

Abstract
Amyotrophic lateral sclerosis (ALS) is an adult-onset degenerative disorder characterized by the death of motor neurons in the cortex, brain stem, and spinal cord. Despite intensive research the basic pathophysiology of ALS remains unclear. Although most cases are sporadic, ∼10% of ALS cases are familial (FALS). Mutations in the Cu/Zn superoxide dismutase (SOD1) gene cause ∼20% of FALS. The gene(s) responsible for the remaining 80% of FALS remain to be found. Using a large European kindred without SOD1 mutation and with classic autosomal dominant adult-onset ALS, we have identified a novel locus by performing a genome scan and linkage analysis. The maximum LOD score is 4.5 at recombination fraction 0.0, for polymorphism D18S39. Haplotype analysis has identified a 7.5-cM, 8-Mb region of chromosome 18q21, flanked by markers D18S846 and D18S1109, as a novel FALS locus.
Amyotrophic lateral sclerosis (ALS) is an adult-onset degenerative disorder characterized by the death of motor neurons in the cortex, brain stem, and spinal cord. The result is progressive muscle weakness, atrophy, and death from respiratory paralysis usually within 3–5 years of symptom onset (Brown 1995). The age-adjusted worldwide incidence of ALS is 0.5–3 per 100,000, without obvious race-related differences (Smith and Appel 1995), and the prevalence is 4–6 per 100,000 (Haverkamp et al. 1995). Approximately 10% of ALS cases are familial (FALS) (Camu et al. 1999), with the majority of cases deemed, in the absence of a positive family history, to be sporadic (SALS). With the exception of some FALS cases with clearly distinct features, the majority of FALS and SALS cases are clinically indistinguishable. A distinct ALS phenotype is seen in the Pacific islands of Guam, where the Chamorro people have had an unusually high incidence of parkinsonism-dementia ALS (de Belleroche et al. 1995).
The diagnosis of ALS requires the presence of both upper and lower motor-neuron features, with disease progression, in the absence of evidence of another disease that may explain these signs. Dementia is found in a portion of patients; estimates vary from 3%–5% (Al Chalabi and Leigh 2000) to 15% (Hudson 1981). At present, there is no cure for ALS, and treatment is primarily palliative.
The underlying cause of ALS remains unknown, and, although it comprises only 10% of cases, FALS is commonly studied, with the aim of mapping the gene(s) that cause or predispose to ALS. Identification of FALS genes may lead to a greater understanding of the mechanisms of cell death in all forms of ALS. This, in turn, may lead to more-effective therapies.
Several FALS loci have been identified. ALS1 (MIM 105400) is an adult-onset autosomal dominant form of the disease. Linkage to chromosome 21q has been established (Siddique et al. 1991), with evidence of genetic heterogeneity (King et al. 1993). Subsequently, mutations have been detected in the Cu/Zn superoxide dismutase gene (SOD1) (Rosen et al. 1993). Adult-onset autosomal dominant FALS neither linked to chromosome 21 nor associated with mutations in the SOD1 gene is designated “ALS3” (Siddique et al. 1991). ALS4 (MIM 602433) is the only identified autosomal dominant juvenile-onset form of ALS and maps to chromosome 9q34 (Chance et al. 1998). A detailed clinical account of several of the family members, including autopsy reports, has determined that these individuals fulfill the ALS criteria but display some additional pathological findings (Rabin et al. 1999).
Autosomal recessive, juvenile-onset ALS (AR-ALS) exists in three forms. A family with type 3 was used to map ALS2 (MIM 205100) to chromosome 2q33 (Hentati et al. 1994). In this form of ALS, symptoms occur during the 1st or 2d decade of life and progress slowly for 10–15 years (Hentati et al. 1994). The causative gene has recently been identified (Hadano et al. 2001). Type 1 is the most prevalent form of AR-ALS and has been linked to chromosome 15q15.1-q21.1 (ALS5 [MIM 602099]) (Hentati et al. 1998). As yet, no locus for type 2 AR-ALS has been identified.
An ALS subtype that includes frontotemporal dementia (ALS FTD [MIM 105550]) has been mapped to chromosome 9q21-q22 (Hosler et al. 2000). Another ALS-FTD locus, which also includes parkinsonian features, is caused by mutations in the tau gene on chromosome 17q21 (Lynch et al. 1994). A 1998 report of an X-linked dominant ALS locus (Siddique et al. 1998) has not been further described.
Approximately 80% of FALS cases cannot be explained by SOD1 mutations; thus, other candidate genes have been tested for a role in disease pathogenesis. The role of neurofilaments in ALS has been intensely examined (Julien et al. 1999). The other isoforms of SOD—that is, MnSOD (SOD2) and extracellular SOD (SOD3)—have been examined, and, although neither is linked to FALS (de Belleroche et al. 1995; Siddique and Deng 1996), mutant SOD1-mediated disease in mice is influenced by the expression of these SOD proteins (Li et al. 1995; Melov et al. 1998, 1999; Andreassen et al. 2000). The neuronal apoptosis–inhibitor gene and the survival motor neuron (SMN) gene, which are implicated in spinal muscular atrophy, also have been studied (de Belleroche et al. 1995; Orrell et al. 1997; Parboosingh et al. 1999), and, recently, the SMN locus has been implicated in the modification of mutant SOD1 disease in a transgenic mouse, a modification that delays disease onset significantly (Kunst et al. 2000). Other genes that have been examined include the apolipoprotein E gene (Bedlack et al. 2000), the apurinic/apyrimidinic exonuclease gene (Hayward et al. 1999), and the persyn gene (Flowers et al. 1999).
The highly cooperative ALS pedigree Fr017 has been collected and assessed by neurologists expert in ALS (J.K., F.S., V.M., and W.C.). In all, there are 51 collected DNA samples. Figure 1 shows the most informative section of the family. This ALS pedigree has 20 affected individuals (definitely affected, by El Escorial ALS diagnostic criteria), and, although, as would be expected, many are deceased, we have DNA from eight affected members, and the structure of the pedigree is such that the genotypes of five deceased affected individuals may be confidently inferred. Clinically, the affected family members have a rather uniform presentation of ALS, beginning, for the majority of the patients, in the legs, with a mean age at onset and a mean duration of 45 years and 5 years, respectively. Clinical presentation is typical of ALS, with progressive weakness involving all four limbs and bulbar level, with upper and lower motor-neuron signs and diffuse denervation on electromyography. There are no atypical features such as pain, dementia, sensory signs, or cerebellar degeneration.
Figure 1.
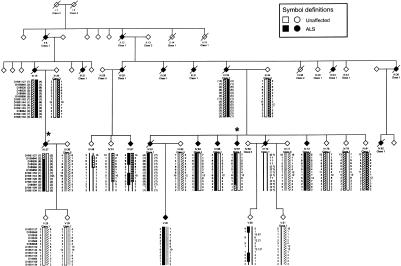
Informative section of pedigree Fr017. Individuals available for typing are indicated by the generated genotype data. The markers examined are shown. Alleles in parentheses are inferred. All inferred haplotypes were reconstructed on the basis of minimal recombinations. A solid black bar indicates the disease haplotype. A thin black line denotes an unknown or unphased haplotype. Key recombinants (IV:37 and IV:67) are indicated by an asterisk (*). Alleles given refer to the Fondation Jean Dausset CEPH alleles, when available. Information is shown only for relevant individuals. To preserve confidentiality, the sex of all individuals has been disguised; in addition, six unaffected haplotype carriers have been omitted from the pedigree.
Blood was collected from all consenting family members. DNA and lymphoblastoid cell lines (Anderson and Gusella 1984) were generated. The study was approved by the Montreal General Hospital Research Ethics Committee and the Assistance Publique des Hôpitaux de Paris Ethics Committee.
SSCP analysis of the SOD1 gene was performed as described elsewhere (Boukaftane et al. 1998), and, since no mutation was detected in the family, a genome scan was performed on the most informative portion of the pedigree (22 individuals, including 7 affected with ALS), by use of a panel of 400 highly polymorphic evenly spaced microsatellite markers. All subsequent markers used were polymorphic microsatellites ordered by the Marshfield genetic maps (Center for Medical Genetics, Marshfield Medical Research Foundation database). Primer sequences were obtained from The Cooperative Human Linkage Center database and from The Genome Database. Genotyping was performed by PCR incorporating [35S]-dATP. Products were electrophoresed on 6% denaturing polyacrylamide gels, and then autoradiography was performed. Alleles were sized by comparison to the M13mp18 sequence ladder. Marker-allele sizes and frequencies were obtained from the Fondation Jean Dausset CEPH database.
The genotype data was analyzed by the MLINK program of the FASTLINK software package (Cottingham et al. 1993). The model used for analysis was autosomal dominant with incomplete and age-dependent penetrance of disease, with three liability classes based on published values: (1) age 0–46 years, 50%; (2) age ⩾47 years, 90%; and (3) spouses, 100%. The ages reflect the age at onset, for affected family members, and the age at last clinical exam, for those unaffected. Simulation analysis using the same model was performed by SLINK software (Ott 1989; Weeks 1990).
Analysis of the data generated by genome scan identified two regions suggestive of linkage—that is, for which the LOD score was >1.5. Further analysis, using all available DNA samples and additional markers flanking the positive regions, excluded linkage to a locus on chromosome 12 and confirmed linkage to a novel locus on chromosome 18.
Once suggestive linkage had been detected, testing for linkage of nearby markers, with the entire set of family samples, was performed. All available markers at this locus (n=30) were then investigated. Simulation-analysis results indicate that the maximum LOD score achievable with this pedigree under this model is 5.1 at recombination fraction (θ) .01. The maximum LOD score obtained in this family is 4.5 at θ = .0, for marker D18S39, a result that is highly significant. Table 1 shows the LOD scores obtained with the most informative markers.
Table 1.
LOD-Score Analysis, for the Most-Informative Markers, for ALS Pedigree Fr017
|
LOD Score at θ = |
|||||||
| Markera (Position [cM]) | .000 | .010 | .050 | .100 | .200 | .300 | .400 |
| D18S846 (77.36) | −6.460 | −1.447 | −.685 | −.354 | −.078 | .015 | .027 |
| D18S39 (77.36) |
4.523 | 4.448 | 4.141 | 3.737 | 2.861 | 1.892 | .861 |
| D18S35 (77.36) |
−.223 | −.205 | −.148 | −.102 | −.059 | −.046 | −.033 |
| D18S858 (80.41) |
3.267 | 3.206 | 2.956 | 2.631 | 1.933 | 1.182 | .435 |
| D18S1103 (83.46) |
1.822 | 1.816 | 1.756 | 1.627 | 1.261 | .819 | .355 |
| D18S64 (84.80) |
1.705 | 1.702 | 1.653 | 1.537 | 1.184 | .738 | .280 |
| D18S1109 (84.80) | −α | −1.395 | −.137 | .283 | .498 | .429 | .233 |
Markers within the disease haplotype are underlined.
Once linkage was confirmed, a disease haplotype was established on the basis of fine-mapping data and was inferred when possible, while a minimum number of recombination events was maintained (fig. 1). Key recombinants in two affected individuals (IV:37 and IV:67) define the smallest common inherited segment. The proximal recombination occurs between markers D18S846 and D18S39 in IV:37. The distal recombination in individual IV:67 is between markers D18S64 and D18S1109. All affected individuals carry the disease-associated haplotype, and, although there are no affected members without the haplotype, it is present in some unaffected members, which probably reflects either reduced or age-dependent penetrance. Extensive genotyping in the region did not reveal increased homozygosity or any other evidence for a large deletion.
Thus, at present, the flanking markers are D18S846 and D18S1109 and span an 7.5-cM, 8-Mb candidate region, which, according to current data, contains ∼50 genes, of which 13 are known and the remainder are predicted (UCSC Human Genome Project Working Draft database). Genes to be screened will be selected and prioritized on the basis of their (a) expression patterns and (b) function and possible involvement in ALS pathogenesis. At present, on the basis of its functional similarity to SOD, the most obvious candidate gene is the thioredoxin-like (TXNL) gene (GenBank accession number NM_004786; MIM 603049). The product of this gene is a small disulfide-containing redox protein and is expressed in all tissues examined, including brain and CNS. Screening of this gene by denaturing high-performance liquid-chromatography analysis is in progress.
We have identified a novel FALS locus on chromosome 18 in a large autosomal dominant adult-onset ALS pedigree, thus representing the most common form of FALS. The disease-gene interval has been mapped to a 7.5-cM region flanked by markers D18S846 and D18S1109. The aim of our future research will be to identify the causative FALS gene in this pedigree, which will have direct consequences in the field of motor-neuron–disease research and treatment.
Another gene involved in ALS pathology may provide crucial information about the biochemical pathways involved, aid in diagnosis, and help to identify those at high risk for the disease. The identification of a novel FALS gene will have profound implications for patients with ALS and for research and will aid in the development of cellular and transgenic models of disease, models that may accurately mimic the neuronal death seen in all forms of ALS. This, in turn, may lead to more-effective therapy.
Acknowledgments
The authors would like to thank the family members for their cooperation, Mrs. Valerie Briolotti for technical assistance, and Dr. André Toulouse for providing helpful comments on the manuscript. We are grateful for the assistance of Drs. Tom Hudson, Andrei Verner, and Carl Brewer, of the McGill University Genome Centre. This work was supported by the Muscular Dystrophy Association (USA); the ALS Association (USA); the Association pour la Recherche sur la Sclerose Laterale Amyotrophique, France; and the Association Française contre les Myopathies, France. V.M.-P. is the recipient of a grant by Singer-Polignac Foundation. G.A.R. is supported by the Canadian Institutes for Health Research.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Center for Medical Genetics, Marshfield Medical Research Foundation, http://research.marshfieldclinic.org/genetics/ (for genetic maps)
- Cooperative Human Linkage Center, The, http://lpg.nci.nih.gov/CHLC/
- Fondation Jean Dausset CEPH, www.cephb.fr [Google Scholar]
- GenBank Overview, http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for TXNL gene [accession number NM_004786])
- Genome Database, The, http://www.gdb.org
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for ALS1 [MIM 105400], ALS2 [MIM 205100], ALS4 [MIM 602433], ALS5 [MIM 602099], ALS FTD [MIM 105550], and TXNL gene [MIM 603049])
- UCSC Human Genome Project Working Draft, http://genome.ucsc.edu/
References
- Al Chalabi A, Leigh PN (2000) Recent advances in amyotrophic lateral sclerosis. Curr Opin Neurol 13:397–405 [DOI] [PubMed] [Google Scholar]
- Anderson MA, Gusella JF (1984) Use of cyclosporin A in establishing Epstein-Barr virus-transformed human lymphoblastoid cell lines. In Vitro 20:856–858 [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante R, Klivenyi P, Klein AM, Shinobu LA, Epstin CJ, Beal MF (2000) Partial deficiency of manganese superoxide dismutase exacerbates a transgenic mouse model of amyotrophic lateral sclerosis. Ann Neurol 47:447–455 [PubMed] [Google Scholar]
- Bedlack RS, Strittmatter WJ, Morgenlander JC (2000) Apolipoprotein E and neuromuscular disease: a critical review of the literature. Arch Neurol 57:1561–1565 [DOI] [PubMed] [Google Scholar]
- Boukaftane Y, Khoris J, Moulard B, Salachas F, Meininger V, Malafosse A, Camu W, Rouleau GA (1998) Identification of six novel SOD1 gene mutations in familial amyotrophic lateral sclerosis. Can J Neurol Sci 25:192–196 [DOI] [PubMed] [Google Scholar]
- Brown RH Jr (1995) Amyotrophic lateral sclerosis: recent insights from genetics and transgenic mice. Cell 80:687–692 [DOI] [PubMed] [Google Scholar]
- Camu W, Khoris J, Moulard B, Salachas F, Briolotti V, Rouleau GA, Meininger V (1999) Genetics of familial ALS and consequences for diagnosis. French ALS Research Group. J Neurol Sci 165 Suppl 1:S21–S26 [DOI] [PubMed] [Google Scholar]
- Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, Griffin JW, Cornblath DR (1998) Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet 62:633–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottingham RW Jr, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed] [Google Scholar]
- de Belleroche J, Orrell R, King A (1995) Familial amyotrophic lateral sclerosis/motor neurone disease (FALS): a review of current developments. J Med Genet 32:841–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flowers JM, Leigh PN, Davies AM, Ninkina NN, Buchman VL, Vaughan J, Wood NW, Powell JF (1999) Mutations in the gene encoding human persyn are not associated with amyotrophic lateral sclerosis or familial Parkinson's disease. Neurosci Lett 274:21–24 [DOI] [PubMed] [Google Scholar]
- Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown RH Jr, Scherer SW, Rouleau GA, Hayden MR, Ikeda JE (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29:166–173 [DOI] [PubMed] [Google Scholar]
- Haverkamp LJ, Appel V, Appel SH (1995) Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 118:707–719 [DOI] [PubMed] [Google Scholar]
- Hayward C, Colville S, Swingler RJ, Brock DJH (1999) Molecular genetic analysis of the APEX nuclease gene in amyotrophic lateral sclerosis. Neurology 52:1899–1901 [DOI] [PubMed] [Google Scholar]
- Hentati A, Bejaoui K, Pericak-Vance MA, Hentati F, Speer MC, Hung WY, Figlewicz DA, Haines J, Rimmler J, Ben Hamida C, Ben Hamida M, Brown RH Jr, Siddique T (1994) Linkage of recessive familial amyotrophic lateral sclerosis to chromosome 2q33-q35. Nat Genet 7:425–428 [DOI] [PubMed] [Google Scholar]
- Hentati A, Ouahchi K, Pericak-Vance MA, Nijhawan D, Ahmed A, Yang Y, Rimmler J, Hung W-Y, Schlotter B, Ahmed A, Ben Hamida M, Hentati F, Siddique T (1998) Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15q15-q22 markers. Neurogenetics 2:55–60 [DOI] [PubMed] [Google Scholar]
- Hosler BA, Siddique T, Sapp PC, Sailor W, Huang MC, Hossain A, Daube JR, Nance M, Fan C, Kaplan J, Hung W-Y, McKenna-Yasek D, Haines JL, Pericak-Vance MA, Horvitz HR, Brown RH Jr (2000) Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-q22. JAMA 284:1664–1669 [DOI] [PubMed] [Google Scholar]
- Hudson AJ (1981) Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain 104:217–247 [DOI] [PubMed] [Google Scholar]
- Julien J-P, Couillard-Després S, Meier J (1998) Transgenic mice in the study of ALS: the role of neurofilaments. Brain Pathol 8:759–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A, Houlden H, Hardy J, Lane R, Chancellor A, de Belleroche J (1993) Absence of linkage between chromosome 21 loci and familial amyotrophic lateral sclerosis. J Med Genet 30:318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunst CB, Messer L, Gordon J, Haines J, Patterson D (2000) Genetic mapping of a mouse modifier gene that can prevent ALS onset. Genomics 70:181–189 [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ (1995) Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11:376–381 [DOI] [PubMed] [Google Scholar]
- Lynch T, Sano M, Marder KS, Bell KL, Foster NL, Defendini RF, Sima AA, et al (1994) Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology 44:1878–1884 [DOI] [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang T-T, Miziorko H, Epstein CJ, Wallace DC (1999) Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA 96:846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Schneide JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC (1998) A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet 18:159–163 [DOI] [PubMed] [Google Scholar]
- Orrell RW, Habgood JJ, de Belleroche JS, Lane RJ (1997) The relationship of spinal muscular atrophy to motor neuron disease: investigation of SMN and NAIP gene deletions in sporadic and familial ALS. J Neurol Sci 145:55–61 [DOI] [PubMed] [Google Scholar]
- Ott J (1989) Computer-simulation methods in human linkage analysis. Proc Natl Acad Sci USA 86:4175–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parboosingh JS, Meininger V, McKenna-Yasek D, Brown RH Jr, Rouleau GA (1999) Deletions causing spinal muscular atrophy do not predispose to amyotrophic lateral sclerosis. Arch Neurol 56:710–712 [DOI] [PubMed] [Google Scholar]
- Rabin BA, Griffin JW, Crain B, Scavina M, Chance PF, Cornblath DR (1999) Autosomal dominant juvenile amyotrophic lateral sclerosis. Brain 122:1539–1550 [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62 [DOI] [PubMed] [Google Scholar]
- Siddique T, Deng HX (1996) Genetics of amyotrophic lateral sclerosis. Hum Mol Genet 5:1465–1470 [DOI] [PubMed] [Google Scholar]
- Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, Sapp P, Hung W-Y, Bebout J, McKenna-Yasek D, Deng G, Horvitz HR, Gusella JF, Brown RH Jr, Roses AD (1991) Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med 324:1381–1384 [DOI] [PubMed] [Google Scholar]
- Siddique T, Hong S, Brooks BR, Hung W-Y, Siddique NA, Rimmler J, Karplan JP, Haines JL, Brown RH Jr, Pericak-Vance MA (1998) X-linked dominant ALS. Paper presented at the annual meeting of the American Academy of Neurology, Minneapolis, April 27–May 2 [Google Scholar]
- Smith RG, Appel SH (1995) Molecular approaches to amyotrophic lateral sclerosis. Annu Rev Med 46:133–145 [DOI] [PubMed] [Google Scholar]
- Weeks DE, Ott J, Lathrop GM (1990) SLINK: a general simulation program for linkage analysis. Am J Hum Genet Suppl 47:A204 [Google Scholar]