

Abstract
Cell behavior on 2-D in vitro cultures is continually being improved to better mimic in vivo physiological conditions by combining niche cues including multiple cell types and substrate stiffness, which are well known to impact cell phenotype. However, no system exists in which a user can systematically examine cell behavior on a substrate with a specific stiffness (elastic modulus) in culture with a different cell type, while maintaining distinct cell populations. We demonstrate the modification of a silicon reconfigurable co-culture system with a covalently linked hydrogel of user-defined stiffness. This device allows the user to control whether two separate cell populations are in contact with each other or only experience paracrine interactions on substrates of controllable stiffness. To illustrate the utility of this device, we examined the role of substrate stiffness combined with myoblast co-culture on adipose derived stem cell (ASC) differentiation and found that the presence of myoblasts and a 10 kPa substrate stiffness increased ASC myogenesis versus co-culture on stiff substrates. As this example highlights, this technology better controls the in vitro microenvironment, allowing the user to develop a more thorough understanding of the combined effects of cell-cell and cell-matrix interactions.
Introduction
The microenvironment that surrounds cells in vivo consists of growth factors, interstitial fluid, and the extracellular matrix (ECM), a protein based scaffold that surrounds and supports cells. These components of the microenvironment provide cells with multiple types of signals that regulate intracellular signaling leading to a change in cell behavior or fate. These variables include but are not limited to paracrine/autocrine signals, cell-cell physical interactions, and ECM stiffness/composition. In the last decade, scientists have demonstrated that it is not accurate to mimic in vivo cell behavior using in vitro substrates which regulate just one of these variables. This is particularly true when attempting to mirror complex processes such as tumorigenesis, morphogenesis, stem cell differentiation and wound healing. 1-6 Embryonic, adult, and induced pluripotent stem cells have all been shown to differentiate based on substrate stiffness and composition and/or cell-cell interactions in vitro,7-10 which further emphasizes the importance of developing technologies that introduce diverse sets of variables for cell culture. In addition to more closely mimicking the microenvironment, these technologies must be easily fabricated, allow for simple readouts such as isolation of mRNA and protein, include the potential to image cell morphology and immunofluorescence (IF), and allow for facile co-culture of different cell types in a controlled manner.
Co-culture is commonly performed when studying cell-cell interaction in vitro. Various co-culture technologies have been developed to monitor contact, paracrine, and autocrine effects. A number of devices use microfabrication, microfluidics, and flow chambers to study cell-cell interactions.11-13 Others pattern cells in specific geometric configurations, use different ECM proteins, or utilize various substrate materials to regulate cell attachment and subsequent cell-cell interaction.1, 2, 14-17 However, most of these approaches require single cell co-culture or low cell density in a complex, microfabricated device that is not always user-friendly.13 Some of these devices cannot use specific proteins and require differential plating of different cell types to achieve a micropatterned co-culture.2, 18 Biochemical readouts can be difficult to produce because of minimal cell material from small cell numbers or involve complex interpretations from the presence of multiple cell populations that are not separable.
Previously, a reconfigurable co-culture device was designed as a tool that integrates the advantages of traditional cell culture, transwell assays, and conditioned media and yet allows organized monitoring of cell interactions.19, 20 The device is composed of two “comb pairs” containing cells that can be paired in either a contact or an 80 μm gap configuration such that the user can systematically understand whether contact or paracrine cues induce change in cell behavior or fate.19 The reconfigurable co-culture device has been shown to be a powerful tool to understand how cell interactions via direct contact or paracrine signaling causes changes in cell behavior.19-21 However, this device requires users to culture cells directly on the hard silicon dioxide comb surface (gigaPascal; gPa), which does not recapitulate the softer microenvironment seen in most tissues in vivo (kiloPascal; kPa). Not only does this stiffness not mimic the in vivo environment, but also recent evidence from a number of groups has demonstrated that substrate stiffness plays an important role in regulating cell behavior, including stem cell differentiation.8, 22-26 Thus far, no tool has been engineered to reproducibly and easily combine substrate stiffness and co-culture to induce changes in a particular cell fate. In order to advance the understanding of cell behavior and better model in vivo environments in 2-D co-culture, we modified the reconfigurable co-culture device with an ECM protein-functionalized hydrogel of tunable stiffness. In this way, the user can monitor cell-cell interactions combined with any permutation of stiffness and ECM protein type. Groups have shown independently that myogenic stiffness (e.g. 10 kPa) and murine myoblast presence can induce myogenic differentiation of ASCs. 10, 26, 27 By using this new tool, we demonstrate that controlled myoblast co-culture on a substrate of myogenic stiffness can cause a synergistic upregulation in myogenic gene expression compared to either method alone.
Results
Gel Polymerization and Characterization of the Surface of the Device
Polyacrylamide (PA) gels have been previously functionalized to borosilicate glass coverslips and slides to control substrate stiffness.8, 17, 26, 28 These protocols were modified specifically for the co-culture device to ensure that complete coverage of each comb finger was achieved. Combs were cleaned, plasma treated with ultraviolet-ozone, and methacrylate functionalized. An azo-photoinitator was used to allow for temporal control of the PA polymerization. UV initiated acrylamide polymerization allows the solution to be carefully placed on the device and to evenly cover the surface of the fingers before the polymerization occurs.
To promote even and consistent polymerization, combs were pre-fit to ensure that there were no gaps in the comb fingers when in contact mode to prevent leakage of the acrylamide solution. Additionally, the combs were placed on dimethyldichlorosilane (DCDMS) coated microscope slides to prevent sticking of residual PA gel to the glass and to easily remove the polymerized comb from the glass surface (Fig. 1a). The solution was carefully added to the center of the comb-pair interface, and a DCDMS coated coverslip was placed on top of the liquid. The liquid spread via surface tension provided by the glass coverslip on the comb (Fig. 1b). Once the solution was applied evenly, the combs were placed (with minimal movement) under a 305 nM UV lamp for 10 minutes. Afterwards, the coverslip was carefully peeled off the comb. Comb pairs were separated from each other with the PA gel in a dehydrated state so that the gel features would be maintained on the comb fingers before swelling in PBS. Once in PBS, several rinses were performed to eliminate any free monomer. Proteins were then attached to the hydrogel surface via N-Sulfosuccinimidyl-6-[4′-azido-2′-nitrophenylamino] hexanoate (sulfo-SANPAH), which reacts with primary amines on the protein and links them to the PA gel. The overall process from comb preparation to cell seeding is depicted in Fig. 1c. All configurations of the device (separated, gap, and contact) are shown in Fig. 1d-f.
Figure 1. Setup and scheme of device modification.

(a) Three cleaned and methacrylate functionalized combs are placed into contact onto a DCDMS coated glass slide to demonstrate the scale of the device. (b) The overall scheme of the device begins with pirhana cleaning and UVO treatment to first activate the device surface. Next, methacrylate groups are functionalized on the surface in order to covalently link polyacrylamide to the surface. The polyacrylamide solution consists of acrylamide monomer, bisacrylamide crosslinker, water, and the azo-photoinitiatior. These are degassed, well mixed and added to the comb as shown in (c). 7.5 μL of polyacrylamide (PA) solution is placed on the comb on the DCDMS coated glass slide. A DCDMS coated coverslip is carefully added to the top of the drop of PA solution to allow even solution spreading over the device. After 10 minutes of exposure to 305 nm UV light, the PA has gelled and the coverslip is carefully peeled off (See inset). The combs are separated and placed in PBS to swell. Then sulfo-SANPAH crosslinker in HEPES buffer is added for 10 minutes under 350 nm UV light. The protein is attached and the device is sterilized with UV. Cells are then added to the surface. The final inset shows a finger of a comb containing ASC’s illuminated by a 40x upright microscope.
PA gels were polymerized from acrylamide solutions with bisacrylamide concentrations of 0.028 w/w%, 0.1 w/w%, and 0.2 w/w%, resulting in soft (1 kPa), intermediate (10 kPa), and stiff PA gels (34 kPa), respectively, as measured by atomic force microscopy (AFM) (Fig. 2a). 7.5 μL of the acrylamide solution was found to create a 30-50 μm tall PA gel, which is above the 20 μm thick threshold that ensures cells feel the gel and not the substrate below.8, 29 Fluorescent microbeads in the polymerization mixture allowed us to determine with confocal microscopy that our gels consistently exceeded the minimum height requirement.
Figure 2. Polyacrylamide stiffness on the device and ASC differentiation.

(a) The elastic modulus of the gel linked to the device in PBS at room temperature is determined by AFM. Monomer and initiator are fixed and bisacrylamide is varied. Positive staining for (b) ßIII-Tubulin on 1 kPa, (c) MyoD on 10 kPa, and (d) CBFA1 on 34 kPa represent differentiation in the appropriate lineages based on previous work with these substrate stiffnesses.
Myogenic Differentiation of ASCs is Increased by Combining Myogenic Stiffness and Paracrine Interactions
Choi et al. recently showed that ASCs differentiate into neuronal, myogenic, and osteogenic lineages on 1, 11, and 34 kPa, respectively.26 We first tested whether these results could be reproduced on the combs because the monomer and crosslinker ratios, initiator, and substrate were different, though the stiffness values were similar. ASCs alone were cultured for six days in growth media and stained for ßIII-Tubulin, MyoD, and CBFA1 as shown in Fig. 2b-d. While ßIII-Tubulin was found in cells on all stiffnesses, cells on 1 kPa combs showed increased branching and a neuronal morphology compared to the two stiffer gels (Fig. 2b). MyoD and CBFA1, transcription factors that are indicative of myogenic and osteogenic differentiation, were present and punctuated in nuclei on the 10 and 34 kPa gels, respectively (Fig. 2c-d); positive staining was not present on gels of other stiffness (Supplementary Fig. 1). Monoculture results on individual combs supported the findings of Choi et al., demonstrating a higher propensity for neuronal, myogenic, and osteogenic differentiation on combs modified with 1 kPa, 10 kPa, and 34 kPa gels, respectively.
We then sought to demonstrate the utility of this device by studying the effect of substrate stiffness and skeletal myoblasts co-culture on ASC differentiation in three distinct configurations. First, combs with adsorbed fibronectin or with a fibronectin-coated myogenic stiffness (10kPa) gel were created. The male half of the comb was seeded with ASCs and the female half with either ASCs or myoblasts (Fig. 3a). The device was kept in gap configuration so that the ASCs experience only the paracrine signals from cells across an 80 μm gap, and not the physical or contact cues from the other cell type. After 6 days, a subset of ASCs on 10 kPa gels in both the presence or absence of myoblasts showed punctate positive staining for MyoD on the comb fingers (Fig. 4a-b) whereas staining was undetectable in ASCs cultured on bare silicon. A human mitochondrial stain confirmed that the stained cells were human and not contaminating murine cells (Fig. 4c-d, Supplemental Figure 2). This data suggests that both myoblast co-culture and myogenic stiffness were promoting ASC myogenesis. MyoD staining was used to confirm that myogenesis had occurred; however extent of myogenesis was quantified via gene expression measurements. RT-PCR gene expression was used to further assess myogenesis of ASCs on only the male halves (ASC halves) of the combs in the ASC-myoblast co-cultures. This was to ensure that only the ASC RNA was isolated and analyzed. Additionally, human specific primers were used as a second means of selecting for only human cells. ASCs were analyzed after six days for expression of Desmin, a later stage muscle-specific intermediate filament protein that proceeds fusion30,31, and Myogenin, an early muscle regulatory factor (MRF) responsible for initiating the cascade of skeletal muscle gene activation 32. Fig. 4e-f shows normalized gene expression changes from qPCR for these two genes; Desmin expression significantly increased when both myoblasts and myogenic stiffness were present (Fig. 4f). Myogenin levels showed a similar increase though not significant when both cells and myogenic stiffness were present (Fig 4e).
Figure 3. Experimental setup. Schematics explaining all cell culture setups to demonstrate utility of the technology.
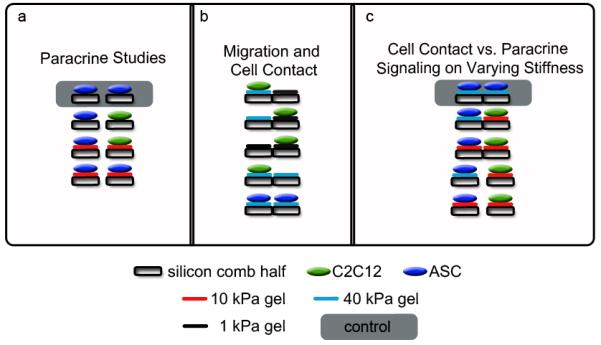
The gray shaded cultures are those to which the qPCR results are normalized. (a) Gap studies comparing 10 kPa gels to bare silicon on the combs with ASC’s (blue) and myoblasts (green). The results are shown in Figure 4. (b) Migration studies with myoblasts were used in the permutations described with various gel types in contact. ASC’s on 40 kPa were also stained to show contact on the same plane on different comb halves. Results are shown in Figure 5. (c) ASC differentiation on various gel stiffness in either contact or gap mode. Gene expression studies using a combination of 10 and 40 kPa gels in gap or contact were analyzed to show changes in myogenic gene expression. IF and qPCR results are shown in Figure 6.
Figure 4. Myoblasts in co-culture with ASCs on 10 kPa gels demonstrate increased myogenesis.
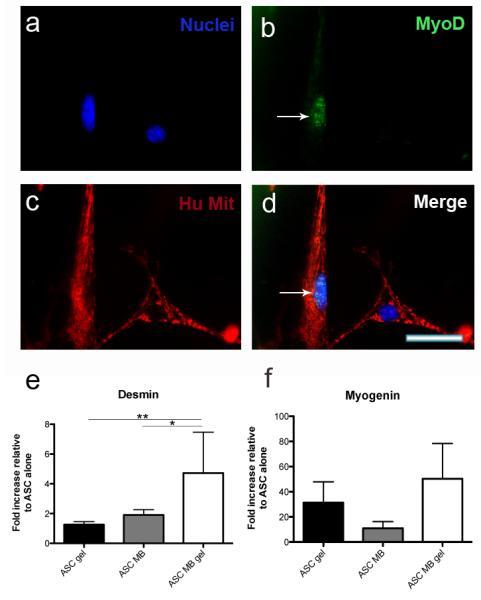
ASC’s and myoblasts were co-cultured in the following groups: ASC-ASC (silicon), ASC-ASC (10 kPa), ASC-myoblast (silicon), ASC-myoblast (10 kPa) for 6 days in growth medium. (a-d) A representative image of myogenic differentiation demonstrates positive staining for MyoD (green) in adipose derived stem cells on 10 kPa gels. The arrow emphasizes the positive green staining. Note the lack of MyoD in the adjacent cell on the right half of the image. MyoD staining was scarcely observed in all cultures except for ASC-ASC (silicon). A Human mitochondrial DNA antibody (red) was used to distinguish the human ASCs from the murine myoblasts. Hoescht stained the nuclei (blue), scale bar = 50 μm. (e) qPCR results for Desmin shows an increase in gene expression when ASCs are plated on either 10 kPa alone or co-cultured with myoblasts on silicon compared to ASCs on bare silicon alone. A synergistic increase is observed when both 10 kPa gels and myoblasts are present in gap compared to 10 kPa monoculture or co-culture on bare silicon (*p<0.05, **p<0.01). (f) The same trend is observed for Myogenin gene expression when both cells and gel are present in culture. Data represents the mean and standard deviation for 6 replicates.
Secondly, the device was used in contact mode to illustrate the influences of cell-cell contact. While gel height does not impact gap mode, contact mode requires gels that are in physical contact, share a border, and are on the same plane. In other words, the gels should have the capability to be seeded separately and once placed into contact allow for cell-cell physical contact across the interface. In our initial gap mode experiments, differential gel swelling as a result of two dissimilar polymer concentrations resulted in different gel heights. For contact mode, it was essential to create gels that maintained near identical heights to ensure only lateral cell-cell contact. As a result, contact mode experiments utilized different acrylamide and bisacrylamide formulations to yield a consistent 30 μm gel height post swelling as graphed in Fig. 5a. The heights were not statistically different (Fig. 5b), and averaged 30.4, 28.2, and 28 μm for the 40, 10 and 1 kPa gels, respectively. The heights of contact formulations were further characterized with AFM topographical maps (Supplementary Fig. 3a-b) to further emphasize that the two disparate gels are on the same plane and in contact with each other. With a contiguous surface, we then asked whether cells migrated from one comb half to another. Myoblasts were grown to confluence on 1 and 40 kPa comb halves, and once adhered, the half was put into contact with another fibronectin functionalized 1 or 40 kPa half without cells (Fig. 3b). Note that myoblasts had not fused into myotubes. All permutations were tested to determine whether migration across the comb would occur; after 4 days, no directed migration, or “durotaxis,”33 was seen in any of the configurations including mismatched ones. Fig. 5c-e demonstrates that these cells do not migrate to the other half regardless of stiffness pairing. When cells were seeded on both halves, ASCs were seen extending projections across the gel interface but still did not exhibit durotaxis (Fig, 5f).
Figure 5. Confirmation of contact formulations and cell-cell contact.

For contact studies, the gels required same heights between soft, medium and stiff moduli in order for cells to experience physical contacts on heterotypic gel stiffness. (a) The elastic modulus determined by AFM for stiff (blue, 40 kPa), medium (red, 10 kPa), and soft (black, 1 kPa) gels. Each gel is created with varying formulations of monomer and crosslinker. (b) The corresponding heights of each of the gel types show an approximate ~30 μm height on the surface of the comb when swollen in PBS. Myoblasts were added to the 1 or 40 kPa gels prior to putting them in contact with a gel that contained only a fibronectin functionalized gel and no cells. These cells were stained for Desmin (green) and Hoescht (blue) for myoblast identification and nuclei, respectively. (c) Cells on a 1 kPa gel in contact with a 40 kPa gel with no cells, (d) Cells on a 1 kPa gel in contact with a 1 kPa gel with no cells, (e) cells on a 40 kPa gel in contact with a 1 kPa gel with no cells (scale bars = 200 μm). No significant migration was observed after four days in culture for each of these formulations. (f-g) ASC’s were plated on different 40 kPa gels and placed into contact for 6 days. Cells were stained with Phalloidin (green) and human mitochondrial DNA (red), and nuclei (blue) The dashed line in (g) represents the interface between the cells that is less obvious than in (f), scale bar = 25 μm.
Finally, we examined the interplay of stiffness, paracrine, and cell-cell contact signals on ASC myogenesis by intentionally mismatching 10 and 40 kPa substrates for given cell types to examine cell fate.. The following groups were cultured (Fig. 3c): ASC (40 kPa) - ASC (40 kPa) in contact, ASC (40kPa) – Myoblasts (10 kPa), and ASC (10 kPa) – Myoblasts (10 kPa) in either gap or contact (5 groups, n=3/group). These cultures (both in gap and contact) were created using the polyacrylamide formulations as previously characterized in Fig. 5a to ensure equal heights between gels in contact. IF images show the two cell types plated in contact on the different gels (Fig. 6a). Desmin positive cells were found when ASCs were cultured on 10kPa gels (Fig. 6b, Supplementary Fig. 4a). This staining was not localized to a specific region on the fingers of the comb. Sparse Desmin positive staining was found on ASC’s seeded on 40 kPa gels co-cultured with myoblasts (Supplementary Fig. 4b). To confirm myogenesis, qPCR was also carried out to determine how disparate gel stiffnesses and configurations affected myogenic gene expression (Fig. 6c). Myogenin was significantly upregulated in all groups co-cultured with myoblasts compared to ASCs on 40 kPa alone. While a similar trend was observed with Desmin, only ASCs on a 10 kPa gel were significantly increased compared to ASCs on 40 kPa alone. Both Desmin and Myogenin were significantly upregulated with ASCs on 10 kPa in contact with myoblasts compared to ASCs on 40 kPa in gap with myoblasts. This suggests that a 10 kPa substrate and contact with myoblasts increases myogenic gene expression.
Figure 6. Differentiation of ASCs in co-cultures on 40 or 10 kPa gels in contact or gap.

Co-cultures were setup with the following groups: ASC (40 kPa)-ASC (40kPa) contact, ASC (40 kPa)-myoblast (10 kPa) contact and gap, and ASC (10 kPa) – myoblasts (10kPa) contact and gap (5 groups, n=3/group). (a) Myoblasts stained positive for Desmin (green) on a 10 kPa gel and ASC’s stained for human mitochondrial DNA (red) on a 40 kPa gel are placed in contact, scale bar = 200 μm. (b) A representative image of a positively stained Desmin cell on an ASC half of a comb on 10 kPa gel, scale bar = 25 μm. (c) qPCR results showing all groups normalized to ASC’s on 40 kPa gels. Fold change on a log 2 scale is used to clearly represent the data in graphical form (*p<0.05, **p<0.01). Myogenin in the ASC 40 kPa control is significantly downregulated compared to all other groups († p<0.01). Error bars have been included in all data contained in the graphs.
Discussion
There is a need to develop novel technologies for 2-D in vitro cell culture, which can examine the different variables that cells experience in vivo such as substrate stiffness, paracrine/autocrine signals, cell-cell physical interactions, and cell-matrix interactions. The technology we have developed has many advantages over traditional co-culture. The reconfigurable co-culture device allows for two cell types to be individually examined in pure populations. This has already been demonstrated previously as a more useful co-culture tool as one can observe contact, paracrine, and temporal changes independently.19, 21 Yet as mentioned, many groups have studied cell behavior on substrates of varying stiffness in the context of stem cell differentiation, wound healing, cancer metastasis, and aging.1-6
Currently, the field lacks methods to examine cell substrate and cell interactions in an organized, independent way. No existing cell culture methods allow one to independently examine substrate stiffness and co-culture with separable cell populations. By covalently linking a polyacrylamide gel on the silicon surface of the reconfigurable co-culture device, we developed a platform to independently vary substrate stiffness of two cell populations, which can be cultured in either contact with each other or with a gap, which only allows paracrine signals between the populations. Moreover, each comb half can be functionalize with different ECM proteins, thereby also allowing one to examine additional cell-matrix interactions. We show that cells on one comb half will interact with cells on the other when placed in contact, yet will remain on their respective half, which allows cell populations to be examined individually. With this system in place, the user can examine: 1) cell contact interactions, 2) cell paracrine interactions, 3) substrate stiffness, 4) cell-ECM protein interactions, and 4) temporal changes by removing one comb half and inserting another. With any co-culture tool it is important that the user can make easy readouts. We have shown that RNA can easily be isolated from polyacrylamide gels on the silicon surface and immunofluorescence can be carried out directly on the device. Cell lysates can be collected in similar fashion though were not shown here.
Adipose derived stem cells (ASCs) have been shown to differentiate into neuronal, myogenic and osteogenic lineages by culturing them on approximately 1, 11, and 30-40 kPa polyacrylamide (PA) gels respectively.26 These gels were previously synthesized by free radical initiation using ammonium persulfate and N,N,N′,N′-Tetramethylethylenediamine covalently linked to amino-silanated glass coverslips.26, 28 In order to functionalize this polymer on the silicon reconfigurable co-culture device, we adapted alternative methods to covalently link the polymer to the substrate and UV initiate the polymerization. In this way, the user can prepare and pipette the solution on the surface carefully and control the initiation of the polymerization. AFM was used to characterize these new gel formulations on the device. We repeated the studies of Choi et al. on these gels by plating ASCs on 1, 10, and 34 kPa gels functionalized on the combs. Positive immunofluorescence staining for neuronal, myogenic, and osteogenic markers on 1, 10, and 34 kPa gels respectively was consistent with previous findings and thus we moved forward to with additional studies to examine the potential of this technology.
Groups have previously shown that paracrine signaling by myoblasts cause spontaneous differentiation of ASCs.10, 34 However, no groups had previously shown the combination of substrate and cell signaling influence on ASC myogenesis. It was hypothesized that by adding myoblasts in culture combined with a myogenic stiffness, ASC myogenesis would increase. We took advantage of the gap configuration of the device to model this paracrine effect and functionalized a 10 kPa PA gel to its surface. Co-culture with myoblasts in combination with a substrate of myogenic stiffness showed a synergistic increase of myogenic differentiation relative to ASC’s on silicon alone, as shown by increased expression of Desmin and Myogenin.
Previous studies have shown myogenic differentiation of ASCs and MSCs with myoblasts in direct co-culture.27, 35, 36 However, it is often unclear as to whether these cells are merely fusing with one another or inducing myogenesis via contact dependent mechanisms. One advantage of the modified reconfigurable co-culture device is that it allows the user to maintain, examine, and perform qPCR/IF readouts on only one population, even when cells are in contact. This cannot be achieved in standard culture setups unless harsh separation methods such as flow cytometry are used. Previous gel formulations in our initial studies performed in the gap configuration varied in height between the different stiffnesses. In order to carry out cultures in contact with disparate stiffnesses, new gel formulations were used to ensure cell-cell contact on the same plane. Advantageously, these cells also were maintained on their respective comb half regardless of gel substrate stiffness. This is most likely due to the fact that the two gels are discrete, or non-continuous, even though they are in physical contact and on the same plane. Choi et al. demonstrates that on gels with gradient stiffness, cells “sense” the stiffer environment and thus move towards stiffer areas.37 However, in our co-culture regime, this cue is eliminated. Though the gels are physically touching on the same plane, they are discontinuous and not physically crosslinked across the interface of the fingers of the comb. Consequently as the cells contract their local matrix, deformations within one gel do not propagate to the other; thus cells cannot sense the other gel via typical mechanotransduction mechanisms.38 Further evidence of this discontinuity can be seen in the representative AFM topographical map, which shows a very slight, 3μm change in the z-plane. The cells would not be able to physically sense the other gel half without cell-cell interaction between the interfaces. This is important in regard to maintaining populations of cells on their respective halves; cells are prevented from migrating to the other gel interface, but can establish physical contacts across the fingers of the comb.
We thus examined the effect of contact of ASCs with myoblasts and observed whether this would increase myogenesis over paracrine signaling alone. Simultaneously, to further show utility of this device, we compared ASCs seeded on 40 kPa to those seeded on 10 kPa. Desmin staining was detected on all cultures with 10 kPa gels and less commonly on 40 kPa gels when myoblasts were present in culture. As mentioned previously, we did not quantify staining due to the rarity of the MyoD/Desmin staining in cultures. However, it is important to note that immunofluorescence quantification can be carried out on this device.21 It has been shown previously that gene expression quantification (RT-PCR and qRT-PCR) for myogenesis of ASCs is an accurate correlation for myogenic protein expression.10, 26, 35, 39 Desmin and Myogenin gene expression of ASCs showed a consistent trend. In general, myoblasts in contact increased gene expression relative to gap alone. However, gene expression for both genes was significantly higher when ASCs were plated on 10 kPa and in contact with myoblasts versus ASCs on 40 kPa in gap with myoblasts. It should be noted, however, that only the cells at the interface of the comb fingers experience direct cell-cell contact. These reasons could explain why contact only marginally increased gene expression relative to gap. ASCs cultured on 10 kPa gels caused an increase of myogenic gene expression relative to those plated on 40 kPa gels. This supports previous findings that 10 kPa gels are in the stiffness regime that is ideal for increased myogenesis, and that this substrate stiffness in combination with myoblast co-culture synergistically upregulates ASC myogenesis.
Experimental
Device Preparation, Polymerization and Protein Functionalization
The silicon reconfigurable co-culture device (combs) was fabricated and developed by previously established methods.19, 20 This device is approximately 15 mm × 11.2 mm when fit into contact with a 350 μm thickness. The combs were initially cleaned by piranha treatment as described previously.20, 21 These combs were first paired together to ensure that they are completely flush in contact mode by visualization under an upright 40 × objective. Once paired, they were UVO treated for 10 minutes to activate the silicon surface. 3-(Trimethoxysilyl)propyl methacrylate (Sigma-Aldrich, St. Louis, MO) was used to further treat the surface in order to covalently attach the polyacrylamide gels to the silicon surface. 3-(Trimethoxysilyl)propyl methacrylate (0.1 mL) was diluted in 20 mL 100% ethanol and 0.6 mL of 1:10 glacial acetic acid was added to make a final methacrylate concentration of 20.3 mM. The solution was immediately mixed and added to the combs. The combs were incubated at room temperature in the solution for 3 minutes and the solution was removed. The combs were rinsed with 100 % ethanol and allowed to air dry for 30 minutes to remove residual ethanol. The combs displayed a visual color change that suggested successful methacrylate coating. Prior to functionalizing with polyacrylamide, 1 mm glass microscope slides (Fisher Scientific, Pittsburgh, PA) and 25mm circle coverslips were treated with dichlorodimethylsilane (DCDMS, Sigma-Aldrich, St. Louis, MO) to create hydrophobic surfaces.28 Each comb pair was placed on a coated microscope slide. For the initial gap studies, acrylamide of 6% w/w of a 40% w/w stock (Fisher Scientific, Pittsburgh, PA), varying amounts of a 2% stock w/w bisacrylamide (Fisher Scientific, Pittsburgh, PA), and Milli-Q water were mixed together to create a total of 5 mL. Acrylamide monomer and bisacrylamide crosslinker concentrations had to be modified and tested in order to achieve similar z-axis heights with varying stiffness for contact scheme. This liquid was degassed by bubbling argon for 10 minutes to remove oxygen. These stocks were then aliquoted and stored at −20 °C. 2,2− -Azobis(2-methylpropionamidine) dihydrochloride (0.5 wt%, Sigma-Aldrich, St. Louis, MO) was added to thawed aliquots of 1 mL of solution. The solution was mixed carefully by rotating the tube several times and sonicating for 5 seconds. 7.5 uL of the solution was added to the interface between the two comb halves in contact. The DCDMS coverslip was added on top of the solution. The comb-polymer solution-coverslip setup was then irradiated with 305 nm UV light 2 inches directly above the setup with a surface intensity of 0.85 mW/cm2 for 10 minutes. After 10 minutes, the DCDMS coverslips and glass were carefully removed from the comb. Peeling the coverslip off required gentle removal to ensure that the PA did not tear off the comb pair. Once the comb pair was separated, it was immediately added into 2 mL of 1X Dulbecco’s Phosphate-Buffered Saline (DPBS, Invitrogen, Carlsbad, CA). These gels were allowed to swell by incubation in PBS at room temperature for 24 hrs. Polyacrylamide mixtures were characterized by atomic force microscopy (AFM) to assess stiffness of the gel.28 0.2 mg/ml of Sufosuccinimidyl-6-(4-azido-2-nitrophenylamino)-hexanoate (Sulfo-SANPAH, Pierce Biotechnology, Rockford, IL) diluted in 50 mM HEPES buffer at pH 8.5 was used to covalently link protein to the polyacrylamide via established methods.28 The Sulfo-SANPAH was washed twice with the HEPES buffer. Next, 10 ug/mL fibronectin (Sigma-Aldrich, St. Louis, MO) diluted in HEPES was added and allowed to attach to the surface overnight at 37 °C. The solution was removed from the combs and combs were rinsed once more with DPBS. The combs were sterilized under UV light in the tissue culture hood for 45 minutes and then used for cell seeding and attachment.
Gel Thickness and Stiffness
Gels were prepared as above with the addition of 10 μl/ml of 0.5 μm fluorescent 505/515-carboxylated microspheres (Invitrogen, Carlsbad, CA) and 0.1% v/v Tween-20. The gels were allowed to swell in DPBS overnight. A 600X CARV II confocal microscope (BD Biosciences) was used to visualize the spheres and determine thickness post-swelling by panning from the bottom of the gel to the top in the z-direction while gels were in PBS.40 Five randomized points on the gel-comb device were taken from 6 comb halves (n=30) per stiffness to determine thickness. Stiffness was quantified via Atomic Force Microscopy (AFM). AFM data was collected by averaging 10 points per comb on 3 separate combs (n=30/group).
Cell Culture
Adipose derived stem cells (ASCs) were harvested from 7 different donor patients. Two groups of three patients’ cells were pooled together. These cells were isolated from donor ages between 20 - 31 years of age by methods described previously26 with the approval of the UCSD human research protections program (Project #101878). All the following experiments were carried out with informed consent and compliance with UC San Diego’s institutional guidelines.
These cells were cultured in adipose cell growth media (AGM) made up of low glucose Dulbecco’s modified Eagle’s medium (low-glucose DMEM) (Invitrogen, Carlsbad, CA), containing 10% fetal bovine serum (FBS, Hyclone), and 100-units/mL penicillin/streptomycin (P/S, Invitrogen, Carlsbad, CA). Cells were passaged at 1:4 and were used for experiments up to passage number 6. Murine C2C12 myoblasts (ATCC, Manassas, VA) were grown as described previously in growth media (GM) with 4.5 g/L glucose-Dulbecco’s modified Eagle’s medium (DMEM), 10% FBS, and 1% P/S.21
Monoculture Differentiation Assays
Each of the fibronectin coated PA comb pairs were placed in a 12-well plate. ASCs were seeded on 1, 10, and 34 kPa gels coated combs. Combs without a gel but with adsorbed fibronection at 10 μg/mL were used as a control. Each comb pair was seeded with 30,000-40,000 cells per well in 2 mL of media. The plate was shaken for 5 seconds after seeding for 30 minutes and 1 hour in order to promote even cell seeding on the surface. After cells were attached, combs were removed and placed into a new 12-well plate with the same AGM. Media was replaced every two days in culture. After 6 days, the cultures were washed with PBS and fixed with 4% paraformaldehyde for 10 minutes for staining. Duplicate stiffness combs were stained for ßIII-Tubulin, MyoD, or CBFA1 to identify neuronal, myogenic and osteogenic differentiation, respectively, as previously demonstrated.26 Staining was carried out as described below.
Gap Co-Culture Differentiation Assays
Combs were prepared as mentioned above with or without PA and functionalized with fibronection. Comb halves were separated and male combs were used for ASC seeding and analysis whereas female halves were seeded with either ASCs or C2C12 myoblasts. The ASC halves were seeded with 30-40,000 cells in AGM and the myoblasts with 750,000 cells in GM per 12-well. The cells were allowed to attach and shaken as in the monoculture studies. Once attached, the cells were taken out of the 12-well, dipped and rinsed in PBS and added into a new 12-well with 2 mL AGM. Combs were fit together in the following configurations in duplicate (4 groups): 1) ASCs in co-culture with ASCs on silicon, 2) ASCs on 10 kPa gels in co-culture with ASCs on 10 kPa gels, 3) ASCs in co-culture with myoblasts on silicon, 4) ASCs on 10 kPa gels in co-culture with myoblasts on 10 kPa gels. All co-cultures were in the gap configuration so that cell contact interactions could not occur. Media was changed every two days. On day 6, the cultures were either fixed with 4% paraformaldehyde for downstream staining or male halves were disengaged from the females and RNA was isolated for downstream qRT-PCR gene expression analysis. Staining was performed as in the monoculture assays with MyoD for the myogenic differentiation marker and with an anti-human mitochondrial DNA antibody (Millipore) that identifies only the human cells. RNA was isolated from only the male halves of the combs and pooled from triplicate cultures, converted to cDNA and analyzed with qPCR. Desmin and Myogenin were analyzed for gene expression in PCR. Fold expression was averaged from six separate experiments.
Migration Studies
Myoblasts were seeded to confluence on comb halves of either 40 kPa or 1 kPa pairs. Once cells adhered, halves were placed into contact with another gel type without any cells in 2 mL of GM. Groups consisted of the following: cells on a 1 kPa gel in contact with a 40 kPa gel with no cells, cells on a 1 kPa gel in contact with a 1 kPa gel with no cells, and cells on a 40 kPa gel in contact with a 1 kPa gel with no cells. Other permutations using 10 kPa (instead of a 1 kPa) and 40 kPa were also tested. Once adhered after 3 hours, the halves were snapped with another corresponding half. After 4 days, the combs were stained with Desmin and Hoescht.
ASC Differentiation on Varying Stiffness Gels in Either Contact or Gap Mode
As with the gap co-culture studies, gels were generated on comb pairs fit into tight contact. In this case, all combs were modified with either 10 kPa or 40 kPa gels. Once polymerized, swollen, and protein functionalized with fibronectin, cells were seeded on comb halves. The cells were allowed to attach and shaken as in the gap co-culture studies. Once attached, the cells were taken out of the 12-well, dipped and rinsed in PBS and added into a new 12-well with 2 mL AGM. Some of the comb pairs were substituted such that groups with 40 kPa male halves and 10 kPa female halves were placed into contact. Because these gels post-swelling measured 30 μm in height, cells could be placed on both halves and maintain z-axis contact. Cells were seeded to confluence on individual halves to create the following groups: ASCs on 40 kPa in contact or gap with myoblasts on 10 kPa, ASCs on 10 kPa in contact or gap with myoblasts on 10 kPa, and ASCs on 40 kPa in contact with ASCs on 40 kPa as a control (5 groups, n=3/group). AGM media was replaced every other day. After 6 days, RNA was collected from the male ASC half of each culture group. qPCR was carried out with Desmin, Myogenin, and GAPDH to test myogenic gene expression.
Immunofluorescence Staining
After fixing in 4% paraformaldehyde (Wako Chemicals, Richmond, VA) for 10 minutes, combs were rinsed with PBS. A solution of 1% Triton X-100 in PBS was added for 15 minutes at room temperature. The combs were then rinsed with a staining solution (SS) made with 1mM MgCl2 in 1X PBS. Primary antibodies: Desmin (1:200, Abcam, Cambridge, MA), neuronal ßIII-Tubulin (1:100, Sigma, St. Louis, MO)), MyoD (1:100, Santa Cruz Biotechnologies, Santa Cruz, CA), CBFA1 (1:100, Alpha Diagnostic International, San Antonio, TX) and for some assays Human Mitochondrial DNA (1:200, Millipore, Temecula, CA) were added to 2% BSA in SS for 30 minutes at 37°C. The solution was removed and washed 3X with SS. Next, secondary antibodies, AlexFluor 488 or AlexaFluor 568 (1:200, Invitrogen, Carlsbad, CA) were added in 2% BSA in SS for 30 minutes at 37°C. The solution was removed and washed three times with SS. Hoescht 33342 (0.1 μg/ml in DI water, Invitrogen, Carlsbad, CA) was incubated for 10 minutes at room temperature. Finally, the samples were rinsed with PBS and combs were individually mounted on microscope slides with Fluoromount-G (Southern Biosciences, Birmingham, AL). Immunofluorescence was carried out using the CARV II Confocal Microscope (BD Biosciences). Images were taken with a Nikon Eclipse TE2000-U microscope with a Cool-Snap HQ Camera and were then analyzed by Metamorph 7.6.
Quantitative Polymerization Chain Reaction (qPCR)
After 6 days, male combs were carefully removed from the female combs and transferred to a new plate. The RNEasy kit (Qiagen, Valencia, CA) was used to extract total RNA from cells. The lysis buffer was directly added to the comb surface and a cell scraper was used to remove and consolidate cells from the comb into the solution. The RNA from two male combs in the same group was pooled together to improve yield. cDNA was synthesized via Superscript III Reverse Transcriptase kit (Applied Biosystems). SYBR Green PCR Master Mix (Applied Biosystems) was used with a 10 μM final concentration of forward and reverse primer. Primers were designed to be human specific (e.g. did not cross-react with the mouse genome) using human and mouse BLAT genome software (genome.ucsc.edu). Myogenin (NM_002479.4) F: 5′-GCCTTG ATGTGCAGCAACAGCTTA-3′, R: 5′-AACTGCTGGGTG CCATTTAAACCC-3′; Desmin (NM_001927.3) F: 5′-CCGCGGGCGGGTTTCGGCTC-3′, R: 5′-GGCCACTCGCGTCGGCTCGC-3′; and GAPDH (NM_002046.4) F: 5′-TGAGCCCGCAGCCTCCCGCTT-3′, R: 5′-TCAGCGCCAGCATCGCCCCACT-3′. Samples were done in technical triplicates on the ABI Prism 7900 HT Fast Real-Time PCR System (Applied Biosystems). The following thermal cycle settings were used: 2min at 50° C, 10 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. The SDS 2.3 Software (Applied Biosystems) was used to record and analyze cycle number and gene expression fold change. All data was normalized to GAPDH. Gap gene expression studies were then normalized to adipose stem cells seeded on non-polyacrylamide silicon combs or adipose stem cells on 40 kPa alone in the final experiment.
Statistical evaluation
Significance was tested for all experiments using a 1-way ANOVA with a two-tailed distribution followed by Tukey post-hoc t-test with a 95% confidence interval. Mean fold changes and standard deviations for gene expression measurements were calculated with at least experimental triplicates. AFM data is represented with averages and s.e.m.
Conclusion
This device has allowed us to probe differences between cell contact and paracrine effects on cells cultured on varying substrate stiffness for this first time. The cells are cultured on separable, spatially organized substrates that achieve a large surface area for cell interaction in 2-D. The culture setup restricts cell migration to allow for gene and protein expression analysis on solely one cell type. This system is not only useful for examining changes in stem cell differentiation but can be applied to study more complex systems in which controlling co-culture and cell microenvironment can elucidate basic mechanisms in cell biology. With numerous potential applications, this device can span multiple disciplines as a useful tool to understanding basic cell-cell and cell-matrix interactions.
Supplementary Material
Acknowledgements
This worked was funded by the National Institutes of Health (NIH) Director’s New Innovator Award Program, part of the NIH Roadmap for Medical Research, through grant number 1-DP2-OD004309 (to K.L.C.) and 1-DP2-OD006460 (to A.J.E). N.R. acknowledges the Training Program in Multi-Scale Analysis of Biological Structure and Function (Interfaces Graduate Training Program), NIH T32 EB009380 and the Training Program in Integrative Bioengineering of Heart, Vessels and Blood; NIH T32 HL105373 for support. G.N.G. acknowledges the American Heart Association for a postdoctoral fellowship (12POST9750018). EH acknowledges the University of California, Irvine ACS/IRG – 98-279-07. K.S. and L.G.V. acknowledge the NSF Graduate Research Fellowship Program.
References
- 1.Goubko CA, Cao XD. Materials Science & Engineering C-Materials for Biological Applications. 2009;29:1855–1868. [Google Scholar]
- 2.Khademhosseini A. Micro and nanoengineering of the cell microenvironment : technologies and applications. Artech House; Boston: 2008. [Google Scholar]
- 3.Calvo F, Sahai E. Curr Opin Cell Biol. 2011;23:621–629. doi: 10.1016/j.ceb.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Duell BL, Cripps AW, Schembri MA, Ulett GC. J Biomed Biotechnol. 2011;2011:852419. doi: 10.1155/2011/852419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salameh TS, Le TT, Nichols MB, Bauer E, Cheng J, Camarillo IG. Int J Cancer. 2013;132:288–296. doi: 10.1002/ijc.27672. [DOI] [PubMed] [Google Scholar]
- 6.Canseco JA, Kojima K, Penvose AR, Ross JD, Obokata H, Gomoll AH, Vacanti CA. Tissue Eng Part A. 2012;18:2549–2558. doi: 10.1089/ten.tea.2012.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keung AJ, Asuri P, Kumar S, Schaffer DV. Integrative Biology. 2012;4:1049–1058. doi: 10.1039/c2ib20083j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 9.Zouani OF, Kalisky J, Ibarboure E, Durrieu MC. Biomaterials. 2013;34:2157–2166. doi: 10.1016/j.biomaterials.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 10.Di Rocco G, Iachininoto MG, Tritarelli A, Straino S, Zacheo A, Germani A, Crea F, Capogrossi MC. J Cell Sci. 2006;119:2945–2952. doi: 10.1242/jcs.03029. [DOI] [PubMed] [Google Scholar]
- 11.Khademhosseini A, Yeh J, Eng G, Karp J, Kaji H, Borenstein J, Farokhzad OC, Langer R. Lab on a Chip. 2005;5:1380–1386. doi: 10.1039/b508096g. [DOI] [PubMed] [Google Scholar]
- 12.Hong S, Pan Q, Lee LP. Integr Biol (Camb) 2012;4:374–380. doi: 10.1039/c2ib00166g. [DOI] [PubMed] [Google Scholar]
- 13.Takayama S, McDonald JC, Ostuni E, Liang MN, Kenis PJA, Ismagilov RF, Whitesides GM. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5545–5548. doi: 10.1073/pnas.96.10.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukuda J, Khademhosseini A, Yeh J, Eng G, Cheng JJ, Farokhzad OC, Langer R. Biomaterials. 2006;27:1479–1486. doi: 10.1016/j.biomaterials.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Kaji H, Camci-Unal G, Langer R, Khademhosseini A. Biochimica Et Biophysica Acta-General Subjects. 2011;1810:239–250. doi: 10.1016/j.bbagen.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christman KL, Enriquez-Rios VD, Maynard HD. Soft Matter. 2006;2:928–939. doi: 10.1039/b611000b. [DOI] [PubMed] [Google Scholar]
- 17.Brafman DA, Chien S, Willert K. Nature Protocols. 2012;7:703–717. doi: 10.1038/nprot.2012.017. [DOI] [PubMed] [Google Scholar]
- 18.Bhatia SN, Balis UJ, Yarmush ML, Toner M. FASEB J. 1999;13:1883–1900. doi: 10.1096/fasebj.13.14.1883. [DOI] [PubMed] [Google Scholar]
- 19.Hui EE, Bhatia SN. Proc Natl Acad Sci U S A. 2007;104:5722–5726. doi: 10.1073/pnas.0608660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.K. S. Hui EE, Bhatia SN. In: Methods In Bioengineering: Microdevices in Biology and Medicine. RS Y. M. a. L., editor. Artech House; 2009. Editon edn. [Google Scholar]
- 21.Rao N, Evans S, Stewart D, Spencer KH, Sheikh F, Hui EE, Christman KL. Biomed Microdevices. 2012 doi: 10.1007/s10544-012-9709-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baker EL, Bonnecaze RT, Zamao MH. Biophysical Journal. 2009;97:1013–1021. doi: 10.1016/j.bpj.2009.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bavister BD. Hum Reprod Update. 1995;1:91–148. doi: 10.1093/humupd/1.2.91. [DOI] [PubMed] [Google Scholar]
- 24.Wells RG. Hepatology. 2008;47:1394–1400. doi: 10.1002/hep.22193. [DOI] [PubMed] [Google Scholar]
- 25.Brandl F, Sommer F, Goepferich A. Biomaterials. 2007;28:134–146. doi: 10.1016/j.biomaterials.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 26.Choi YS, Vincent LG, Lee AR, Dobke MK, Engler AJ. Biomaterials. 2012;33:2482–2491. doi: 10.1016/j.biomaterials.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vieira NM, Brandalise V, Zucconi E, Jazedje T, Secco M, Nunes VA, Strauss BE, Vainzof M, Zatz M. Biol Cell. 2008;100:231–241. doi: 10.1042/BC20070102. [DOI] [PubMed] [Google Scholar]
- 28.Tse JR, Engler AJ. Curr Protoc Cell Biol. 2010 doi: 10.1002/0471143030.cb1016s47. Chapter 10, Unit 10 16. [DOI] [PubMed] [Google Scholar]
- 29.Maloney JM, Walton EB, Bruce CM, Van Vliet KJ. Physical Review E. 2008:78. doi: 10.1103/PhysRevE.78.041923. [DOI] [PubMed] [Google Scholar]
- 30.Paulin D, Li Z. Exp Cell Res. 2004;301:1–7. doi: 10.1016/j.yexcr.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Londhe P, Davie JK. Skelet Muscle. 2011;1:14. doi: 10.1186/2044-5040-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Black BL, Olson EN. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 33.Tse JR, Engler AJ. PLoS One. 2011;6:e15978. doi: 10.1371/journal.pone.0015978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pecanha R, Bagno LL, Ribeiro MB, Robottom Ferreira AB, Moraes MO, Zapata-Sudo G, Kasai-Brunswick TH, Campos-de-Carvalho AC, Goldenberg RC, Saar Werneck-de-Castro JP. J Bone Joint Surg Am. 2012;94:609–617. doi: 10.2106/JBJS.K.00351. [DOI] [PubMed] [Google Scholar]
- 35.Beier JP, Bitto FF, Lange C, Klumpp D, Arkudas A, Bleiziffer O, Boos AM, Horch RE, Kneser U. Cell Biology International. 2011;35:397–406. doi: 10.1042/CBI20100417. [DOI] [PubMed] [Google Scholar]
- 36.Eom YW, Lee JE, Yang MS, Jang IK, Kim HE, Lee DH, Kim YJ, Park WJ, Kong JH, Shim KY, Lee JI, Kim HS. Biochem Biophys Res Commun. 2011;408:167–173. doi: 10.1016/j.bbrc.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 37.Choi YS, Vincent LG, Lee AR, Kretchmer KC, Chirasatitsin S, Dobke MK, Engler AJ. Biomaterials. 2012;33:6943–6951. doi: 10.1016/j.biomaterials.2012.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holle AW, Engler AJ. Current Opinion in Biotechnology. 2011;22:648–654. doi: 10.1016/j.copbio.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meligy FY, Shigemura K, Behnsawy HM, Fujisawa M, Kawabata M, Shirakawa T. In Vitro Cell Dev Biol Anim. 2012;48:203–215. doi: 10.1007/s11626-012-9488-x. [DOI] [PubMed] [Google Scholar]
- 40.Leng L, McAllister A, Zhang BY, Radisic M, Gunther A. Advanced Materials. 2012;24:3650–3658. doi: 10.1002/adma.201201442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.