre-treatment) was measured for each time point and defined as the 100 % baseline value. Error bars represent the S.E.M. of three to nine independent experiments. * = p < 0.05 compared with control.
Mononuclear cells require p38 MAPK activation for gp120-induced neurotoxin production
In an alternative approach to assess the role of monocytic lineage cells and of HIV-1 coreceptors in the neurotoxicity of gp120, we used human mononuclear THP-1 cells and primary MDM as models for macrophages and microglia. Like primary human MDM, monocytic THP-1 cells express besides CD4, CXCR4 and CCR5, are permissive to HIV infection, and produce neurotoxins in response to HIV-1 virus and gp120 [12,36,40]. In the present study both THP-1 cells and MDM were incubated with 200 pM or 1 nM HIV-1 gp120SF2 for 4 d and then the cell-free supernatant/conditioned media (CM) was transferred into microglia-depleted rat cerebrocortical cultures (at 10 % cerebrocortical culture media volume) for 3 d. HIV-1 gp120 induced neurotoxin production in both THP-1 cells (Fig. 6) and MDM (Fig. 7). THP-1+gp120 CM reduced neuronal survival in an envelope-dose dependent fashion by approximately 40–60 % of rat cerebrocortical cultures exposed to control CM or not treated with CM (Fig. 6A). Control CM did not significantly affect neuronal survival. In contrast, control supernatants from BSA/vehicle-exposed THP-1 cells or control THP-1 CM spiked with HIV gp120 did not significantly increase neuronal death in microglia-depleted cerebrocortical cell cultures.
FIGURE 6.

Inhibition of p38 MAPK in monocytic THP-1 cells abrogates the HIV-1 gp120-induced release of neurotoxic factors. Cell-free conditioned media (CM) from THP-1 cells was collected after treatment for 4 d with 200 pM or 1 nM gp120SF2 or 0.001 % BSA. Microglia-depleted rat cerebrocortical cultures were then treated with control (THP-1 Control) CM or neurotoxic (THP-1 gp120 @ 0.2, or 1 nM) CM for 3 d and analyzed for neuronal survival as shown in (A). Neuronal survival was assessed using fluorescence microscopy of neuronal MAP-2 and nuclear DNA as described in the legend to Fig. 2. After incubation with gp120SF2 or BSA vehicle control for 4 d to generate conditioned media, THP-1 cells were lysed and 25 μg protein of each sample was analyzed by immunoblotting using antibodies against the active and total form of p38 MAPK, respectively, and an antibody against loading control protein actin as indicated (B, upper panel). Following depletion of microglia and exposure to THP-1CM for 1 and 3 d rat cerebrocortical cells were lysed and analyzed by Western blotting in the same way as the THP-1 cells except that GAPDH was used as loading control (B, lower panel). Note the increase of activated, phospho-p38 MAPK in both THP-1 cells after gp120 stimulation and microglia-deficient cerebrocortical cells after exposure to cell-free CM from gp120-exposed THP-1 cells. Introduction of human monocytic THP-1 cells into CCR5−/−/CXCR4−/− mouse cerebrocortical cell cultures confers susceptibility to gp120 neurotoxicity (C). The viral envelope protein fails to induce neurotoxicity in cerebrocortical cells comprising neurons, astrocytes and microglia that lack the chemokine receptors [16]. THP-1 cells were added to the mixed neuronal-glial cell cultures following prior treatment with LME and washes to remove endogenous microglia. Cultures with and without THP-1 cells were incubated for 24 hrs with 200 pM of gp120 from HIV-1SF162 or BSA vehicle as control. Neuronal survival was assessed by fluorescence microscopic quantification of MAP-2+ neurons and nuclear DNA as described above for panel (A). Kinetic of p38 MAPK activation in THP-1 cells exposed to gp120 (D). The monocytic cells were incubated with gp120 (200 pM) for the indicated time periods prior to immunoprecipitation and kinase assay analysis as described in the legend to Fig. 1. (E) THP-1 cells were transduced with a dominant negative inactive p38 MAPK mutant (AdV DNp38αAF) or a GFP control construct (AdV GFP), at a MOI of 10, 2 d prior to inoculation for 4 d with or without 200 pM gp120SF2. Rat cerebrocortical cultures were then exposed for 3 d to a 10 % concentration of THP-1 CM as shown in schematic diagram and assessed for neuronal survival (E). Error bars represent the S.E.M. of three to six independent experiments, * = p < 0.001 (A and E), < 0.04 (C), < 0.05 (D) compared to values for control (ANOVA with Fisher’s post hoc test).
FIGURE 7.

HIV-1 gp120 stimulates secretion of neurotoxins from primary human macrophages. Cell-free conditioned media (CM) from human monocyte-derived macrophages (MDM) was collected after treatment for 1 d (‘1d’) and 4 d (‘4d’) with 200 pM HIV-1 gp120SF2 or gp120SF162 or with 0.001 % BSA. Microglia-depleted rat cerebrocortical cultures were then treated with a 10 % concentration of control (MDM Control) CM or neurotoxic (MDM gp120SF2 or gp120SF162) CM for 3 d and analyzed for neuronal survival (A). Neuronal survival was assessed using fluorescence microscopy of neuronal MAP-2 and nuclear DNA as described in the legend to Figure 2. (B) Equal numbers of MDM in each sample were lysed in SDS-containing gel-loading buffer and protein was analyzed by immunoblotting using antibodies against the active and total form of p38 MAPK, respectively, and an antibody against loading control protein actin as indicated. (C) Inhibition of p38 MAPK activity in primary human MDM abrogates the HIV-1 gp120-induced release of neurotoxic factors. MDM were transduced with dominant negative inactive p38 MAPK mutant (AdV DNp38αAF) or a GFP control construct (AdV GFP), at a MOI of 10, 2 d prior to inoculation for 4 d with or without 200 pM gp120SF2 or gp120SF162. Rat cerebrocortical cultures depleted of microglia were then exposed for 3 d to a 10 % concentration of MDM CM and assessed for neuronal survival. Error bars represent the S.E.M. of three independent experiments, * = p < 0.001 compared to values for control (ANOVA with Fisher’s post hoc test).
Interestingly, THP-1 cells exposed to gp120SF2 also showed increased levels of phosphorylated p38 MAPK in a dose-dependent manner after 4 d, suggesting that activation of the kinase in mononuclear cells correlated with neurotoxin production (Fig. 6B, upper panel). Furthermore, we found that cell-free neurotoxic THP-1 media generated by incubation of the cells with 200 pM gp120 caused in comparison with control CM an increase of active p38 MAPK in microglia-depleted cerebrocortical neuronal cells after 1 and 3 days exposure (Fig. 6B, lower panel).
Since the media conditioned by THP-1 cells and MDM in the presence of gp120 must be expected to carry over viral envelope protein onto microglia-free cerebrocortical cells, it could be possible that gp120 acted directly on neuron and astrocytes contributing to neurotoxicity in synergy with a factor secreted by gp120-stimulated but not control-treated mononuclear cells. In order to assess a potential role for HIV coreceptors in such a hypothetical scenario, we introduced 15,000 monocytic THP-1 cells into 300,000 murine cerebrocortical cells deficient in both HIV coreceptors (CCR5−/−/CXCR4−/−) that were previously pretreated with LME to deplete endogenous microglia (5 % final concentration of THP-1 cells as replacement for microglia). For introduction into murine microglia-free cerebrocortical cells, THP-1 cells were first suspended in conditioned media of the same mouse cell culture at 1.5 × 106 cells/ml. This cell suspension was then distributed at 10 μl/well into the cerebrocortical cells that were previously treated with LME and washed to remove potentially remaining microglial cell debris and excess LME. THP-1 cells, however, did not adhere to cerebrocortical cultures even in the presence of HIV/gp120 (data not shown). Of note, cerebrocortical cells comprising neurons, astrocytes and microglia but lacking both major HIV co-receptors are themselves resistant to induction of gp120 neurotoxicity [16]. The chemokine receptor-deficient cerebrocortical cultures with and without THP-1 cells were then exposed for 24 h to gp120 or BSA as a control. Neuronal survival was determined after fixation, permeabilization and immunolabeling of neurons with MAP-2 in combination with nuclear staining using H33342 by fluorescence microscopy (Fig. 6C). Exposure of cerebrocortical cultures to gp120 in the absence of THP-1 cells, or to THP-1 cells alone (after pretreatment with LME) did not significantly change neuronal survival. However, addition of gp120 in the presence of THP-1 cells caused a significant demise of neurons. This finding suggested that the presence of mononuclear cells expressing CXCR4 and CCR5 (besides CD4) sufficed to restore susceptibility to gp120 toxicity in combined CCR5-CXCR4-deficient cerebrocortical cells and further supported a critical role of monocyte-lineage cells and their HIV coreceptors in gp120-induced neurotoxicity. On the other hand, the findings also showed that an interaction with neuronal or astrocytic HIV coreceptors of any potentially carried-over gp120 in the CM of mononuclear cells was dispensable for neurotoxicity to occur. Finally, this experimental approach revealed that a 24 h incubation of THP-1 cells with HIV gp120 sufficed to produce significant neurotoxicity.
Therefore, we next analyzed whether gp120 triggered in THP-1 cells activation of p38 MAPK during a 24 h exposure. The monocytic cells were exposed for 5, 15 and 30 min, and 24 h to gp120 or BSA vehicle as control and p38 MAPK activity was subsequently assessed in cell lysates using an immunocomplex kinase assay as described above for rat cerebrocortical cells. The kinase assay showed that exposure of monocytic cells to gp120 resulted in a significant increase of active p38 MAPK at all four time points (Fig. 6D).
Similar to THP-1 cells, MDM+gp120 CM reduced neuronal survival to between 70 and 80 % of rat cerebrocortical cultures treated with control MDM CM (Fig. 7A). Of note, MDM+gp120 CM retrieved after 4 days was not significantly more toxic than CM of the same sample collected earlier, after only 24 h incubation. Control MDM CM collected at 24 h and 4 d, or spiked with gp120 before addition to microglia–depleted cerebrocortical cells, and CM from MDM incubated with CCL4 or CCL5 all lacked neurotoxicity thus confirming the specificity of the toxic effect triggered by HIV gp120.
Comparable to THP-1 cells, although less pronounced, MDM exposed to 200 pM gp120 of HIV-1 SF2 and SF162 showed increased levels of phosphorylated p38 MAPK after 4 d, again suggesting that activity of the kinase in mononuclear cells correlated with neurotoxin production (Fig. 7B).
Next, we assessed the importance of p38 MAPK signaling in monocyte lineage cells after gp120 treatment. To analyze whether or not p38 MAPK in mononuclear cells contributes to gp120-induced neurotoxicity, we successfully infected THP-1 cells and MDM each at a multiplicity of infection (MOI) of 10 with adenoviruses expressing GFP (AdV-GFP) or dominant-negative p38αAF MAPK (AdV-DNp38α) [37] 2 d prior to HIV-1 gp120 exposure. The dominant-interfering p38αAF MAPK, but not AdV-GFP, strongly reduced the release of neurotoxins by both THP-1 cells and MDM due to HIV-1 gp120SF2 treatment increasing neuronal survival (Fig. 6E and Fig. 7C, respectively). Reduction of neurotoxin release was due to the presence of the DNp38α kinase mutant and not the result of decreased mononuclear cell viability, as the viability of both the control (AdV-GFP)- and AdV-DNp38α-infected cells, in THP-1 cells and MDM, respectively, were equivalent (data not shown).
Altogether, the experiments showed equally in THP-1 cells and MDM that HIV gp120 triggered neurotoxin production which in turn could be prevented by inhibiting active p38 MAPK and thus supported the suitability of THP-1 cells as a model for macrophage-mediate neurotoxicity of the viral envelope protein. Hence, in order to further investigate the role of endogenous p38 MAPK in gp120-induced neurotoxicity, we utilized small interfering RNA (siRNA) technology to knockdown expression of p38α MAPK in THP-1 cells prior to incubation with HIV-1 gp120SF162. We were able to efficiently reduce expression of endogenous p38 MAPK protein by 50–75 % compared to non-targeting (NT) siRNA (* p < 0.001) using three different and separate p38α MAPK siRNA duplexes, sip38α-1, α-2, and α-3, with or without gp120SF162 treatment (Fig. 8A and 8B). Conditioned media collected from THP-1 cells nucleofected with p38α MAPK siRNA for 48 h (efficient knockdown between 48–72 h, data not shown) and treated with gp120SF162 for an additional 24 h (p38α siRNA:THP-1 gp120SF162 CM) was significantly less neurotoxic than CM from THP-1 cells nucleofected with NT siRNA and treated with gp120SF162 (NT siRNA:THP-1 gp120SF162 CM; Fig. 8C). The incubation with gp120 was limited to 24 h in order to stay within the time frame of maximal reduction of p38 MAPK protein expression in the presence of the specific siRNAs. Importantly, these experiments also confirmed that incubation with gp120 for 24 h was sufficient to obtain significantly neurotoxic conditioned media. Direct neuronal interaction with gp120 did not appear to cause significant amounts of neuronal death in the absence of microglia, as observed after addition of gp120 to microglia-depleted cultures. In contrast, as a control 20 min exposure to 300 μM NMDA prior to 24 hr incubation in control CM was still able to induce excitotoxic neuronal death (Fig. 8C).
FIGURE 8.

Reduction of endogenous p38 MAPK expression with siRNA in monocytic THP-1 cells prevents induction of neurotoxicity upon exposure to gp120. Three separate small interfering RNAs (siRNA) sequences of p38α MAPK (MAPK14) (sip38α-1, α-2, α-3) were utilized to knockdown expression of p38 MAPK by 50–75 % compared to non-targeting (NT) siRNA in THP-1 cells after 48 h via Amaxa nucleofection techniques, followed by subsequent 200 pM gp120SF162 24 h treatment (A and B). Representative western blots of THP-1cell lysates treated 24 h with or without 200 pM gp120SF162 beginning 48 h after siRNA nucleofection (-, NT or non-targeting; or +, sip38α-1); p38 MAPK, CCR5, CXCR4, Akt, JNK1/2, and GAPDH protein levels are shown (A). Densitometry analysis of cell lysates from three independent siRNA experiments showed a significant reduction in protein amount only for p38 MAPK (B) but no significant change for CCR5 or any other tested protein. Cell-free conditioned media (CM) from THP-1 cells was collected 2 d after nucleofection of siRNA (NT or sip38α-1, α-2, α-3) followed by treatment for 24 h with 200 pM gp120SF162 or 0.001 % BSA (vehicle control). Microglia-depleted rat cerebrocortical cultures then received the THP-1 CM for 3 d and subsequently were analyzed for neuronal survival (C). Neuronal survival was assessed via microscopic quantification of neuronal MAP-2 and nuclear DNA and compared using NTsiRNA:THP-1 Control CM-treated cultures as overall control condition (C). Direct treatment of microglia-depleted cerebrocortical cultures with gp120 did not produce significant neuronal death (NT siRNA : THP-1 Control CM + gp120), however direct NMDA treatment (NT siRNA : THP-1 Control CM + NMDA; 300 μM, 20 min) showed unimpaired neuronal response to excitotoxic insult (C). Error bars represent the S.E.M. of three independent experiments, * = p < 0.001 compared to values for NT siRNA controls (ANOVA with Fisher’s post hoc test).
Inhibition of p38 MAPK activation in cerebrocortical neurons suffices to prevent neuronal death stimulated by gp120-induced neurotoxic media from mononuclear THP-1 cells
We next tested whether inhibition of p38 MAPK activity in neurons would suffice to prevent neuronal death upon neurotoxic stimuli from THP-1 cells treated with HIV-1 gp120. Therefore, prior to treatment of microglia-depleted rat cerebrocortical cultures with gp120-induced neurotoxic THP-1 conditioned media, we pretreated the cerebrocortical cultures with 10 μM SB203580 or DMSO as vehicle control for 15 min [15,16]. As before, THP-1 cells were separately incubated with HIV-1 gp120SF2 or gp120SF162 for 4 d in order to obtain neurotoxic and non-toxic control supernatants. The THP-1 conditioned media (CM) was then transferred onto SB203580-pretreated microglia-depleted rat cerebrocortical cultures (at 10 % cerebrocortical culture media volume) for 3 d. Interestingly, inhibition of p38 MAPK by 10 μM SB203580 in microglia-depleted rat cerebrocortical cultures prior to the addition of gp120-induced neurotoxic THP-1 CM prevented neuronal apoptotic death (Fig. 9) indicating that p38 MAPK is a crucial mediator not only in toxin-producing macrophages/microglia but also in toxin-sensitive neurons. Again, spiking non-toxic THP-1 CM with additional 200 pM gp120SF2 or gp120SF162 before application to microglia-depleted rat cerebrocortical cultures failed to produce neurotoxicity; confirming the requirement of microglia for gp120-induced neuronal death (Fig. 9A, THP-1CM control + SF2 or SF162).
FIGURE 9.

Inhibition of p38 MAPK in microglia-depleted rat cerebrocortical cultures rescues neurons from gp120-activated THP-1 neurotoxicity. Cell-free conditioned media (CM) from THP-1 was collected after treatment for 4 d with gp120SF2, gp120SF162 or 0.001 % BSA. Microglia-depleted rat cerebrocortical cultures were then pre-treated with 10 μM SB203580 for 15 min prior to treatment with control or neurotoxic THP-1 CM for 3 d as shown in schematic diagram and analyzed for neuronal survival (A). Neuronal survival was assessed using quantification of apoptotic, TUNEL+ nuclei (green), neuronal MAP-2 immunostaining (red), and nuclear DNA (blue); representative examples of images used for quantification are shown (B). Direct treatment of microglia-depleted cerebrocortical cultures with gp120 did not produce significant neuronal death (THP-1Control CM + SF2 or SF162) (A). Error bars represent the S.E.M. of three independent experiments, * = p < 0.05 (ANOVA with Fisher’s post hoc test).
Discussion
The interaction of HIV gp120 with CD4 and the viral coreceptors, CCR5 and/or CXCR4, has been shown to be vital to productive infection and to stimulation of intracellular signaling cascades presumably responsible for most pathological effects of viral-host interactions [3,10,14–17,19,20,24–26,32,41,42]. Previously, we and others showed that the stress-related p38 MAPK can apparently mediate gp120-induced neuronal death [10,15,16]. Furthermore, activation of p38 MAPK has been observed following gp120 treatment in macrophages and thus implicated in HIV pathogenesis and the development of AIDS [25,43]. However, the present study supplies evidence that p38 MAPK and its activity are essential to not only the reduction of neuronal survival by HIV-1 gp120, but also the neurotoxic phenotype of gp120-exposed microglia and macrophages. In addition, both consequences of exposure to HIV-1 gp120, the overall increased activation of p38 MAPK in neurons and neuronal death, are dependent on the presence of microglia. Therefore, this is the first study to show that p38 MAPK in two different cell types may be culpable and necessary for gp120-induced neurotoxicity in a mixed glial-neuronal environment.
Activation of p38 MAPK above baseline by HIV envelope protein gp120 has previously been reported for striatal neurons for a time frame of up to 4 h of exposure [10]. However, our results presented here show that p38 MAPK activity fluctuates at earlier time points after a short-lived initial rise, and after a decrease at 1 h displays an overall increasing trend over a 24 h time course. This difference may be due to the fact that Singh et al. [10] used striatal neuron-rich cultures as their experimental model and treated with more than twice the amount of gp120 (500 pM) than applied in our study (200 pM) [15,16,32]. On the other hand, the early increase of p38 MAPK activation at 5 min gp120 exposure was represented in both studies. This early peak in p38 MAPK activity does not correlate with neuronal death in our study as it was not abolished by pre-treatment with CCL4 or by microglial depletion from mixed cultures, which has been shown in our studies to inhibit gp120-induced reduction in neuronal survival [15,16]. The initial peak of p38 MAPK activity at 5 min due to CCL4 alone or gp120 alone or CCL4+gp120 is most likely due to neuronal activation since it is not abolished after microglial depletion but absent in neuron-deficient cultures exposed to gp120. The question of why the temporary rise of p38 MAPK activity at 5 min is apparently not associated with neurotoxicity and the underlying question of how the rise in kinase activity at 5 min is different from the rise between 1 and 3 h and up to 24 h remains unknown at this time and is subject of ongoing studies. One possible explanation for the gradual up and down of p38 MAPK activity in surviving neurons may be a yet to be defined mechanism leading to a disturbance of the regulatory balance between phosphorylation and de-phosphorylation by phosphatases. However, CCL4 has previously been shown to immediately activate p38 MAPK in C6 glioblastoma cell lines [44] and this was also the case in our neuronal cultures (Fig. 3). We were also able to show that CCL4-stimulated p38 MAPK activity diminished to basal levels within 1 h exposure and stayed similar to control up to 24 h. Thus, our study is the first to show CCL4-induced p38 MAPK activity in neuronal cultures over a 24 h time period and how it is different from gp120-induced p38 MAPK activity. The question of why CCL4 was unable to block gp120-induced p38 MAPK at 5 min when incubated together also remains to be investigated, although we have previously shown that CCL4 can prevent neuronal death caused by the viral envelope [16]. That finding could be explained by direct competition at the receptor level and, in the case of CXCR4-prefering gp120, via heterologous desensitization, which we demonstrated in the same study. Both mechanisms may occur at the level of macrophage chemokine receptors, which is further supported by our present study.
Other related questions for future investigations are first why CCL4, in contrast to gp120, fails to produce a lasting increase in active p38 MAPK, and second, how a CCR5-binding viral envelope, but not the β-chemokine, can cause neurotoxicity. While our present study cannot provide the answers to those two questions, CCL4 and gp120 represent different ligands for the same receptor, and as such may elicit different or even opposing effects. Importantly, gp120 binds with greater affinity to chemokine receptors if CD4 is also present (as in macrophages/microglia and lymphocytes) and previous studies by other groups have shown that even different natural ligands for the same receptor, such as CCR5 and its ligands CCL3, -4, -5, -8 and -14, can have different activities and trigger diverse signaling within the same cell population [45,46].
Although p38 MAPK activation is known to be important for gp120-induced neuronal death [10,15,16], the distribution of phosphorylated p38 MAPK had not been investigated. Our report shows that phosphorylated p38 MAPK is localized in both neurons and microglia in mixed cerebrocortical cell cultures. Importantly, based on detection by immunofluorescence, active p38 MAPK in microglia occurs at early time points upon treatment with gp120 and may be sustained over 24 h, while neuronal p38 MAPK activation appears to occur downstream of microglial kinase activation after longer exposure to gp120 or possible neurotoxins from activated microglia. The number of neurons counted positive for phosphorylated p38 MAPK staining does not appear to be different from control at 5 min, 1 h and 3 h, but is significantly reduced at 24 h, in line with reduction in neuronal survival at 24 h. However, besides counting of cells positive for phosphorylated p38 MAPK a quantitative readout for p38 MAPK activity between control and gp120 treatments is provided by our kinase assay results. Baseline activity of p38 MAPK appeared to mostly reside within neurons as shown by the low remaining activity in glial cells after deletion of the neuronal population, which made up about 30 % of the total cell population in the rat cerebrocortical cultures. Furthermore, our results suggest that the majority of the gp120-induced p38 MAPK activity originates in the neuronal cell population. In addition, the combination of immunofluorescence detection and immunocomplex kinase assay indicated that the increase or decrease of p38 MAPK activity during the first 3 h was not based on a significant change in the number of cells positive for active kinase but rather reflected a change of p38 MAPK activity within a given number of cells. Therefore, neurons surviving 24 h after gp120 exposure appear to represent a smaller cell population that either is still in the process of dying or tolerates a higher p38 MAPK activity than that occurring under the respective control conditions. Given the fact that inhibition of p38 MAPK activity was protective, the variable measurements of kinase activity between 3 h and 24 h presumably reflect a temporary deactivation of the active kinase by phosphatases as well as the death and consequent drop-out of neurons that previously contributed to an overall increasing kinase activity in the cell culture as a whole. An additional and not mutually exclusive explanation is that primary, mixed neuronal-glial cell cultures do not grow in a synchronized fashion as for example cell lines do, and therefore different neurons in a given culture may respond to neurotoxins and eventually enter into a cell death pathway (apoptosis) at different times. Thus, the fluctuations of p38 MAPK activity may result from the dynamic interplay of activation and inactivation of the kinase as well as the gradual demise of neurons.
We also performed Western blotting experiments to address the question of active p38 MAPK in cerebrocortical neurons exposed to conditioned media from THP-1 cells (Fig. 6). We detected increased p38 MAPK activity in the microglia-depleted cerebrocortical cultures at 24 h and 3 d after treatment with neurotoxic THP-1 conditioned media. Of note, the results shown in Figs. 6 and 8 indicated that at 24 h the demise of a significant part of neurons has already occurred. Thus, our findings suggested that the remaining, surviving neurons (and astrocytes) may indeed have adapted, at least for the time of the experiment, to an increased p38 MAPK activity. The possibility of such an adaptation in surviving neurons is a novel hypothesis that warrants future investigations.
Other groups have shown that gp120 triggers p38 MAPK activation in macrophages and monocytes, which can enter the brain soon after HIV infection and presumably contribute to the development of HAND [25,43]. In concurrence with these previous studies, gp120 elicited p38 MAPK activation in rat microglia and in our models for microglia, human monocytic THP-1 cells and primary MDM. These mononuclear cells produced significant neurotoxicity upon treatment with gp120 for 24 h to 4 d. Activation of p38 MAPK by HIV-1 gp120 in these phagocytic cells must occur before the activation of the same kinase increases in neurons as evidenced by the kinetic of p38 MAPK in gp120-treated monocytic THP-1 cells and the reduction of neuronal death [15,16] and decrease in kinase activity signal in cerebrocortical cells after microglial depletion. In addition, suppressing p38 MAPK activity in monocytic THP-1 cells and primary MDM via the dominant-negative, inactive p38 MAPK mutant caused a significant reduction in neurotoxicity. Thus, the dominant-negative inactive p38α MAPK mutant suggests a critical role of p38 MAPK signaling in macrophages and microglia for gp120-induced neurotoxicity. Furthermore, this finding was consistent with our observation of p38 MAPK-dependent gp120 neurotoxicity in the mixed rat cerebrocortical cultures that were obtained using the same adenoviral constructs [15,16] and various dosages of the pharmacological inhibitor SB203580 (Fig. 2).
In addition, the similarity of the results obtained in this study with primary human MDM and THP-1 cells, strongly supports the notion that the latter cell type is a very suitable model of microglia and macrophages. Therefore, additional experiments were performed using monocytic THP-1 cells. In particular, knockdown of endogenous p38α MAPK via three separate siRNA duplexes all derived from exon 2, similar to studies using a macrophage-specific deletion of p38αMAPK in vivo [47], significantly and specifically reduced expression of the protein by 50–75 % in mononuclear THP-1 cells. The α isoform of p38 MAPK is thought to be the most important isoform involved in inflammatory regulation [47,48] and a possible therapeutic target for CNS disorders beyond HAND and HIV-associated dementia, such as Alzheimer’s disease [49]. Importantly, in our hands knockdown of p38α MAPK by 50–75 % sufficed to abrogate gp120-induced neurotoxicity similar to the significant reduction seen with over-expression of the dominant-negative mutant of the kinase; supporting the theory that p38 MAPK is vital for macrophage-mediated neurotoxicity of HIV gp120, but does not necessarily involve all p38 MAPK molecules available in the cell. This interpretation is in line with our observation that besides partial depletion of p38 MAPK protein, lower concentrations of the p38 MAPK pharmacological inhibitor SB203580 than what we previously assessed [15,16,32] can both sufficiently and specifically suppress HIV-1 gp120-induced neurotoxicity.
In our studies using mixed neuronal-glial cell cultures, direct neuronal interaction with gp120 did not appear to cause significant amounts of neuronal death in the absence of microglia, as observed when gp120 was added to microglia-deficient cultures directly or via spiking of control THP-1 supernatants. However, it could potentially be possible that gp120 acted directly on neuron and astrocytes contributing to neurotoxicity in synergy with a factor secreted by gp120-stimulated but not control-treated mononuclear cells. Therefore, we performed neurotoxicity assays in mouse cerebrocortical cultures deficient in both HIV coreceptors (double CCR5/CXCR4 knockouts) which are even in the presence of endogenous microglia themselves resistant to gp120-induced neurotoxicity [16]. Therefore, we replaced endogenous microglia with monocytic THP-1 cells in order to selectively provide monocytic cells possessing HIV-coreceptors. The results showed that without HIVgp120 co-receptors present in neurons and astrocytes, gp120-treated THP-1 cells (and their supernatants) remained neurotoxic. Hence, the presence of mononuclear cells expressing CXCR4 and CCR5 (besides CD4) sufficed to restore susceptibility to gp120 toxicity in combined CCR5-CXCR4-deficient cerebrocortical cells, and a direct interaction of gp120 with neurons or astrocytes, at least via chemokine receptors, was dispensable for induction of neuronal death.
On the other hand, inhibition of p38 MAPK in rat cerebrocortical neurons and astrocytes prior to the addition of gp120-induced mononuclear cell-derived neurotoxins prevented neuronal apoptosis as shown in Fig. 9. This data suggested that inhibition of stress-related signaling via p38 MAPK, at the level of neurons (and possibly astrocytes) also succeeded in protecting against gp120-induced macrophage/microglia neurotoxicity and thus revealed a potential mechanism to rescue mature neurons from HIV gp120-induced injury and death even if neurotoxins are present. However, our findings also emphasize the potential of limiting or abrogating the upstream involvement of macrophages/microglia in HIV-1 gp120 neurotoxicity. Therefore, further investigation into the mechanism of macrophage/microglia-mediated gp120 neurotoxicity is warranted.
Importantly, our results revealed that p38 MAPK provides a causal link in the neurotoxic mechanism triggered by HIV-1 gp120 in two disparate cell types similar to the behavior of the transcription factor p53 as previously shown [21]. This comparable requirement for p38 MAPK and p53 warrants further investigation since it has been reported that direct phosphorylation of p53 by p38 MAPK is a critical step in the induction of apoptosis in HIV-1/gp120 induced syncytia [27].
Although p38 MAPK activity in both cell types is important for the overall neurotoxic effect of HIV-1 gp120 and β-chemokines and pharmacological inhibitors of p38 MAPK affect neuronal and glial cells, the blockade of p38 MAPK activity in microglia or neurons, respectively, suffices to ameliorate toxicity of gp120. Thus our finding that blockade of the kinase in macrophages and microglia, the primary productively HIV-infected cell type in the brain, suffices to prevent the indirect neurotoxicity of the viral envelope protein may offer new cell-type specific therapeutic strategies for protection of the CNS against HIV-associated injury and functional impairment.
Acknowledgments
We thank Dr. Jiahuai Han, The Scripps Research Institute, La Jolla, CA for adenoviral vectors, Dr. Stuart Lipton (Sanford-Burnham Medical Research Institute) for support during the initial phase of the project, and Dr. Pedro Aza-Blanc (Sanford-Burnham Medical Research Institute) for advice on the selection of siRNAs. We also thank Rebecca Ruf and Cyrus De Rozieres for technical assistance in Western blotting and tissue culture and Cari Cox and Irene Catalan for help with tissue culture and image analysis.
Abbreviations used in this paper
- HAND
HIV-1 associated neurocognitive disorders
- HAD
HIV-associated dementia
- MND
HIV-1-associated mild neurocognitive disorder
- ANI
HIV-1-associated asymptomatic neurocognitive impairment
- MDM
monocyte-derived macrophages
Footnotes
This work was supported by National Institutes of Health R01 Grants NS050621 and MH087332 (M.K.).
Reference List
- 1.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–1799. doi: 10.1212/01.WNL.0000287431.88658.8b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 3.Porcheray F, Samah B, Leone C, Dereuddre-Bosquet N, Gras G. Macrophage activation and human immunodeficiency virus infection: HIV replication directs macrophages towards a pro-inflammatory phenotype while previous activation modulates macrophage susceptibility to infection and viral production. Virology. 2006;349:112–120. doi: 10.1016/j.virol.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi: 10.1038/nri1527. [DOI] [PubMed] [Google Scholar]
- 5.Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- 6.Masliah E, Ge N, Achim CL, Hansen LA, Wiley CA. Selective neuronal vulnerability in HIV encephalitis. J Neuropathol Exp Neurol. 1992;51:585–593. doi: 10.1097/00005072-199211000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- 8.Glass JD, Fedor H, Wesselingh SL, McArthur JC. Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol. 1995;38:755–762. doi: 10.1002/ana.410380510. [DOI] [PubMed] [Google Scholar]
- 9.Adle-Biassette H, Chretien F, Wingertsmann L, Hery C, Ereau T, Scaravilli F, Tardieu M, Gray F. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–133. doi: 10.1046/j.1365-2990.1999.00167.x. [DOI] [PubMed] [Google Scholar]
- 10.Singh IN, El-Hage N, Campbell ME, Lutz SE, Knapp PE, Nath A, Hauser KF. Differential involvement of p38 and JNK MAP kinases in HIV-1 Tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience. 2005;135:781–790. doi: 10.1016/j.neuroscience.2005.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi B, De GU, He J, Wang S, Lorenzo A, Busciglio J, Gabuzda D. Apoptosis induced by HIV-1 infection of the central nervous system. J Clin Invest. 1996;98:1979–1990. doi: 10.1172/JCI119002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giulian D, Wendt E, Vaca K, Noonan CA. The envelope glycoprotein of human immunodeficiency virus type 1 stimulates release of neurotoxins from monocytes. Proc Natl Acad Sci U S A. 1993;90:2769–2773. doi: 10.1073/pnas.90.7.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 14.Meucci O, Fatatis A, Simen AA, Bushell TJ, Gray PW, Miller RJ. Chemokines regulate hippocampal neuronal signaling and gp120 neurotoxicity. Proc Natl Acad Sci U S A. 1998;95:14500–14505. doi: 10.1073/pnas.95.24.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaul M, Ma Q, Medders KE, Desai MK, Lipton SA. HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 2007;14:296–305. doi: 10.1038/sj.cdd.4402006. [DOI] [PubMed] [Google Scholar]
- 17.Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL, Horuk R. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1 alpha is mediated by the chemokine receptor CXCR4. Curr Biol. 1998;8:595–598. doi: 10.1016/s0960-9822(98)70230-1. [DOI] [PubMed] [Google Scholar]
- 18.Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL, Taub D, Horuk R. CD4-independent association between HIV-1 gp120 and CXCR4: functional chemokine receptors are expressed in human neurons. Curr Biol. 1997;7:112–121. doi: 10.1016/s0960-9822(06)00055-8. [DOI] [PubMed] [Google Scholar]
- 19.O’Donnell LA, Agrawal A, Jordan-Sciutto KL, Dichter MA, Lynch DR, Kolson DL. Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes. J Neurosci. 2006;26:981–990. doi: 10.1523/JNEUROSCI.4617-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung R, Ravyn V, Wang L, Ptasznik A, Collman RG. Signaling mechanism of HIV-1 gp120 and virion-induced IL-1beta release in primary human macrophages. J Immunol. 2008;180:6675–6684. doi: 10.4049/jimmunol.180.10.6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garden GA, Guo W, Jayadev S, Tun C, Balcaitis S, Choi J, Montine TJ, Moller T, Morrison RS. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004;18:1141–1143. doi: 10.1096/fj.04-1676fje. [DOI] [PubMed] [Google Scholar]
- 22.Cocchi F, Devico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV- suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 23.Kerr SJ, Armati PJ, Pemberton LA, Smythe G, Tattam B, Brew BJ. Kynurenine pathway inhibition reduces neurotoxicity of HIV-1- infected macrophages. Neurology. 1997;49:1671–1681. doi: 10.1212/wnl.49.6.1671. [DOI] [PubMed] [Google Scholar]
- 24.Zheng J, Ghorpade A, Niemann D, Cotter RL, Thylin MR, Epstein L, Swartz JM, Shepard RB, Liu X, Nukuna A, Gendelman HE. Lymphotropic virions affect chemokine receptor-mediated neural signaling and apoptosis: implications for human immunodeficiency virus type 1-associated dementia. J Virol. 1999;73:8256–8267. doi: 10.1128/jvi.73.10.8256-8267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Del Corno M, Liu QH, Schols D, De Clercq E, Gessani S, Freedman BD, Collman RG. HIV-1 gp120 and chemokine activation of Pyk2 and mitogen-activated protein kinases in primary macrophages mediated by calcium-dependent, pertussis toxin-insensitive chemokine receptor signaling. Blood. 2001;98:2909–2916. doi: 10.1182/blood.v98.10.2909. [DOI] [PubMed] [Google Scholar]
- 26.Fantuzzi L, Spadaro F, Purificato C, Cecchetti S, Podo F, Belardelli F, Gessani S, Ramoni C. Phosphatidylcholine-specific phospholipase C activation is required for CCR5-dependent, NF-kB-driven CCL2 secretion elicited in response to HIV-1 gp120 in human primary macrophages. Blood. 2008;111:3355–3363. doi: 10.1182/blood-2007-08-104901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perfettini JL, Castedo M, Nardacci R, Ciccosanti F, Boya P, Roumier T, Larochette N, Piacentini M, Kroemer G. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J Exp Med. 2005;201:279–289. doi: 10.1084/jem.20041502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ono K, Han J. The p38 signal transduction pathway: activation and function. Cell Signal. 2000;12:1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 29.Johnson GV, Bailey CD. The p38 MAP kinase signaling pathway in Alzheimer’s disease. Exp Neurol. 2003;183:263–268. doi: 10.1016/s0014-4886(03)00268-1. [DOI] [PubMed] [Google Scholar]
- 30.Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ, Erhardt JA, Ohlstein EH, Hunter AJ, Harrison DC, Philpott K, Smith BR, Adams JL, Parsons AA. Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev. 2001;21:129–145. doi: 10.1002/1098-1128(200103)21:2<129::aid-med1003>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 31.Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164:355–362. doi: 10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi WT, Kaul M, Kumar S, Wang J, Kumar IM, Dong CZ, An J, Lipton SA, Huang Z. Neuronal apoptotic signaling pathways probed and intervened by synthetically and modularly modified (SMM) chemokines. J Biol Chem. 2007;282:7154–7163. doi: 10.1074/jbc.M611599200. [DOI] [PubMed] [Google Scholar]
- 33.Kaul M, Loos M. Collagen-like complement component C1q is a membrane protein of human monocyte-derived macrophages that mediates endocytosis. J Immunol. 1995;155:5795–5802. [PubMed] [Google Scholar]
- 34.Terheyden P, Loos M, Storkel S, Kaul M. Human macrophages simultaneously express membrane-C1q and Fc-receptors for IgG. Immunol Lett. 2005;101:202–209. doi: 10.1016/j.imlet.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 35.Kaplan G, Gaudernack G. In vitro differentiation of human monocytes Differences in monocyte phenotypes induced by cultivation on glass or on collagen. J Exp Med. 1982;156:1101–1114. doi: 10.1084/jem.156.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science. 1990;250:1593–1596. doi: 10.1126/science.2148832. [DOI] [PubMed] [Google Scholar]
- 37.Zhao M, New L, Kravchenko VV, Kato Y, Gram H, Di Padova F, Olson EN, Ulevitch RJ, Han J. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999;19:21–30. doi: 10.1128/mcb.19.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JC, Kassis S, Kumar S, Badger A, Adams JL. p38 mitogen-activated protein kinase inhibitors--mechanisms and therapeutic potentials. Pharmacol Ther. 1999;82:389–397. doi: 10.1016/s0163-7258(99)00008-x. [DOI] [PubMed] [Google Scholar]
- 39.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, Mackay CR, Sodroski J, Gabuzda D. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature. 1997;385:645–649. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- 41.Ohagen A, Ghosh S, He J, Huang K, Chen Y, Yuan M, Osathanondh R, Gartner S, Shi B, Shaw G, Gabuzda D. Apoptosis induced by infection of primary brain cultures with diverse human immunodeficiency virus type 1 isolates: evidence for a role of the envelope. J Virol. 1999;73:897–906. doi: 10.1128/jvi.73.2.897-906.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kitai R, Zhao ML, Zhang N, Hua LL, Lee SC. Role of MIP-1beta and RANTES in HIV-1 infection of microglia: inhibition of infection and induction by IFNbeta. J Neuroimmunol. 2000;110:230–239. doi: 10.1016/s0165-5728(00)00315-5. [DOI] [PubMed] [Google Scholar]
- 43.Lee C, Liu QH, Tomkowicz B, Yi Y, Freedman BD, Collman RG. Macrophage activation through CCR5- and CXCR4-mediated gp120-elicited signaling pathways. J Leukoc Biol. 2003;74:676–682. doi: 10.1189/jlb.0503206. [DOI] [PubMed] [Google Scholar]
- 44.Misse D, Esteve PO, Renneboog B, Vidal M, Cerutti M, St Pierre Y, Yssel H, Parmentier M, Veas F. HIV-1 glycoprotein 120 induces the MMP-9 cytopathogenic factor production that is abolished by inhibition of the p38 mitogen-activated protein kinase signaling pathway. Blood. 2001;98:541–547. doi: 10.1182/blood.v98.3.541. [DOI] [PubMed] [Google Scholar]
- 45.Mueller A, Mahmoud NG, Goedecke MC, McKeating JA, Strange PG. Pharmacological characterization of the chemokine receptor, CCR5. Br J Pharmacol. 2002;135:1033–1043. doi: 10.1038/sj.bjp.0704540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oppermann M. Chemokine receptor CCR5: insights into structure, function, and regulation. Cell Signal. 2004;16:1201–1210. doi: 10.1016/j.cellsig.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, Lawrence T, Otsu K, Brissette JL, Arthur JS, Park JM. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008;9:1019–1027. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang YJ, Chen J, Otsuka M, Mols J, Ren S, Wang Y, Han J. Macrophage deletion of p38alpha partially impairs lipopolysaccharide-induced cellular activation. J Immunol. 2008;180:5075–5082. doi: 10.4049/jimmunol.180.7.5075. [DOI] [PubMed] [Google Scholar]
- 49.Munoz L, Ranaivo HR, Roy SM, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterson DM. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J Neuroinflammation. 2007;4:21. doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]